





This study investigated whether Helicobacter pylori could activate the nucleotide-binding oligomerization domain-like receptor (NLR) family, pyrin domain-containing 3 (NLRP3) inflammasome in human macrophages and the involvement of reactive oxygen species (ROS) in inflammasome activation. Phorbol-12-myristate-13-acetate (PMA)-differentiated human acute monocytic leukemia cell line THP-1 was infected with H. pylori. The levels of pro-inflammatory cytokines interleukin (IL)-1β and IL-18 in supernatant were measured by ELISA. Intracellular ROS level was analyzed by flow cytometry. Quantitative real-time PCR and western blot analysis were employed to determine the mRNA and protein expression levels of NLRP3 and caspase-1 in THP-1 cells, respectively. Our results showed that H. pylori infection could induce IL-1β and IL-18 production in PMA-differentiated THP-1 cells in a dose- and time-dependent manner. Moreover, secretion of IL-1β and IL-18 in THP-1 cells following H. pylori infection was remarkably reduced by NLRP3-specific small interfering RNA treatment. In addition, the intracellular ROS level was elevated by H. pylori infection, which could be eliminated by the ROS scavenger N-acetylcysteine (NAC). Furthermore, NAC treatment could inhibit NLRP3 inflammasome formation and caspase-1 activation and suppress the release of IL-1β and IL-18 from H. pylori-infected THP-1 cells. These findings provide novel insights into the innate immune response against H. pylori infection, which could potentially be used for the prevention and treatment of H. pylori-related diseases.
INTRODUCTION
Helicobacter pylori infection causes chronic gastritis and has been recognized as a risk factor for the development of peptic ulcers, gastric cancer and gastric mucosa-associated lymphoid tissue lymphoma (IARC 1994; Peek and Blaser 2002). Results from animal models and clinical studies have demonstrated that pathogenesis of H. pylori is associated with enhanced expression of pro-inflammatory cytokines, including interleukin (IL)-1β and IL-18 (Troost et al., 2003; Lamb and Chen 2013). Studies have found that IL-1β is secreted in response to H. pylori infection in human dendritic cells (DCs), macrophages and monocytes (Basak et al., 2005; Fehlings et al., 2012; Hocès de la Guardia et al., 2013). Moreover, in H. pylori-stimulated DCs and an IL-1β-null mouse model, excessive IL-1β was shown to play an important role in H. pylori-associated gastric carcinogenesis (Mitchell et al., 2012; Shigematsu et al., 2013). Previously, increased IL-18 expression was found in H. pylori-infected human gastric mucosa and gastric epithelial cells in vitro and therefore might represent a novel therapeutic target (Okamura et al., 1995; Micallef et al., 1996; Ushio et al., 1996; Tomita et al., 2001; Bombardieri, McInnes and Pitzalis 2007; Jelusic, Lukic and Batinic 2007; Shimada et al., 2008; Yamauchi et al.2008).
In innate immune responses, pathogen-associated molecular patterns are recognized by pattern recognition receptors (PRRs) in hosts (Janeway and Medzhitov 2002), including Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (Franchi et al., 2009; Takeuchi and Akira 2010). NLRs, such as NOD1/2, could sense the presence of microbial components in cytosol and drive the activation of downstream target genes (Kanneganti, Lamkanfi and Núñez 2007; Kawai and Akira 2010). Other NLRs, including NLRC4, NLRP1 and NLRP3, are involved in the assembly of the inflammasome complex, resulting in caspase-1 activation and pro-inflammatory cytokine maturation (Martinon, Burns and Tschopp 2002; Schroder and Tschopp 2010; Franchi, Muñoz-Planillo and Núñez 2012). NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is abundantly expressed in macrophages, which can be activated by reactive oxygen species (ROS) (Khare et al., 2010; Jin and Flavell 2010; Leemans, Cassel and Sutterwala 2011). On the other hand, previous studies have shown that H. pylori infection exerts similar function in the aspect of caspase-1 activation and cytokine maturation (Basak et al., 2005; Hitzler et al., 2012; Kim et al., 2013). However, little is known about the role of inflammasome activation in H. pylori infection and the involved signaling pathways.
In this study, we demonstrated that H. pylori potently activated NLRP3-dependent signaling pathways to elicit IL-1β and IL-18 secretion in human acute monocytic leukemia THP-1 cells and that ROS production was required for the activation of the NLRP3 inflammasome. Our findings elucidate the molecular mechanism in inflammatory responses against H. pylori infection and might contribute to the understanding and treatment of H. pylori-related diseases.
MATERIALS AND METHODS
Cell line and cell culture
Human acute monocytic leukemia cell line THP-1 (China Center for Type Culture Collection, Wuhan University, Wuhan, Hubei, China) was cultured in RPMI 1640 medium (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS; Hyclone), 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin in a 5% CO2, 37°C humidified incubator. To induce differentiation, THP-1 cells were seeded onto 24-well plates (Costar; Corning, Corning, NY, USA) at a density of 4 × 105 cells well−1 and stimulated with 50 nM phorbol-12-myristate-13-acetate (PMA; Sigma, St Louis, MO, USA) for 48 h (Park et al., 2007).
H. pylori strain and infection
H. pylori reference strain 26695 (expressing the s1/m1 form of VacA; GenBank accession number NC_000915) was routinely grown on Columbia agar plates containing 5% defibrinated sheep blood (Oxoid, Basingstoke, UK) at 37°C for 48–72 h under microaerobic conditions (5% O2, 10% CO2 and 85% N2). Bacteria were suspended in PBS and the number of bacteria per milliliter was determined spectrophotometrically (OD600 = 1 × 108 CFU ml−1).
For the infection, H. pylori were suspended in antibiotic-free RPMI 1640 containing 10% FBS. PMA-differentiated THP-1 cells in the exponential growth phase were seeded onto 6-well plates at a density of 1 × 106 cells well−1 and incubated in 1% FBS overnight. After washing with PBS, cells were inoculated with H. pylori at a multiplicity of infection (MOI) of 100 (based on trial results with an MOI series from 25 to 200) for another 12 h. PMA-differentiated THP-1 cells cultured in RPMI 1640 (containing 10% FBS) and stimulated with 100 ng ml−1 lipopolysaccharide (LPS) from Escherichia coli O55:B5 (Sigma) were used as the negative and positive controls, respectively. At indicated time points (3, 6, 12 and 24 h post-infection), extracellular bacteria were killed by gentamicin. Cell supernatants were assayed for cytokine secretion by ELISA and cells were collected and subjected to intracellular ROS measurement, real-time PCR and western blot analysis.
Small interfering RNA silencing
Cells were transfected with control small interfering RNA (siRNA) or NLRP3-specific siRNA (100 nM/106 cells) using Lipofectamine 2000 (Invitrogen). Control non-target siRNA (sense UUCUCCGAACGUGUCACGUtt; antisense ACGUGACACGUUCGGAGGAGAAtt) and siRNA targeting NLRP3 (sense GGUGUUGGAAUUAGACAAC; antisense GUUGUCUAAUUCCAACACC) (Eitel et al., 2012) were synthesized by RiboBio (Guangzhou, Guangdong, China). At 24 h post-transfection, cells were infected with H. pylori as described above.
Cytokine secretion detection
Secreted IL-1β and IL-18 in culture supernatants were quantified using ELISA kits (NeoBioscience, Shenzhen, Guangdong, China) according to the manufacturer's instructions. Absorbance at 450 nm was read by a microplate reader (Bio-Rad, Hercules, CA, USA). The cytokine concentration was determined according to a standard curve set up with the recombinant cytokines from the kits. Experiments were performed in triplicate.
Intracellular ROS measurement
The intracellular ROS level was measured by flow cytometry using the probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Sigma). Cells were seeded onto 6-well plates and infected with H. pylori. At 3, 6, 12 and 24 h post-infection, cells were treated with 10 μM DCFH-DA at 37°C for 30 min. After washing, the fluorescence was examined by flow cytometry. For the inhibition assay, cells were pre-incubated with 25 mM N-acetylcysteine (NAC; BioXtra grade), a general ROS scavenger, for 30 min and then subjected to H. pylori infection. Three independent experiments were performed and the averaged results were obtained.
Quantitative real-time PCR
RNA was extracted using the RNeasy Mini kit (Qiagen, Düsseldorf, Germany). Reverse transcription was performed with a reverse transcription system (Promega, Madison, WI, USA). Real-time PCR was performed using SYBR Green PCR Master Mix (Promega) according to the manufacturer's instructions on the ABI 7300 instrument. The PCR conditions were as follows: 95°C for 10 min; 95°C for 15 s, 60°C for 1 min, for 40 cycles. The primer sequences were as follows: human NLRP3, forward 5′-AACAGCCACCTCACTTCCAG-3′ and reverse 5′-CCAACCACAATCTCCGAATG-3′; human caspase-1, forward 5′-GCACAAGACCTCTGACAGCA-3′ and reverse 5′-TTGGGCAGTTCTTGGTATTC-3′; and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward 5′-ATGGCCTTCCGTGTTCCTAC-3′ and reverse 5′-GCTTCACCACCTTCTTGATGTC-3′. The mRNA expression level was determined using ABI software (Applied Biosystems, Grand Island, NY, USA) and normalized to GAPDH. Experiments were performed in triplicate.
Western blot analysis
Samples were lysed using RIPA lysis buffer (Pierce, Rockford, IL, USA) and the protein concentrations were determined by the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins were separated on 15% SDS–PAGE and then transferred onto a polyvinylidene difluoride membrane (Millipore, Milford, MA, USA). The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline with Tween 20 at room temperature for 1 h. To detect the active caspase-1 subunit (p10), the blot was incubated with rabbit anti-human anti-caspase-1 polyclonal antibody (1:1000 dilution; Pierce) or anti-β-actin polyclonal antibody (1:10 000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and subsequently incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5000 dilution; Invitrogen). For NLRP3 detection, 9% SDS–PAGE was performed and the blot was incubated with rabbit anti-human NLRP3 antibody (1:200 dilution; Atlas Antibodies, Stockholm, Sweden) for 1 h. Sample bands were visualized by the enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ, USA). Three independent experiments were performed and representative data are shown.
Statistical analysis
Data were expressed as the mean ± SD. Statistical analysis was performed using SPSS version 18.0 (SPSS, Chicago, IL, USA). Repeated measures analysis of variance (ANOVA) was used for the comparison. P < 0.05 was considered statistically significant.
RESULTS
H. pylori enhances the production of IL-1β and IL-18 in THP-1 cells
To investigate the effects of H. pylori infection on the IL-1β and IL-18 secretion of THP-1 cells, ELISA was performed. PMA-differentiated THP-1 cells were infected with H. pylori at MOI series from 25 to 200. The maximum response was observed at 100 MOI, which was therefore chosen for the following experiments. These cells were infected with H. pylori at 100 MOI for 0, 3, 6, 12 and 24 h, respectively. Results from ELISA showed that, compared with the negative control group, the secretion of IL-1β and IL-18 by THP-1 cells was markedly increased after H. pylori infection, in a time-dependent manner (Fig. 1) (P < 0.01). Notably, THP-1 cells infected with H. pylori produced significantly more IL-1β and IL-18 than cells stimulated with LPS. These results suggest that H. pylori infection induces an inflammatory response in THP-1 cells in a dose- and time-dependent manner.
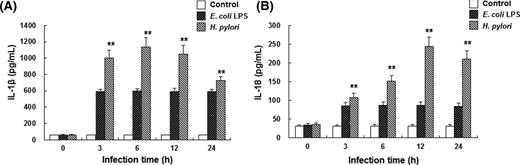
H. pylori enhances the production of IL-1β and IL-18 in THP-1 cells. PMA-differentiated THP-1 cells were infected with H. pylori at 100 MOI for 3, 6, 12 and 24 h, respectively. Cell culture supernatant was analyzed for human IL-1β (A) and IL-18 (B) secretion by ELISA (n = 3). PMA-differentiated THP-1 cells cultured with RPMI 1640 medium and stimulated with 100 ng ml−1 LPS from E. coli were used as the negative and positive controls, respectively. Compared with negative control group, **P < 0.01.
NLRP3 inflammasome activation mediates the pro-inflammatory cytokine elevation in H. pylori-infected THP-1 cells
To investigate the mechanism through which pro-inflammatory cytokines were elevated by H. pylori infection in THP-1 cells, the expression levels of inflammasome-related components, NLRP3 and caspase-1, were detected by quantitative real-time PCR and western blot analysis. Results from real-time PCR showed that mRNA expression levels of NLRP3 and caspase-1 were enhanced in THP-1 cells infected with H. pylori (Fig. 2A and B) (P < 0.01). Their mRNA levels peaked at 6 h post-infection, increased by 4.8- and 5.0-fold, respectively, compared with the control group. Upon inflammasome assembly, pro-caspase-1 undergoes autocleavage and activation to obtain 20 kDa (p20) and 10 kDa (p10) subunits. Western blot analysis showed that caspase-1 p10 was observed at 3 h after H. pylori infection, suggesting inflammasome activation in these cells (Fig. 2C).
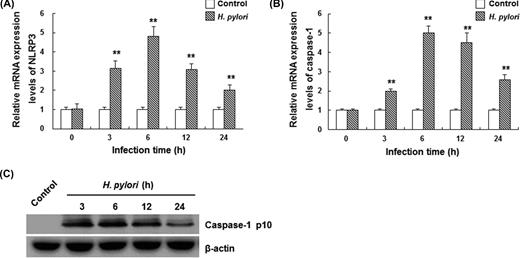
H. pylori infection promotes NLRP3 and caspase-1 activations in THP-1 cells. PMA-differentiated THP-1 cells were infected with H. pylori at 100 MOI for 3, 6, 12 and 24 h, respectively. (A and B) The mRNA expression levels of NLRP3 (A) and caspase-1 (B) were detected by quantitative real-time PCR (n = 3). Data were normalized to GAPDH. Compared with the control group, **P < 0.01. (C) Western blot analysis of mature (p10) form of caspase-1. One representative blot from three independent experiments is shown.
To further investigate the necessity of NLRP3 in enhanced cytokine secretion, NLRP3 expression was silenced by RNA interference in THP-1 cells. Compared with wild-type THP-1 cells or cells treated with negative control siRNA, NLRP3-specific siRNA led to a significant reduction in NLRP3 mRNA and protein expression levels (Fig. 3A and B) (P < 0.01). When these cells were infected with H. pylori for 12 h, the secretion of IL-1β and IL-18 was also significantly declined, indicating that NLRP3 was necessary for the enhanced IL-1β and IL-18 secretion after H. pylori infection (Fig. 3C and D). These results suggest that the elevated secretion of pro-inflammatory cytokines in H. pylori-infected THP-1 cells is mediated by NLRP3 inflammasome activation.
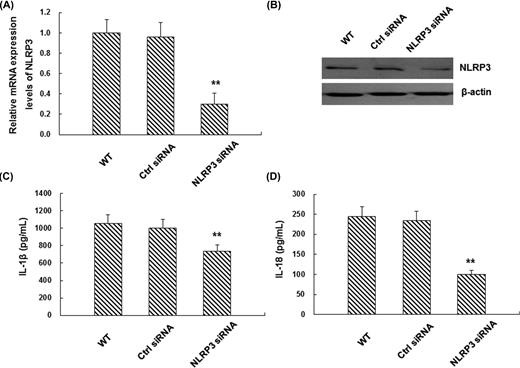
NLRP3 is necessary for H. pylori-induced IL-1β and IL-18 production in THP-1 cells. (A and B) THP-1 cells were transfected with non-target control siRNA (ctrl) or NLRP3-specific siRNA. The mRNA (A) and protein (B) expression levels of NLRP3 were detected by real-time PCR (n = 3) and western blot analysis (one representative blot from three independent experiments is shown), respectively. (C and D) 24 h later, siRNA-transfected THP-1 cells were infected with H. pylori at 100 MOI for another 12 h and the levels of IL-1β (C) and IL-18 (D) were measured by ELISA (n = 3). Compared with the wild-type (WT) control group, **P < 0.01.
Intracellular ROS generation is essential for H. pylori-induced inflammasome activation in THP-1 cells
ROS is recognized as one of the triggers of NLRP3 inflammasome activation under physiopathological circumstances. Therefore, the involvement of intracellular ROS generation was detected with DCFH-DA in THP-1 cells infected with H. pylori. Our results demonstrated that H. pylori treatment led to a significant increase in the intracellular ROS level, compared with the control group (Fig. 4A) (P < 0.01). The intracellular ROS level began to increase at 3 h and peaked at 6 h after H. pylori infection. However, when these cells were treated with 25 mM NAC (ROS scavenger) for 30 min, the H. pylori-induced elevation in the mRNA expression levels of NLRP3 and caspase-1 in THP-1 cells was obviously eliminated (Fig. 4B and C) (P < 0.05). Furthermore, H. pylori-induced caspase-1 activation as well as increased IL-1β and IL-18 secretion were inhibited by NAC treatment (Fig. 4D–F). These results suggest that intracellular ROS plays an important role in H. pylori-induced inflammasome activation and pro-inflammatory cytokine elevation in THP-1 cells.
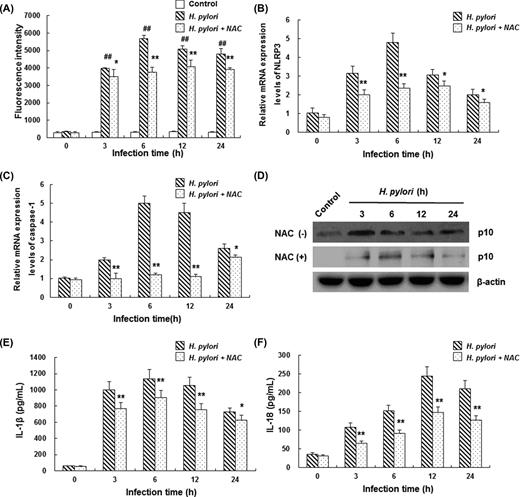
Intracellular ROS generation is essential for H. pylori-induced inflammasome activation in THP-1 cells. (A) PMA-differentiated THP-1 cells were infected with H. pylori at 100 MOI for 3, 6, 12 and 24 h, respectively. Then, the intracellular ROS level was detected by flow cytometry with DCFH-DA. In the H. pylori + NAC group, PMA-differentiated THP-1 cells were pre-treated with 25 mM NAC for 30 min and subsequently infected with H. pylori. Mean fluorescence intensity values indicate ROS levels in THP-1 cells (n = 3). (B–F) PMA-differentiated THP-1 cells were pre-treated with 25 mM NAC for 30 min and subsequently infected with H. pylori at 100 MOI. (B and C) The mRNA expression levels of NLRP3 (B) and caspase-1 (C) were determined by real-time PCR (n = 3). (D) Caspase-1 activation in THP-1 cells was detected by western blot analysis (images were acquired under equal exposure times and one representative blot is shown). (E and F) Secretion of IL-1β (E) and IL-18 (F) in cell culture supernatant was detected by ELISA (n = 3). Compared with the control group, ##P < 0.01; compared with H. pylori infection alone, *P < 0.05, **P < 0.01.
DISCUSSION
Pro-inflammatory cytokines play important roles in the pathogenesis of H. pylori infection and H. pylori-associated complications, including cancers (Lamb and Chen 2013). Mucosal levels of pro-inflammatory cytokines (IL-1β, IL-6 and tumor necrosis factor-α) have been shown to be significantly elevated in H. pylori-positive gastric specimens (Lindholm et al., 1998; McGee and Mobley 2000). IL-1β and IL-18 increase the secretion of other cytokines and regulate the expression levels of adhesion molecules in inflammatory cells (Okamura et al., 1995; Ushio et al., 1996; Dinarello 2005). Moreover, previous studies have demonstrated that H. pylori induces IL-1β and IL-18 production in DCs, macrophages and monocytes (Basak et al., 2005; Yamauchi et al., 2008; Fehlings et al., 2012). However, the mechanism through which IL-1β and IL-18 are produced in response to H. pylori infection remains poorly understood.
The inflammasome mediates the rapid response to pathogens through TLR pathways (Ogura, Sutterwala and Flavell 2006; Franchi et al., 2008; Lamkanfi and Dixit 2011). In fact, processing and secretion of IL-1β and IL-18 depend on the activity of caspase-1, which is activated by the inflammasome (Martinon, Burns and Tschopp 2002; Schroder and Tschopp 2010; Leemans, Cassel and Sutterwala 2011; Franchi, Muñoz-Planillo and Núñez 2012). Two signals are required in the activation of caspase-1 by the NLRP3 inflammasome. The priming signal involves the stimulation of PRRs by microbial molecules or endogenous cytokines, which is required for the up-regulation of NLRP3 and cytokine precursors. The second signal could be induced by ATP, certain bacterial toxins or particulate matter, leading to activation of the NLRP3 inflammasome (Martinon, Burns and Tschopp 2002; Schroder and Tschopp 2010; Leemans, Cassel and Sutterwala 2011; Franchi, Muñoz-Planillo and Núñez 2012). H. pylori possesses several virulence factors that are associated with antigen presentation and bacterial recognition (Pachathundikandi, Tegtmeyer and Backert 2013). Recent studies suggest that the host innate immune receptors TLR2 and NOD2 provide the priming signal through the induction of NLRP3 and pro-IL-1β processing in H. pylori-infected DCs (Kim et al., 2013), which is consistent with the fact that these PRRs can induce gene expression via nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) activation (Kawai and Akira 2010). However, the second signal required for inflammasome activation remains elusive.
Monocyte-derived cells, including macrophages and DCs, play critical roles in the innate immune response to pathogens. The NLRP3 inflammasome complex has been implicated in inflammatory responses to numerous pathogens in these cells (Ogura, Sutterwala and Flavell 2006; Franchi et al., 2008; Duncan et al., 2009; Gross et al., 2009; Abdul-Sater et al., 2010; Lamkanfi and Dixit 2011; Negash et al., 2013). Recent studies show that H. pylori infection activates caspase-1 via NLRP3 inflammasome activation and induces mature IL-1β secretion in mouse DCs (Kim et al., 2013). In the present study, THP-1 cells were infected with H. pylori and the involvement of the NLRP3 inflammasome complex in these cells was investigated. Our results showed that NLRP3 and caspase-1 mRNA expression levels were significantly elevated upon H. pylori infection, along with the enhanced secretion of IL-1β and IL-18. The activation of caspase-1 following H. pylori infection was further confirmed by the detection of the 10 kDa subunit. These results suggest that H. pylori infection activates the NLRP3 inflammasome activation in vitro, which is in agreement with previous findings (Kim et al., 2013; Hitzler et al., 2012).
Most NLRP3 inflammasome activation pathways share common mechanisms, including potassium (K+) efflux, ROS production and cathepsin B release (Jin and Flavell 2010; Khare et al., 2010). Increasing evidence shows that H. pylori induces intracellular ROS generation in different human cell lines, causing injuries in hosts (Ding et al., 2007; Luo et al., 2013). However, to date, it is unclear whether ROS could act as the second signal in triggering inflammasome activation after H. pylori infection. Our results showed that H. pylori infection significantly enhanced intracellular ROS production in THP-1 cells and that the ROS scavenger NAC significantly suppressed H. pylori-induced up-regulation of inflammasome component transcripts, NLRP3 and caspase-1. Scavenging of ROS led to a reduction in cytokine secretion and caspase-1 activation following H. pylori infection, implying the direct involvement of ROS in NLRP3 inflammasome activation and/or related cytokine secretion. Taken together, these results demonstrate that NLRP3 inflammasome activation in H. pylori infection depends on a mechanism involving, at least partially, ROS production, in which ROS provides the second signal required for the inflammasome activation, in line with previously published findings (Semper et al., 2014). Of course, further studies are still needed to identify whether there are additional mechanisms that might be involved in H. pylori-induced NLRP3 inflammasome activation. Collectively, our data imply the involvement of the NLRP3 inflammasome in H. pylori-induced inflammatory responses. To further verify this, the expression of NLRP3, the crucial component of NLRP3 inflammasome, was silenced in THP-1 cells. NLRP3 silencing significantly inhibited H. pylori-induced IL-1β and IL-18 secretion in THP-1 cells, suggesting that the NLRP3 inflammasome mediates the pro-inflammatory cytokine elevation following H. pylori infection.
Although the NLRP3 inflammasome can be activated by various bacteria and their components (Kim et al., 2013; Kim and Jo, 2013), the role of inflammasome activation in H. pylori infection is largely unknown in the literature. Recently, Benoit et al., utilizing apoptosis-associated speck-like domain containing a caspase recruitment domain (ASC)-deficient mice, reported that loss of ASC protein could abolish the expression of IL-1β and IL-18 and drastically reduce interferon expression after H. pylori infection, resulting in attenuated inflammatory response and defected H. pylori clearance (Benoit et al., 2009). A later study provides evidence that the H. pylori type IV secretion system can induce NLRP3 inflammasome activation in host mice, regulating the innate immune function. This process requires cooperative interaction among host innate immune receptors TLR2, NOD2 and NLRP3 in regulating the activation of caspase-1 and IL-1β in DCs (Kim et al., 2013). However, Kim et al., found that NLRP3-deficient mice are not impaired in the clearance of H. pylori (Kim et al., 2013). In contrast, Semper et al., showed that mice lacking NLRP3 are colonized with H. pylori at higher levels compared with wild-type animals (Semper et al., 2014). Our in vitro data indicated that IL-1β and IL-18 secretion following H. pylori infection depended on the activation of the NLRP3 inflammasome. Further studies are still needed to elucidate the precise role of the NLRP3 inflammasome in H. pylori colonization and inflammation-associated pathology.
In summary, our results showed that H. pylori-induced elevation of IL-1β and IL-18 secretion required NLRP3 inflammasome activation in PMA-differentiated THP-1 cells. Activation of the NLRP3 inflammasome was mediated by intracellular ROS production. Our study provides novel insights into the innate immune response to H. pylori infection, which might involve ROS-induced inflammasome activation. Therefore, drugs targeting ROS production could potentially be used for the prevention and treatment of H. pylori-related diseases.
This work was supported by the Construct Program of the Key Discipline in Hunan Province, Hunan Provincial Key Laboratory for Special Pathogen Prevention and Control [Hunan Provincial Science & Technology Department document (approval number: 2014-5) and Hunan Provincial Education Department document (approval number: 2012-312)], the Key Project of Hunan Provincial Education Department (no. 11A103) and the Research Project of Hunan Provincial Science & Technology Department (no. 2013TT2021).
DISCLOSURES
All authors declare no financial competing interests.
All authors declare no non-financial competing interests.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}