





Abstract
DNA phylogenetic comparisons have shown that morphology-based species recognition often underestimates fungal diversity. Therefore, the need for accurate DNA sequence data, tied to both correct taxonomic names and clearly annotated specimen data, has never been greater. Furthermore, the growing number of molecular ecology and microbiome projects using high-throughput sequencing require fast and effective methods for en masse species assignments. In this article, we focus on selecting and re-annotating a set of marker reference sequences that represent each currently accepted order of Fungi. The particular focus is on sequences from the internal transcribed spacer region in the nuclear ribosomal cistron, derived from type specimens and/or ex-type cultures. Re-annotated and verified sequences were deposited in a curated public database at the National Center for Biotechnology Information (NCBI), namely the RefSeq Targeted Loci (RTL) database, and will be visible during routine sequence similarity searches with NR_prefixed accession numbers. A set of standards and protocols is proposed to improve the data quality of new sequences, and we suggest how type and other reference sequences can be used to improve identification of Fungi.
Database URL:http://www.ncbi.nlm.nih.gov/bioproject/PRJNA177353
Introduction
Fungi encompass a diverse group of organisms ranging from microscopic single-celled yeasts to macroscopic multicellular mushrooms. This implies that many of the challenges necessary to document fungal diversity overlap with those faced by researchers in other fields. Although yeast researchers share the challenges of other microbiologists to obtain viable cultures to study, macrofungal researchers often document species from dried specimens and face obstacles comparable with those of botanists. The majority of described fungal species still lacks any DNA sequence data, but it is also apparent that the vast majority of fungal diversity will have to be assessed solely by comparing DNA sequences, without accompanying cultures or physical specimens (1).
DNA sequence comparisons have demonstrated that many traditionally used phenotypic characters in Fungi are the result of convergent evolutionary processes and do not necessarily predict relatedness. Therefore, cryptic species continue to be discovered with phylogenetic methods even after examining well-studied species. Since the 19th century, it has also been accepted that fungi can occur in several morphological forms (morphs) arising from sexual, asexual or vegetative reproduction. Because these morphs often do not occur together in time and space, DNA characters greatly enhance the efficiency to confirm that separate morphs constitute a single species. This contributed to the declaration that different species names that have traditionally been applied to sexual and asexual morphs of the same fungal species are redundant (2). This redundancy is reflected in the most recent set of the rules guiding how fungal species are named, the International Code of Nomenclature for algae, fungi and plants (ICN) (3).
Improvements in how electronic data are disseminated also prompted changes in the ICN, namely, a requirement to register all new fungal taxonomic names at one or more online repositories. Recently, three candidates, Fungal Names, Index Fungorum or MycoBank were proposed (4). These databases provide an invaluable source of important information on vouchers that facilitate fungal identification. This, in turn, will aid the large-scale reassessment of taxonomic names required as part of the transition to use one name for each fungal species (5, 6). It will also improve the integration to a sequence-based classification (7).
For effective DNA-based identification to be implemented, the scientific community needs a continuously expanding, public and well-annotated set of DNA sequences. Each of these sequences needs to be associated with accurate specimen data and a current species name. Just as the current ICN addresses the requirements for a common nomenclature of species names, improved standards related to DNA sequences and specimens will improve the ability to communicate diversity effectively in ecological and microbiome studies. This infrastructure will provide the framework required to further our understanding of biology across all groups of Fungi.
Current state of sequence databases
GenBank, together with its collaborative partners in the International Nucleotide Sequence Databases Collaboration (INSDC), i.e. the DNA Data Bank of Japan and the European Nucleotide Archive (ENA) has long been the most comprehensive resource of nucleotide data (8). It is tasked with archiving the world’s genetic data as an open resource to all researchers. In spite of an extensive review of user submissions, GenBank essentially relies on users to accurately name their sequences. This results in a significant number of sequences deposited under erroneous or imprecise names, so-called ‘dark taxa’ (9, 10). This complicates efforts to clearly assign taxonomic names to unknowns. Mycologists have long been a vocal group in arguing for improving the accuracy of names used in GenBank (11, 12). In addition, biologists have expressed concern about the lack of associated voucher data in many GenBank entries (13). Although a specimen voucher qualifier has been available and promoted by GenBank since 1998 (14), this remains poorly used by submitters. To improve this, GenBank now recommends applying a version of the Darwin Core standards (15), which intends to facilitate the sharing of information about biological diversity through reference definitions (e.g. a standardized specimen voucher format) for relevant data. Where feasible, this will apply to any biorepository data shown in the ‘specimen_ voucher’, ‘culture_collection’ and ‘bio_material’ qualifiers of a GenBank sequence accession (14). This format also allows for vouchers to be linked directly from a sequence accession to a dedicated specimen or culture page at the relevant biorepository (where available), and it improves traceability across different databases.
A number of additional specialized databases focused on specific marker sequences have been built to further enhance sequence accuracy. Mycologists have used DNA sequence data for testing species-rank hypotheses for over 20 years. The internal transcribed spacer (ITS) region containing two spacers (ITS1 and ITS2) flanking the nuclear ribosomal 5.8S gene has been an especially popular marker (16). A curated ITS database focused on human and animal pathogenic fungi was established at www.mycologylab.org for the International Society of Human and Animal Mycology (ISHAM). Initially, the UNITE database (http://unite.ut.ee/) had a similar functional focus on ectomycorrhizal ITS sequences (17). Since then, it has expanded to provide tools for assessing sequence quality and Web-based third-party sequence annotation (PlutoF) to published sequences for all Fungi. The UNITE database now acts as a GenBank mirror for all fungal ITS sequences and has a particular focus on integrating sequences from environmental samples into reproducible taxonomic frameworks (18). Among other databases with similar aims, ITSoneDB focuses on ITS1 sequences (19), whereas the ITS2 database houses ITS2 sequences and their 2D structures (20). A number of additional publicly available online databases favor other sequence markers for fungal identification, e.g. the large and small nuclear ribosomal subunits (18S, 28S) and fragments from the translation elongation factor 1-alpha gene (21, 22). Several of these databases are focused on specific taxonomic groups (23–25). The DNA barcoding movement made an important impact on sequence accuracy by promoting a clear set of standards for DNA barcodes: raw sequence reads and reliable sequence data combined with a correct taxonomic name as well as collection and voucher information (26, 27). The Barcode of Life Data System (BOLD; 28) has a significant amount of sequence data that overlap with GenBank and was explicitly set up for DNA barcoding. The CBS-KNAW Fungal Biodiversity Centre, MycoBank and the recently launched BOLD mirror, EUBOLD, are also proposing online identification tools that can compare unknown sequences simultaneously against several reference databases.
Despite its long history of usage, mycologists have only recently proposed the ribosomal ITS region as a universal DNA barcode marker for Fungi (29). This means that regardless of several limitations (30), ITS will likely remain the main marker of choice for fungal identification in the immediate future. Since 2012, the ITS region has specifically been used for species identification in numerous DNA barcoding studies on a variety of fungal groups ranging from mucoralean fungi (31) to common molds such as Aspergillus and Penicillium (32). Broader-scale studies have evaluated the utility of generating fungal barcodes for a wide variety of fungal specimens (33–35). Extracted DNA can reliably be amplified by means of the polymerase chain reaction (PCR) and sequenced for most dried fungal specimens up to 30 years old. In several cases, much older specimens have been successfully sequenced (36–39) opening the possibilities of generating fungal barcodes from some legacy type specimens. The current age record for a fungal sequence from a type specimen stands at 220 years for a mushroom species, Hygrophorus cossus, collected in 1794 (40).
The use of multigene analysis has now become common in defining phylogenetic species boundaries within mycology. The standard for species delimitation in mycology remains the genealogical concordance species recognition concept first advocated by Taylor et al. (41). This relies on character comparisons from at least three unlinked loci. In comparison, the DNA barcode approach relies on less rigorous analysis techniques using universally sampled sequences from only one or rarely two markers. The focus in DNA barcoding is on obtaining limited sequence data from the largest possible number of specimens. It is most efficient in specimen identification, used in concert with a well-validated database containing accurate species delimitations (42). However, where information on species boundaries are lacking, it can also be used for initial species discovery. In line with this, it is our intention to maximize the accuracy of the sequences available for specimen identification and to emphasize where more sampling is required during species discovery.
Selecting reference sequences
DNA barcodes per definition, have to be backed up by publicly accessible raw data files (trace data), and, if they are linked to type material, have the potential to act as reference sequences that also provide a higher confidence in sequence accuracy. However, many other important sequences are already available in the public databases that would not be qualified as official barcodes. The need to communicate specific levels of confidence in sequence accuracy has yielded proposals for a quality scale in sequences (43), but establishment of such a system remains elusive. Since 1 January 1958, any validly published species name is connected to a type, which should be treated as primary reference. A type can principally be an original depiction of a species, though then it is good practice to designate a separate specimen as a neotype, or epitype where appropriate. However, in the majority of cases, a type will be a physical specimen. Type specimens are the only specimens to which one can reliably apply the original name, thereby providing a physical link to all other associated information. Having an ITS sequence or other marker sequences connected to a reliable publicly accessible representative of the species thus provides researchers with a reference point to a specific name. This makes it possible to unambiguously communicate findings with the research community and provide the opportunity to generate additional related data and expand current collections.
At GenBank, a particular challenge has always been the annotation of sequences derived from type material. Until recently, there was no standard means to specify type-related information during the process of data submission. Notes can be added to individual accessions, but they remain cumbersome to uncover in queries. In this article, we describe efforts to address this shortfall by expanding fields to indicate type material in the National Center for Biotechnology Information (NCBI) Taxonomy database. A number of ITS sequences were re-annotated and formatted in a separate curated database at NCBI, RefSeq Targeted Loci (RTL). This database was originally set up as a repository for bacterial type sequences obtained from the databases RDP, SILVA and Greengenes. It was subsequently expanded to Fungi—initially using a divergent set of sequences generated by the collaborative Assembling the Fungal Tree of Life project (AFToL) (44).
We release an initial reference sequence set of nearly 2600 ITS accessions covering ∼2500 species for inclusion in RTL. These records have been extensively verified with input from collaborators at Index Fungorum, MycoBank and UNITE, as well as a large group of taxonomic specialists. The existing set was chosen to represent most currently accepted orders (45) with eventual expansion to lower hierarchical taxa. It is intended that this new reference sequence set will continue to be widely used, adapted and expanded by the research community.
Materials and Methods
Verification steps for RefSeq data set:
Verification was done in the following order with each step building on the information of the previous step (Figure 1).
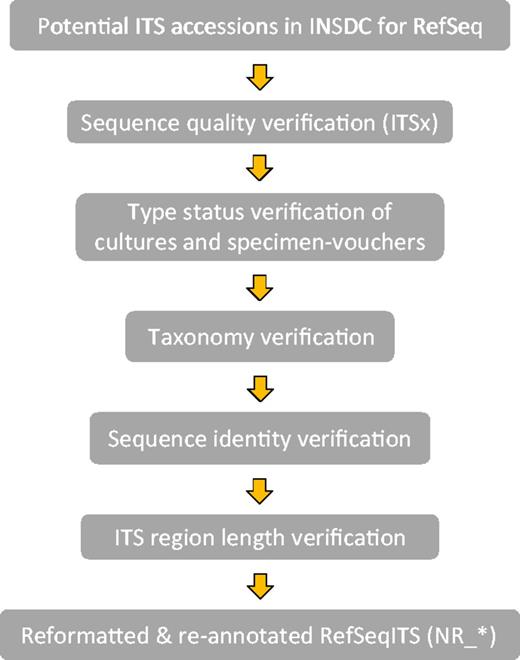
Workflow of the ITS verification for RTL ITS.
1) Collecting ITS records for evaluation. Lists of potential ITS accessions from type and verified specimens for display in RefSeq were generated in several ways via Entrez queries in the NCBI Nucleotide database, daily taxonomy curation and collaboration with experts in the fungal research community.
2) Sequence quality. All accessions were verified with the Perl script ITSx (46) to ensure sequence continuity including the ITS1, 5.8S and ITS2 regions. A record was excluded if it had an incomplete ITS2 region (as inferred by ITSx) and incomplete ITS1 region, which had no conserved CATTA-like motif at the 5′ end (within 40 bases of the end), and the length was <80% of the average complete ITS region (annotation as inferred by ITSx) for the taxonomic order to which it belonged. If there was no order defined then the class statistic was used, and if no class was defined then the statistic of the complete ITS region at kingdom rank (Fungi) was considered. In addition, sequences were also verified for non-ATGC characters [i.e. IUPAC DNA ambiguity symbols (47)], which often indicates poor quality. Their presence was limited to <0.5% of the ITS region. In some cases, exceptions to this rule and the length requirement were made for sequences representing underrepresented lineages.
3) Type material definitions. Type: The ICN defines a type as ‘that element to which the name of a taxon is permanently attached’ (Article 7.1). In addition, it states that types are not necessarily defined as the best representatives of the taxon (Article 7.2). We thus attempted to distinguish between the various types and annotate type status in the organism note field in each sequence record. We only considered one of the following types per species: holotype, isotype, lectotype, neotype, epitype, syntype and paratype. The holotype is a single specimen designated by the original author at the time of a species description. The other types indicate a variety of relationships to that specimen or can serve as replacements in certain circumstances (see glossary of the ICN for details: http://www.iapt-taxon.org/nomen/main.php?page=glo). Where we could not clearly distinguish the kind of type, these are annotated only as type. For the verification of type status, we relied mainly on the information at culture collection databases listed in Table 1 and the nomenclatural databases, MycoBank and Index Fungorum, as well as experts in the fungal research community. The main source of type status information was publications. Type status information can currently not be extracted from publications in a high-throughput manner, and the documents themselves are not always freely accessible, making curation efforts time consuming and heavily dependent on manual curation. Where possible, types tied to the original species description (protolog) of the currently accepted name were selected.
List of collection databases with specimen pages to which links were established from records in GenBank
Unique acronyms were taken from the GenBank collections database and, where possible, agree with labels used by Index Herbariorum, WFCC and GRBio
List of collection databases with specimen pages to which links were established from records in GenBank
Unique acronyms were taken from the GenBank collections database and, where possible, agree with labels used by Index Herbariorum, WFCC and GRBio
Ex-type: Living cultures do not have the formal nomenclatural status of a type specimen, but sequences obtained from cultures that were derived from type specimens were also indicated; where possible, it was indicated from what kind of type collection these originated. Details on such ex-type cultures and type specimens are both included under ‘type material’ in the NCBI Taxonomy database. Type identifiers in the NCBI Taxonomy database can include both heterotypic synonyms (also referred to as taxonomic or facultative synonyms) and homotypic synonyms (also referred to as nomenclatural or obligate synonyms). A simplified description of homotypic and heterotypic synonyms are indicated in Supplementary Material (Supplementary Figure S1).
Verified: This label was used to label placeholder sequences for important lineages in the fungal tree of life until sequences derived from type material are available. We relied on the advice from acknowledged taxonomic experts and input from large collaborative projects such as the AFToL project.
4) Current taxonomic name. ITS records from type specimens were selected only for current names where a single type applies, i.e. homotypic names. This means all associated obligate synonyms can effectively be traced to a single type specimen. Records associated with types from names that were synonymized subjectively were excluded where possible (heterotypic names). However when possible, we combined and annotated heterotypic types from asexual and sexual morphs (anamorphs and teleomorphs) from the same species in order to promote nomenclatural stability. An example is indicated in Supplementary Material (Supplementary Figure S1). The taxonomic names in current use were identified by consulting the latest publications, acknowledged taxonomic experts, culture collection databases as well as MycoBank and Index Fungorum. Where possible, a script using cURL (http://curl.haxx.se/) was used to extract type status and names from databases such as CBS and MycoBank.
5) Sequence identity. This is not the first attempt at verifying data in INSDC, and thus we relied on the data from the UNITE (version 5) and ISHAM databases to help verify sequences. Also, the sequence identity of selected INSDC records that could potentially be represented in RefSeq was compared with other sequences from type specimens. These were identified via type specimen identifiers obtained from MycoBank (compared with the isolate, strain, collection and specimen voucher fields) and from daily taxonomy curation. Finally, any type material data were uploaded as permanent name types in the NCBI Taxonomy database.
Sequence identity was considered accurate, and a sequence was considered to be associated with the type specimen if one of these conditions were met:
There was >99.5% identity over >90% of the ITS region in the potential RefSeq sequence compared with another type specimen sequence of the same TaxID in GenBank using megablast alignments. Instead of using 100% identity, we used 99.5% to accommodate for a small number of non-ATGC characters. Each sequence record is associated with one TaxID, and the TaxID represents one taxon that in NCBI taxonomy can accommodate several synonymous names (e.g. http://www.ncbi.nlm.nih.gov/Ta x onomy/Browser/ wwwtax. cgi ? mode= Info&id= 48 4 9 0 & lvl=3&lin=f&keep = 1& srchm o de = 1& unlock).
The same accession was in UNITE’s list of representative sequences (RepSs) or reference sequences (RefSs) with the same TaxID. Any synonymous taxon names used in UNITE were resolved, and the TaxID were identified with the name status tool in NCBI taxonomy (http://www.ncbi.nlm.nih.gov/Taxonomy/TaxIdentifier/tax_identifier.cgi).
There was >99.5% identity over >90% of the ITS region in the potential RefSeq sequence compared with RepS or RefS with the same TaxID from UNITE using megablast alignments.
Possible misidentifications/labeling of accessions (not verified above) were investigated for the following:
Sequences that were >99.5% identical over >90% of the ITS region to more than one type sequence identified in GenBank or RefS/RepS from UNITE of a different TaxID using megablast alignments.
Sequences that were <98.5% identical over >90% of the ITS region to RefS/RepS from UNITE of the same TaxID using megablast alignments.
Same accessions associated with different TaxIDs in GenBank and UNITE.
Further investigation was necessary if more than one ITS accession were available for a type, and one or more copies were <99.6% identical to the sequence selected for representation in RefSeq. This was done to ensure that the selected sequence was not the outlier in the group, which may be the result of low sequence quality or mislabeling. Sequence copies were aligned using MAFFT (http://www.ebi.ac.uk/Tools/msa/mafft/) and viewed in BioEdit (48) to determine which sequence contained the bases that are at odds with the rest. If only two sequence copies from the specimen were available to compare, then additional sequences from the same TaxID were aligned. If the uncertainty could not be resolved, then no sequence was selected for RefSeq.
Reformating of accessions for RefSeq
Each ITS record was re-annotated with ribosomal RNA (rRNA) and miscellaneous RNA (misc_RNA) features representing the boundaries of the rRNA and the ITSs as predicted by the ITSx Perl script. Lists of ITS records with metadata provided by experts were compared with metadata in the original GenBank submission. Source features were reformatted, corrected and augmented with information where needed. Culture collection specimens entered in the strain or isolate fields were moved to the culture collection field. Similarly, any herbarium specimen information was moved to the specimen voucher field. If the original GenBank submission contained no identifier from a collection in the NCBI Collections database, then the appropriate public collection identifier obtained from the original species description was added to the RefSeq record. Collection codes used in the culture collection and specimen voucher fields followed the acronym format used by GenBank indexing (the NCBI Collections database available at http://www.ncbi.nlm.nih.gov/projects/BioCollection/search_collection.cgi). Both these fields were formatted appropriately with the code separated with a colon from the collection’s correct identifier. If a dedicated specimen page was available online, with the collection identifier in the URL, direct links to the culture collection database could be made available when the correct format was used. Google searches for the presence of online databases of all collections associated with this data set were performed and specimen specific pages identified. The note field for each record was augmented with type status information, which included the type category (holotype, isotype, etc.) and the species name associated with the first description of the specimen.
Centrality analysis and clustering
A centrality analysis was performed with BioloMICS (from BioAware, Hannut, Belgium) to find the most central sequence to a given group, which is the sequence having the highest average similarity to other members of the group (49). Because sequences selected for RefSeq were limited to only one record per species (a few species with known internal variation had multiple records from one specimen), the group was not defined at species rank but at genus rank. The centrality analysis shows the diversity in a designated group.
A multidimensional cluster analysis was performed to visualize the distribution of the data. The distance between every pair of sequences was calculated based on similarity, and a distance matrix was created. Using the multidimensional scaling tool (BioloMICS) with the distance matrix, the data points were visualized in 3D and colored according to the classification rank specified.
Results
Sequence quality
Of a set of ∼3100 accessions considered for inclusion, we removed 16% for a variety of reasons, and currently, 2593 accessions were selected for RefSeq. The most commonly encountered problem was lack of sufficient and reliable metadata associated with a sequence record to confirm a type specimen in a timely manner. During the process of verifying sequence quality, some records were identified by ITSx as being problematic and were excluded, e.g. containing assembly chimeras, incomplete sequences (e.g. a missing 5.8S gene), sequences not from the ITS region or not of fungal origin. Figure 2 shows the length variation of accessions destined for RefSeq and with a complete ITS region (which accounted for ∼70% of the RefSeq ITS records). When the nuclear ribosomal 18S end or 28S start was within the first or last ∼25 bases of the sequence in the record, it became difficult for the ITSx script to identify it with confidence. This was due to the fact that the probability of a hidden Markov model score influenced by chance alone became much higher. Sequences with a CATTA motif within 40 bases of the 5′ end of a sequence but with an ITSx annotation of ‘ITS1 partial’ were considered complete. Some sequences contained more than one CATTA motif. These were compared with closely related sequences with a complete ITS region as defined by ITSx to confirm that these were complete for the ITS1 spacer. The boundary of the 18S was mostly (in 84% of the sequences with an 18S fragment) defined by the CATTA motif, although not all sequences contained this motif. Rather, variations of the CATTA motif were observed in some sequences, such as CATTC (e.g. in Mortierellaceae), CATTG (e.g. in Diaporthales), CACTA (e.g. in Cystofilobasidiales) or CAGTA (e.g. in Tremellales). The submitted sequence toward the end of the ITS2 spacer was frequently long enough to identify the 28S start with confidence, and no additional effort was made to identify conserved motifs within the last few bases. The majority (95% of 2593 sequences) of the RefSeq-selected ITS sequences had no undetermined bases, and the rest mostly had only one non-ATGC character, but none had more than four non-ATGC characters.
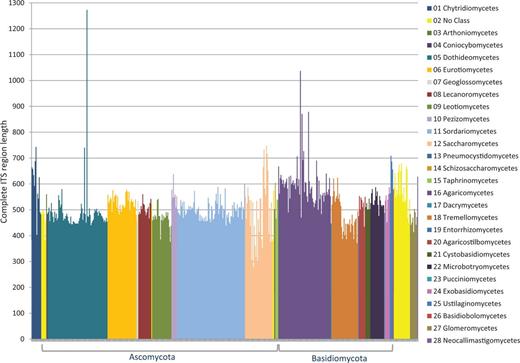
ITS length variation of complete ITS regions in the RTL data set according to class.
Type status
Metadata associated with accessions in lists provided by mycology experts were compared with source metadata of these accessions in GenBank at NCBI. Conflicting information (e.g. collection/specimen identifiers) was resolved by updating the GenBank record (if submitted to GenBank and the original submitter was involved) or the RefSeq record. When the correct information was not rapidly discernable, records were excluded from further analysis. Type status information of culture collection identifiers or specimen vouchers provided by the community was also compared with taxonomy/collection databases and publications. This curation process involved the research community and curators at culture/herbarium collections to resolve conflicting or missing information. Similar to the process in GenBank, culture/herbarium information from recently published research is not released publicly until collection curators receive notification or find the associated publication in the public domain. In addition, even though more and more culture/herbarium information is digitized, backlogs often exist. Thus, the absence of culture/herbarium numbers at an accessible online database does not necessarily imply a dead, contaminated or misidentified specimen. Only a small portion of the conflicting type specimen identifier information or absence was attributed to typographical errors or dead/contaminated cultures (14 identifiers). Where possible, we excluded types of heterotypic synonyms (see Supplementary Figure S1), as explained in following curation steps.
Current taxonomic name
Identifying homotypic synonyms involved collecting the name by which a type specimen was first described, the name with which this specimen is currently associated and finally the original name (basionym) of that current name. Names provided by experts in the research community were compared with those in GenBank records, as well as other collection and taxonomy databases. The original name of the type specimen had to be the same as the current name or basionym and, if not, it needed to be a homotypic synonym of the current name to be considered for inclusion in this RefSeq set. The majority of this current name information (92%) was easily accessible with a script from MycoBank, and ∼56% of the original names of type specimens were accessible from collection databases. However, any remaining information required a manual labor-intensive effort to obtain or verify names from publications or less accessible databases. This step in the curation process revealed many discrepancies between databases (and publications), which included orthographic variants, a need for taxonomic updates and spelling or labeling mistakes. Most discrepancies were resolved by addressing these issues at NCBI Taxonomy, external taxonomy and collection databases involving the respective curators. At publication time, 94% of RefSeq ITS records used the same current name that MycoBank or Index Fungorum used, and the rest used published names that were not public at both databases (1%) and different/not designated as current name at Index Fungorum (5%).
Sequence identity
Sequence identity of UNITE's curated list of RepSs or RefSs were compared with those in GenBank selected for RefSeq curation. The UNITE database uses a centrality test to verify sequence identity and evaluate curation. However, because of filtering steps at UNITE and newly described species with a unique ITS sequence, not all RefSeq accessions are present in UNITE or associated with a species hypothesis (SH) (Figure 3). Comparisons against selected type sequences in GenBank verified the sequences selected for RefSeq at type specimen level making sure the best sequence for the specimen was selected. Comparisons identified classification discrepancies between MycoBank, Index Fungorum (used by UNITE) and NCBI Taxonomy, which have been communicated among the different curation databases. This comparison step also identified discrepancies in voucher or species names between the GenBank record and the publication, which could then be corrected. Sequences that were >99.5% identical and had over 90% overlap of the ITS region with more than one type sequence in GenBank or RefS/RepS from UNITE under a different TaxID were investigated. Discrepancies mostly revealed the existence of closely related species, which have been noted in a publication or by experts in the fungal research community. Thus, for these cases, there was no problem with the identity of the specimen under the classification point. Sequences that were <98.5% identical and had over 90% overlap of the ITS region to RefSs/RepSs from UNITE of the same TaxID were also investigated. Discrepancies mostly revealed the unclear indication of types from heterotypic synonyms, effect of non-ATGC characters, incorrect type specimens or sequences incorrectly associated with the culture or specimen. In two records, the difference between sequences from the same type material (same collection) was as great as 13 bases in a taxon not known for variation within ITS copies, and these records were excluded. Intragenomic variation is a known phenomenon in the ITS region (50). Such variation may typically be encountered when sequences were derived from cloned PCR products. Where needed, multiple ITS records for a single species were added. For example, Fungi from Glomeromycota are often represented with more than one ITS record. A few similar cases with multiple ITS sequences were also indicated in Basidiomycota (Megacollybia subfurfuracea, Mucidula mucida and Ponticulomyces kedrovayae).
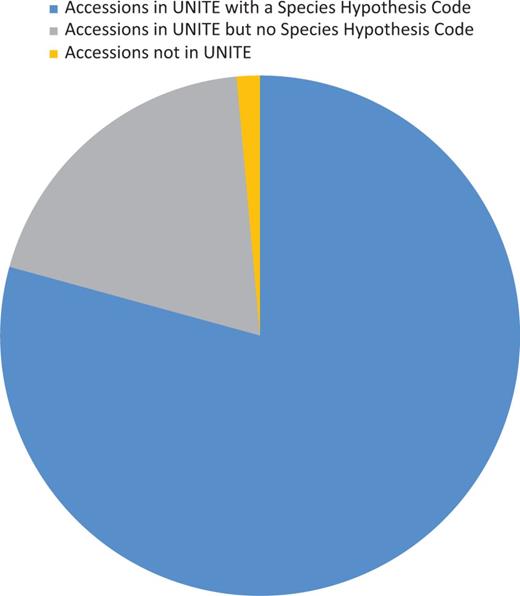
Diagram showing the proportion of accessions associated with UNITE (version 6) data.
Reformatting accessions for RefSeq
All ITS accession numbers in RefSeq start with NR_, and are associated with an RTL Bioproject number. This allows RefSeq users to easily find all curated records (http://www.ncbi.nlm.nih.gov/nuccore/ ? term= PRJNA1 77 3 53) and view a summary of the project (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA177353). The definition line (which appears in the output of the sequence similarity search tool BLAST in GenBank) has been simplified to the following format: ‘[species name] [culture collection/specimen voucher identifier] ITS region; from TYPE/verified material’ (for example: Penicillium expansum ATCC 7861 ITS region; from TYPE material). All records were re-annotated, and the 34% that did not have annotation in INSDC now have annotation in the RefSeq version. Only ∼25% of the selected INSDC records had culture collection or/and specimen voucher information that was correctly fielded and formatted. Culture collection information was moved to the culture collection field and formatted correctly for just over 1000 records. By doing so, these records could potentially be linked to more metadata at a collection's database. The ‘rRNA’ feature key was used to indicate the boundaries of the 18S, 5.8S and 28S rRNA and the ‘misc_RNA’ feature key to indicate the position of the two ITSs (Figure 4).

Anatomy of an RTL record. The marked areas indicate most common additions to the original nucleotide record. (A) New RTL accession number; (B) new simplified definition line; (C) Bioproject number for the ITS-targeted loci project; (D) GenBank synonym of current taxonomic name (used in cases of common usage); (E) label indicating that this is a RefSeq record; (F) comment regarding the source of the record; (G) the culture collection or specimen voucher presented as a validated structured triplet or doublet that can link directly to a relevant outside culture or specimen page; (H) additional information on the type and basionym name; (I) the ITS entry of all records was re-annotated to indicate the spacers and ribosomal genes.
About 250 records were edited to correct collection/specimen voucher information or to add a collection number from a public collection. After curation, all records contained a culture collection or/and specimen voucher identifier in the correct field. Sequences originated from material kept at 159 collections of which only 32% had a searchable public database. However, most (∼75%) of the records were associated with material from collections with a public database (Figure 5). A small number of collections had a specimen page URL that includes the collection number and to which a link can easily be formatted. Before curation started, links existed to five biorepositories. Additional links were added, and the full list of biorepositories with their acronyms indicated is in Table 1. More links will be added as this becomes possible. Recently, LinkOut features linking to SH pages (maintained by UNITE) also became available for individual NCBI ITS sequence records.
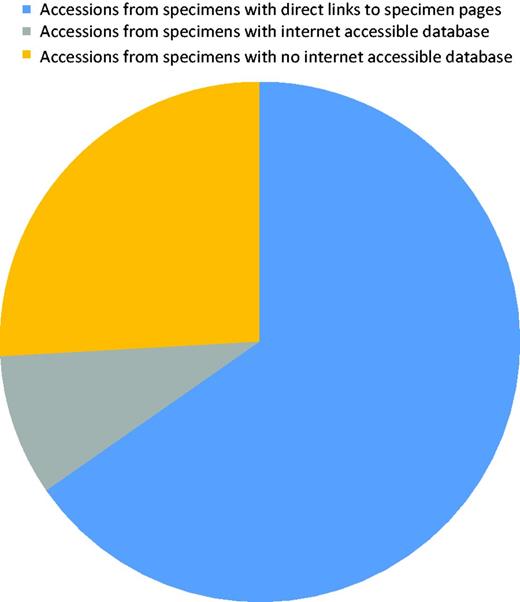
Diagram showing the proportion of accessions that originated from specimens associated with a collection that has an online database.
Results of centrality analysis
To visualize the taxonomic diversity in our currently selected data set, we present a profile of the RefSeq data set at class in using multidimensional scaling clustering (Figure 6). The centrality analysis at genus rank of the curated RefSeq ITS data set has shown that the ITS variation around a central sequence differs greatly among genera as visualized for those with ≥20 sequences (Figure 7). The centrality score range from 0 to 1, where a score of 1 reflected a sequence identical to the calculated central sequence. Based on this score, most species in some genera (e.g. Penicillium, Colletotrichum) form a tight group in relation to their central sequences (Figure 7). It was clear that in some genera, species cannot be distinguished by comparing ITS sequences only. Centrality scores of 1 indicated where the ITS region did not show variation to distinguish it from the central sequence, and these included a number of taxa, mainly from Cladosporium. The inability of the ITS region to distinguish between many, but not all, species has been reported before in several species, including Cercospora (51) and Cladosporium (52, 53). However, the distribution of centrality scores (Figure 7) shows that some genera are either diverse in terms of ITS sequence similarity or are in need of taxonomic revision (e.g. Candida, Cryptococcus). It is already well known that the large genera of asexual species Candida and Cryptococcus are polyphyletic (54). Other large genera, like Mortierella (55) and Mucor (56), also require revision. Thus, given the poorly defined boundaries of some genera and lack of ITS variability in several species, classifying an unknown ITS sequence to species, and sometimes genus rank will not always produce a definitive answer.
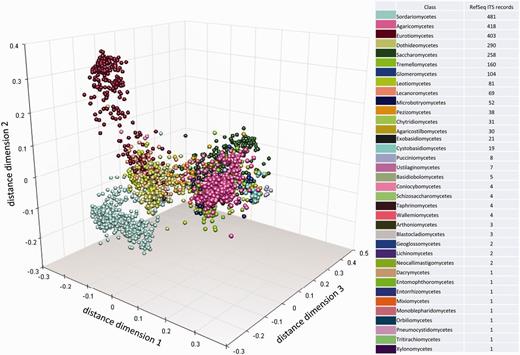
Multidimensional scaling clustering of RTL ITS sequences and coloring, according to the NCBI Taxonomy classification at class rank. Each marker represents an individual sequence.
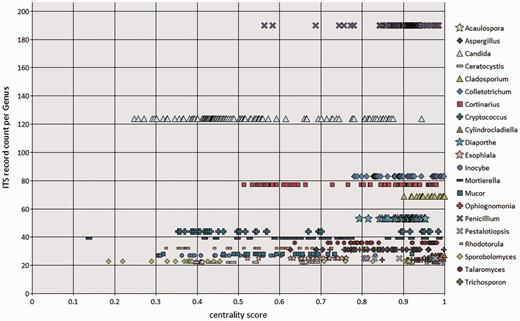
BioloMICS centrality scores of ITS sequences at genus rank, showing genera with ≥20 ITS records in the RefSeq data set. Each marker represents an individual sequence.
Discussion
Changes to NCBI databases
The NCBI Taxonomy database (http://www.ncbi.nlm.nih.gov/taxonomy) acts as the standard nomenclature and classification repository for the INSDC. It is a central core where taxonomic information for the entries in other databases—such as GenBank—is stored. NCBI Taxonomy uses an array of name classes, e.g. ‘scientific name’, ‘synonym’, ‘equivalent name’ to express various taxonomic attributes (57). An example of a taxonomic record is shown in the Supplementary Material (Supplementary Figure S2). Two specific name types unique to fungi, ‘anamorph’ and ‘teleomorph’, but falling out of favor (58), will slowly be phased out as fungal classification adapts to a new nomenclatural system. An additional name type was recently added to the taxonomy database, ‘type material’. This information is indexed so that related sequence records annotated with type specimen or ex-type culture identifiers with synonymous (homotypic and heterotypic) species names can be found with an Entrez query.
The following Entrez query ‘sequence from type[filter] AND fungi[orgn]’ will list all fungal taxonomic entries (from all genes and genetic markers) with type material attributes. The same query can be used to do a limited BLAST search on sequences from type material. Currently (March 2014), this covers over 150 000-nt sequence accessions in INSDC databases, including several additional regions besides ITS. This includes genome sequences and RefSeq messenger RNA records. In the era of phylogenomics, researchers may also be interested to know which genomes are from type specimens and the associated proteins. In the future, the Entrez Protein Clusters database will also include fungal protein accessions, and those originating from type specimens can be marked as such. In addition to fungal type data, there are now >500 type-associated entries for metazoa and already >500 000 for bacteria.
NCBI Taxonomy currently lists >28 000 binomial fungal names at species rank, and 56% had good quality (not chimeric or broken) ITS records in GenBank (including synonyms). Our current data set of RefSeq sequences represents 16% of binomials with clean ITS data. In terms of classification (regardless of presence or absence of ITS data), the RefSeq set covers 660 of 4387 possible fungal genera, 249 of 514 possible families, 120 of 153 possible orders and 36 of 37 possible classes. With continued curation, more types will be identified and the associated sequences added to the RefSeq database.
The presence of curated type material improved the efficiency of taxonomic updates at NCBI. The validation of sequences and type material released in RefSeq allowed >300 taxonomic names to be merged, rectified or updated in NCBI Taxonomy. Several taxonomic names that were submitted with a genus and strain identifier only and not updated upon publication could easily be verified and updated by relying on accurate specimen data present in their sequence accessions. Similarly, curating and knowledge of synonyms are important because it can greatly influence the accuracy of any microbiome or ecological study. Recent studies on the oral microbiome provide a good example (59). Several researchers still continue to use large polyphyletic genus names to discuss species of clinical importance (60).
Standards for traceability of specimen vouchers
The most time-consuming step in this curation process was to identify and verify type specimens and cultures. It was useful to import type identifiers from, for example, MycoBank, but identifiers must still agree with the metadata in the GenBank records. Using the same identifier from a specific collection (especially when specimen vouchers from herbarium material are involved) and a standard structure among various sources such as taxonomy databases, collections, publications and sequence records will contribute tremendously to improve this process. Listing type specimen identifiers in the abstract of a paper represents another helpful measure to avoid having type information hidden behind a paywall. Using an Entrez query such as this: (collection cbs[prop] OR cbs[title] AND fungi[orgn]) AND (2014/01/01: 3000[PDAT]) can help CBS collection curators, for example, to identify newly released sequence records since the beginning of 2014. The search term ‘CBS’ is just an example and can be replaced with any other acronym in the NCBI collections.
NCBI Collections
The ability to provide direct links between GenBank and biorepositories (herbaria and fungaria, natural history collections, zoos, botanical gardens, biobanks, culture collections and others) relies on using unique identifiers to denote cultures and specimens. The potential pages to target with links have been expanded by several projects aimed at increasing the digital presence of a number of institutions. For example, the Mycology Collections data Portal (MyCoPortal; http://mycoportal.org/) provides direct access to digitized specimens records provided by The Macrofungi Collection Consortium, a collaboration of 35 institutions in 24 states in the USA (http://mycoportal.org/portal/index.php). The Global Plants Initiative (GPI) is another such effort, housed at the Royal Botanic Gardens, Kew. This is an international partnership of more than 300 herbaria in 72 countries. GPI’s goal is to digitize and provide access to type specimens of plants, fungi and algae through community-contributed JSTOR Global Plants online database (http://plants.jstor.org). Other resources include Straininfo, which databases information related to cultures and strains (61).
NCBI has retained a record of all biorepositories to assist indexing of submissions in the NCBI Collections database (http://www.ncbi.nlm.nih.gov/projects/BioC ollec tion/search_collection.cgi). The majority of fungal-related acronyms rely on unique identifiers of herbaria indexed at Index Herbariorum (62), whereas the majority of culture collections are listed in the directory of the World Federation for Culture Collections (WFCC). At NCBI, these unique identifiers are also used for museum collections. These multiple sources often contain redundant identifiers, so it is necessary to provide unique versions. This was achieved by adding a country abbreviation in angular brackets. For example, BR<BEL> was used to distinguish the National Botanic Garden of Belgium from the Embrapa Agrobiology Diazothrophic Microbial Culture Collection, BR<BRA>. For the present, it is more practical for NCBI to continue usage of this resource for its own curating and indexing functions. Another effort, the Global Registry of Biorepositories (GRBio) has been supported by the Consortium for the Barcode of Life (CBOL). This currently lists >7000 biorepository records by combining data from CBOL, Index Herbariorum and the Biodiversity Collections Index. It also lists >20 personal collections and allows for registrations online.
Application of Darwin Core and other standards
Darwin Core is a data standard for publishing and integrating biodiversity information (15). The Darwin Core standard triplet format for specimen data consists of a structured string containing an institutional ID, collection code and catalog ID, all separated by colons. Currently, NCBI uses unique labels from the Collections database (14). In many cases, a secondary collection code (such as a collection devoted to Fungi or Plants at a specific institution) is not necessary. In the example given above, the ex-type culture of Colletotrichum brevisporum is indicated as a doublet only, e.g. /specimen_ voucher =” BR <BEL>: 70109”.
It is now possible to register typification events at MycoBank. MycoBank Typification numbers for the designation of lectotypes, epitypes and neotypes can be obtained and referred to in publication (63). The challenge remains to standardize voucher data, so it can be tracked consistently among multiple databases. In a future release of the MycoBank Web site scheduled for 2014, GenBank sequence identifiers will be requested upon deposition of new fungal names and/or associated type specimens. Some changes to the ICN to clarify the circumstances for epitypification have also been proposed (64).
DNA barcoding and standards for GenBank submissions
The ITSx script (46) has been a helpful and time-saving tool in curating the ITS records. It has also provided an important quality control tool for anyone downloading and submitting ITS sequences. The script is efficient to confirm complete ITS regions if enough nucleotides are present in the 18S and 28S region, otherwise curation time needs to be spent to verify the coverage. In addition, sequences were also screened for non-ATGC characters, but ideally one would also like to be able to view sequence traces and be assured about the quality of the base calls. Currently, this information is not available for any ITS sequence. The standard for a DNA barcode (http://www.barcoding.si.edu/PDF/DWG_data_standards-Final.pdf) contains a set of sequence quality requirements in addition to increased scrutiny of specimen data. Part of this involves the deposit of trace data in addition to the sequence deposit at the INSDC. Currently, GenBank will assign a BARCODE keyword to sequences that meet these standards. However, many sequences continue to be referred to as DNA barcodes in the literature without meeting all these requirements. The deposit of sequence traces is a crucial missing element, and it is not likely to see an increase in the foreseeable future. Many highly significant sequences from types and other important specimens already exist in the INSDC databases. Sequences selected as part of this article should meet all the standards for a DNA barcode except for the deposition of trace data. It should therefore be also possible to use these sequences as ‘barcode-like’ or reference sequences, although they would not formally qualify for barcode status.
Effectiveness of ITS as barcode marker
The nuclear rRNA cistron consists of multiple copies ranging from a single copy to >200 in Fungi (65, 66). A number of processes can cause within-individual sequence heterogeneity in the ribosomal repeat, which complicates any analysis using ITS sequences. This includes intra- and intertaxon hybridization accompanied by lack of homogenization (concerted evolution) of the ribosomal repeat at some level in a wide range of species (67–72). Often the rate at which homogenization occurs and whether this varies from taxon to taxon is unknown. However, the process can be rapid (73). In genetically diverse interbreeding populations, however, the ribosomal repeat may never completely homogenize. Other heterogeneity can be due to variation between chromosomes in diploid or heterokaryotic specimens. It is also feasible that more than one ribosomal repeat could exist in some taxa as a consequence of hybridization or horizontal gene transfer (74). Collections selected as types or as exemplars for a species are often not completely homogenic. When heterogeneity is low, this has been handled by creating a consensus barcode using ambiguity codes as is commonly done for members of the Glomeromycota (75). In many cases, however, the level of genetic divergence between haplotypes or between copies of the ribosomal repeat can be significant (≥3% sequence divergence) (50, 76).
In addition to overestimating diversity, the ITS region can also underestimate diversity for several species groups (77). The search for alternative regions has already yielded several markers with equal or improved performance in specific lineages. During the last decade, phylogenetics has moved on from analyzing multiple genes to full genomes in a search for the true species phylogeny (42, 78–82). DNA barcodes have different criteria from phylogenetic markers, although they can often be used interchangeably (83). So the search for a single marker sequence that could represent an idealized phylogeny will most likely also yield a good candidate for a DNA barcode that could identify all Fungi.
Defining types and reference sequences
Currently, the public sequence databases include a mix of sequences derived from type and non-type strains and with various degrees of curation and certainty. An improved and expanded nomenclature for sequences has been proposed elsewhere (41), based on an earlier proposal for ‘gene types’ (84). This work was done with a zoological perspective, addressing concepts formulated under the International Code of Zoological Nomenclature. To continue this discussion and present a system applicable to species codified under the ICN, we propose a simplified framework for consideration. Following this concept, species can be divided into several categories according to the combination of the type/reference strain status and of the sequence length/quality.
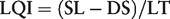
According to this LQI parameter, all the sequences currently presented in the RefSeq database exceed the minimal requirement for robust identification. In general, sequence databases could be managed according to simple rules, defining a hierarchy of sequences according to their origin, for example:
Type/reference sequences with high LQI are used for any purpose and serve as potential targets for the RefSeq database.
Type/reference sequences with low LQI are used for identification with a warning on the identification quality until they can be replaced with better sequences.
Non-type/reference sequences with high LQI can be used for any purpose, other than species identification.
Non-type/reference sequences with low LQI until better sequences are obtained.
The UNITE database for molecular identification of fungi represents another approach to improve sequence accuracy and fungal species identification. It comprises all fungal ITS sequences in INSDC and offers extended functionalities for their curation and analysis (http://unite.ut.ee) (18). All sequences are clustered into SHs variously designated at 97–100% similarity (at 0.5% intervals) to seek to reflect the species rank. The SHs are assigned unique identifiers of the accession number type—e.g. SH133781.05FU—and are resolved with URLs such as ‘http://unite.ut.ee/sh/SH158651.06FU’. All INSDC sequences that belong to an SH are hyperlinked from GenBank/ENA directly to that SH in UNITE through a LinkOut feature. More than 205 000 ITS accessions in the UNITE database can be accessed by using the query ‘loprovunite[filter]’ in GenBank. The SH concept is also implemented in the next-generation sequencing pipelines QIIME (85, 86), mothur (87), SCATA (http://scata.mykopat.slu.se/), CREST (88) and in the recently launched EU BOLD mirror (www.eubold.org). A total of ∼21 000 SH or operational taxonomic units (OTUs) (excluding singletons) at 98.5% similarity are indexed in the current (sixth) release of UNITE. By default, a sequence from the most common sequence type in each SH is chosen to represent the SH and to form part of its name. It is also possible to change the chosen representative where there is a need to exercise extended control. Sequences from type material, in particular, are given priority whenever available and of satisfactory length and technical quality (18, 89).
Conclusions
The Linnaean binomial has been a constant anchor in biology, and it remains central to communication in biology (90). It is intuitive to the way humans process information regarding the natural world, if not always in concert with shifting evolutionary concepts. Given the huge genetic diversity found within the kingdom Fungi, coupled with often cryptic and convergent morphologies, attempts to clearly delineate species boundaries remains a substantial challenge. This has led to a view that taxonomists might be better served by not focusing on fungal species names until more is known about their general biology (91). Although this might be a provocative view, even with ample DNA sequence data, debates about species boundaries will likely persist. A single name, linked to a specific specimen without dispute, following the rules and standards set down in the ICN will remain essential. It follows logically that if the same link can be made for DNA sequences, these sequences can provide reliable reference points for names in computational comparisons.
In this article, we have focused on the re-annotation of a taxonomically diverse set of marker sequences such that a clear link between a species name, a specimen or culture and its sequences can be established with a high level of certainty. The most important part of this process is the increased focus on specimen and culture annotations using a standardized format that can be traced across multiple databases. We used a number of redundant steps in the curation process to remove errors. Yet as is true for any database, some will remain. Because RefSeq is a fully curated database, relying on selections made by taxonomists at NCBI in consultation with a range of experts, it will also be simple to remove questionable sequences as soon as we are aware of them. Feedback about incorrect RefSeq records can be received here: http://www.ncbi.nlm.nih.gov/projects/RefSeq/update.cgi.
There is a substantial and growing increase in the number of sequences being deposited in public sequence databases without scientific binomials (10). To better distinguish truly novel lineages from poorly identified ones, an accurate set of reference sequences will be essential. The manual curation performed in this study relied on mining the information from a variety of resources, including the associated literature. This scales poorly beyond a few thousand entries. For this reason, we focused on a manageable subset of reference sequences focused on ties to type material. It is hoped that machine learning techniques, as already applied to taxonomic names from literature (92), can also improve specimen and, specifically, type material annotation in the future. Our initial data set of ∼2600 ITS records should provide a valuable training set for such techniques.
It is projected that there are ∼400 000 fungal names already in existence. Although only 100 000 are accepted taxonomically, it still makes updates to the existing taxonomic structure a continuous task. It is also clear that these named fungi represent only a fraction of the estimated total, 1–6 million fungal species (93–95). Moving forward, as new species are being described, this process must be documented in a more efficient manner, keeping track of the type specimen information in association with its sequence data. Submitters of newly generated fungal ITS sequences are also asked to consider previously published guidelines (96, 97).
We propose the following steps in submitting future type-related data as part of a normal submissions process to GenBank and the nomenclature databases. It is important to emphasize that Refseq selections will only happen after submission to INSDC databases and does not require a separate user-directed process.
Where possible, submitters can alert GenBank indexers to the presence of type material and include a table during their submissions:
<species name>\t <type strain/specimen>\t <type of type>, for example:
Aspergillus niger \t ATCC 16888 \t ex-neotype
Agaricus chartaceus \t PERTH 07582757 \t holotype
Saccharomyces cerevisiae \t CBS 1171 \t ex-type
If using ITS sequence data, use Figure 4 annotation as an exemplar during GenBank submission, applying annotations determined by the ITSx script.
Register typification with correctly annotated specimen numbers in an available database, e.g. MycoBank.
Verify that the format for specimens and ex-type cultures in GenBank match that in the published record as far as possible.
Extend the principles for traceable specimen and culture data during species descriptions in mycological journals.
Correctly formatted type specimen identifiers from public culture/herbarium collections should not be limited only to ITS records but also any other sequence records including genome records. Genome sequencing centers use ITS regions to confirm the identity of the fungus being sequenced. It is a good practice to include this with a genome assembly. However, the ribosomal cistron is often omitted because of difficulty to establish the exact copy number and the positions of the multiple copies in the genome. The RefSeq ITS set has already been applied in improving genome assembly quality at NCBI by identifying contamination in genome assemblies, especially in obligate biotroph genomes.
The increasing digitization of the biological literature and the growing availability of tools to search the literature and biorepositories are improving ways to link and contextualize sequences and biological data (9). The ability to semantically enhance journals will also allow future taxonomic papers to be mined for valuable taxonomic information (98). Type information is often found in a variety of formats that makes it challenging for machine reading. PubMed Central already has an initial species description extension in XML that could serve as a purpose for linking taxonomic data to additional metadata. This could include barcode data, and some shortened machine-searchable version could be placed in abstracts, so it is easily indexed in various openly accessible literature services like PubMed, PubMed Central and others without residing behind a paywall (99). We also advocate a newly available option to comment on papers in PubMed, PubMed Commons, by registering third-party opinions on sequences and species contained within the relevant publications (http://www.ncbi.nlm.nih.gov/pubmedcommons/).
In the immediate future, we will explore ways to streamline the expansion of RTL for Fungi. We currently only rely on selection by NCBI Taxonomy and RefSeq curators in consultation with numerous taxonomists. Cooperation with the nomenclature databases, MycoBank and Index Fungorum, as well as annotation and specimen databases such as UNITE and MyCoPortal should be expanded where possible. The focus on specimen and culture designations can be extended to include reliable standardized geographical data. We have also collected a smaller set of sequence accessions from the 28S nuclear ribosomal gene and will work to expand the set of accessions to include this and several other markers, using the re-annotated bio collection data where possible. Finally, working with partners to collect and sequence rare species in developing countries should be explored as well, ensuring the availability of annotated reference sequences to all potential users.
It seems likely that nomenclature will face increasingly radical changes in the future. DNA sequencing technology is rapidly revealing biodiversity information. Sequences obtained from environmental sampling can potentially be named under the current ICN with a DNA sample as a physical specimen, but this will not apply in many cases. This will require additional means to standardize labeling and to improve communication. Addressing this unsampled diversity may be ‘the next major challenge for fungal taxonomy’ (6). However, as we show here, much needs to be done to improve the way sampled diversity data are currently disseminated. During the next few years, several conversations will commence on ways to label sequences in public databases to facilitate sequence-based taxonomy (7). We hope the topics covered in this article will contribute to those discussions.
Acknowledgements
B.R. and C.L.S. acknowledge support from the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Funding
Funding for open access charge: The Intramural Research Programs of the National Center for Biotechnology Information, National Library of Medicine and the National Human Genome Research Institute, both at the National Institutes of Health.
Conflict of interest. None declared.
References
Author notes
†These authors contributed equally to this work.
Citation details: Schoch,C.L., Robbertse,B., Robert,V. et al. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database (2014) Vol. 2014: article ID bau061; doi:10.1093/database/bau061

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}