




Abstract
Alcohol consumption during pregnancy causes fetal alcohol spectrum disorder, which includes neuroapoptosis and neurobehavioral deficits. The neuroapoptotic effects of alcohol have been hypothesized to involve suppression of brain activity. However, in vitro studies suggest that ethanol acts as a potent stimulant of cortical activity. We explored the effects of alcohol (1–6 g/kg) on electrical activity in the rat somatosensory cortex in vivo at postnatal days P1–23 and compared them with its apoptotic actions. At P4–7, when the peak of alcohol-induced apoptosis was observed, alcohol strongly suppressed spontaneous gamma and spindle-bursts and almost completely silenced neurons in a dose-dependent manner. The dose-dependence of suppression of neuronal activity strongly correlated with the alcohol-induced neuroapoptosis. Alcohol also profoundly inhibited sensory-evoked bursts and suppressed motor activity, a physiological trigger of cortical activity bursts in newborns. The suppressive effects of ethanol on neuronal activity waned during the second and third postnatal weeks, when instead of silencing the cortex, alcohol evoked delta-wave electrographic activity. Thus, the effects of alcohol on brain activity are strongly age-dependent, and during the first postnatal week alcohol profoundly inhibits brain activity. Our findings suggest that the adverse effects of alcohol in the developing brain involve suppression of neuronal activity.
Introduction
Alcohol consumption during pregnancy is deleterious to the fetal brain. It results in a number of neurological and behavioral deficits characteristic of the fetal alcohol spectrum disorder with the most extreme presentation corresponding to fetal alcohol syndrome. The deleterious effects of alcohol in fetal primates and neonatal rodents involve massive stimulation of neuronal and glial apoptosis, this effect is specific for the immature brain and most pronounced in rodents during the first postnatal week (Ikonomidou et al. 2000; Farber et al. 2010; Creeley and Olney 2013; Olney 2014). The apoptotic effects of alcohol on developing neurons have been suggested to involve its inhibitory action on neuronal activity through a suppression of N-methyl-d-aspartate (NMDA) receptors and a potentiation of γ-aminobutyric acid (GABA)(A) receptors, which are the 2 major alcohol targets in the brain (Lovinger et al. 1989; Masood et al. 1994; Wright and Schoepp 1996; Glykys et al. 2007; Mody et al. 2007). However, the effects of alcohol on brain activity during the critical developmental window of the apoptogenic alcohol actions remain elusive.
Early in development, the brain displays unique activity patterns that are thought to participate in the development of neuronal circuits (Katz and Shatz 1996; Khazipov and Luhmann 2006; Blankenship and Feller 2010). In neonatal rodents, cortical activity is organized in intermittent oscillatory activity bursts in the alpha–beta (spindle-bursts) and gamma (early gamma oscillations, EGOs) frequency ranges (Khazipov et al. 2004; Yang et al. 2009; Mohns and Blumberg 2010; Minlebaev et al. 2011; Colonnese and Khazipov 2012; Khazipov et al. 2013; Yang et al. 2013). These bursts are internally generated in thalamocortical circuits, but they can also be triggered by inputs from the sensory periphery. In the somatosensory (S1) cortex, they are driven by sensory feedback resulting from myoclonic twitches (Khazipov et al. 2004; Mohns and Blumberg 2010; Tiriac et al. 2012). In the visual system, before the onset of vision, they are driven by spontaneous retinal waves (Hanganu et al. 2006; Hanganu et al. 2007; Colonnese and Khazipov 2010; Colonnese et al. 2010). Similar activity patterns are also expressed in the human brain during the second half of gestation (Milh et al. 2007; Andre et al. 2010; Colonnese et al. 2010; Stjerna et al. 2012; Chipaux et al. 2013). Generation of these early oscillatory patterns primarily involves the activation of ionotropic glutamate AMPA/kainate and NMDA receptors, and GABAergic interneurons support these oscillations in a developmental manner compartmentalizing the activated cortical areas via surround inhibition (Minlebaev et al. 2007; Minlebaev et al. 2009; Minlebaev and Khazipov 2011; Kirmse et al. 2015). Early oscillations provide conditions for the temporal binding of topographically linked thalamic and cortical neurons, and support synaptic plasticity, thus participating in the activity-dependent formation of thalamocortical circuits during the critical developmental period (Minlebaev et al. 2011; Yang et al. 2013).
Considerable evidence indicates that in addition to its roles in synaptic plasticity, the ongoing neuronal activity also promotes neuronal survival by preventing developmental apoptosis (Mennerick and Zorumski 2000; Heck et al. 2008; Leveille et al. 2010; Hagenston and Bading 2011). This is supported by massive apoptosis induced in the rodent pup and fetal primate brain by the drugs inhibiting NMDA receptors and enhancing the function of GABA(A) receptors, including ethanol and some other drugs—ketamine, benzodiazepines and barbiturates, anesthetic agents, and antiepileptic drugs (Ikonomidou et al. 1999; Ikonomidou et al. 2000; Bittigau et al. 2002; Jevtovic-Todorovic et al. 2003; Creeley and Olney 2013; Olney 2014). Previous study has pointed to the age-dependent effects of the general anesthetic isoflurane on brain activity, characterized by total suppression of activity during the first postnatal week and developmental emergence of the burst-suppression activity pattern during the second postnatal week (Sitdikova et al. 2014). This developmental profile of the effects of isoflurane on brain activity correlated with the neuroapoptotic and neurobehavioral effects of isoflurane in the immature brain (Jevtovic-Todorovic et al. 2003) supporting the hypothesis that the basis for the critical period of susceptibility of the neonatal rat brain to this agent involves suppression of activity. It has been hypothesized that the neuroapoptotic effects of alcohol also involve suppression of brain activity (Ikonomidou et al. 2000). Yet, the only available evidence on the effects of alcohol on neuronal network activity in the immature brain, which has been obtained using hippocampal slices of neonatal rats in vitro, indicates that alcohol not only fails to suppress the immature activity pattern of giant depolarizing potentials (GDPs) but that it actually increases network activity through an increase in GDP frequency, this has led to a conclusion that alcohol is a potent stimulant of immature neuronal networks (Galindo et al. 2005). This observation evidently goes against the hypothesis that the apoptogenic actions of alcohol in the immature brain involve suppression of brain activity. However, how alcohol acts on the immature brain activity in vivo remains elusive.
In the present study, we used intracortical recordings of electrical activity from the primary somatosensory cortex of head-restrained, nonanesthetized rats aged from postnatal day P1–P23 to characterize the age- and dose-dependence in the effects of alcohol on spontaneous and sensory-evoked cortical activity and to compare them with the apoptotic alcohol actions. Our main finding is that at postnatal days P4–7, when the peak of alcohol-induced apoptosis is observed, alcohol strongly suppressed spontaneous network activity and almost completely silenced neurons in a dose-dependent manner that correlated well with the dose-dependent neuroapoptosis, and that the suppressive effects of ethanol waned during the second and third postnatal weeks. These findings point to an age-dependence in the effects of alcohol on brain activity and support the idea that the adverse consequences of maternal alcohol consumption on the fetal brain involve profound suppression of the cortical activity, which may subsequently cause alterations in early developmental processes including activity-dependent apoptosis, synaptogenesis and formation of neuronal networks.
Materials and Methods
Ethical Approval
This work has been carried out in accordance with EU Directive 2010/63/EU for animal experiments and all animal-use protocols were approved by the French National Institute of Health and Medical Research (INSERM, protocol N007.08.01) and Kazan Federal University on the use of laboratory animals (ethical approval by the Institutional Animal Care and Use Committee of Kazan State Medical University N9-2013).
Surgery
Wistar rats of either sex from postnatal day P1 (P0 = day of birth) till postnatal day P23 were used. Surgery was performed under isoflurane anesthesia. In brief, the skull of the animal was cleaned of skin and periosteum. Two plastic or metal bars were fixed to the nasal and occipital bones of the rat's head by dental cement. The skull was covered by dental cement except for a 4–9 mm window above the primary somatosensory cortex. After surgery, the animals were warmed, and left for 1 h to recover from anesthesia. During recordings, the head was fixed to the frame of the stereotaxic apparatus by the attached bars; animals were surrounded by a cotton nest and heated via a thermal pad (35–37°C). A chlorided silver wire, placed in the cerebellum or visual cortex, served as a ground electrode.
Extracellular Recordings
Extracellular local field potentials (LFP) and multiple unit activity (MUA) were recorded using 16-site linear silicon probes (100-μm separation distance between recording sites, Neuronexus Technologies, MI) placed vertically into the S1 cortex to a depth of 0–1.5 mm from the cortical surface to trace the columnar activity from all cortical layers. Receptive fields of the recorded cortical column were identified by the shortest latency MUA responses in layer 4 evoked by brief (10 ms) local touch of the limbs' skin with the blunt end of a needle attached to a piezo actuator or by single whisker deflection. Recordings were organized in alternating 15-min sessions of recordings of spontaneous activity and sensory-evoked activity. Values for L4, L5/6, and L2/3 before P4 (when L4 layer differentiates from the cortical plate) correspond to the depths with the short latency responses and 200–300 μm below and above, respectively. The signals were amplified and filtered (1000×; bandpass 0.1–9 kHz) using a Neuralynx amplifier (Neuralynx, MO), digitized (32 kHz sampling frequency, 0.5 μV voltage resolution) and saved on the PC for post hoc analysis. Spontaneous and sensory-evoked activity was recorded for 1 h, and then the rats were injected intraperitoneally (i.p.) with a 20% solution of ethanol in normal saline and recorded for 4 h in 15-min long alternating sessions of recordings of spontaneous and sensory-evoked activity. In a separate set of animas (n = 5; P4-6), the same volumes of normal saline were injected intraperitoneally and the parameters of activity obtained at different time points after injection served for the comparison with the data obtained after injection of ethanol.
Data Analysis
Raw data were preprocessed using a custom-developed suite of programs in Matlab (MathWorks, USA) environment. The wide-band signal was down sampled to 1000 Hz and used as the LFP signal. Positive polarity is shown up in all figures. For spike detection, the wide-band signal was filtered (bandpass 300–3000 Hz) and negative events exceeding 6 standard deviations in amplitude were considered as spikes. Sensory-evoked potentials (SEPs) were detected as the first troughs of sensory-evoked responses in L4. For the analysis of sensory-evoked oscillations, 500 ms periods following the SEPs were used. For baseline activity assessment, the recordings 200 ms prior to the stimulus were used. LFPs and extracellular units were analyzed by custom-written, MATLAB-based programs. Spectral analysis was carried out using Chronux toolbox procedures. Spectral power and coherence was estimated using direct multi-taper estimators (10 Hz bandwidth, 3 tapers, 200 ms spectral window padded to double its length with zeros) or continuous wavelet transformation with the Morlet mother wavelet. To remove slow frequency envelope, the LFP was filtered at 10–100 Hz. Gamma and spindle-burst oscillation peaks were detected with the following steps: 1) the LFP signal was bandpass filtered (30–80 Hz for gamma and 8–30 Hz for spindle-bursts), 2) the times of negative troughs with amplitude >10 µV were detected from the filtered signal, 3) gamma and spindle-burst oscillations were considered as a minimum of 3 cycles with periods <30 and 120 ms each, respectively, both associated with spikes. Current-source density (CSD) analysis across cortical depth was used to eliminate volume conduction and localize synaptic currents. CSD was computed for each recording site according to a differential scheme for the second derivative and smoothed with a triangular kernel of length.
Toxicology
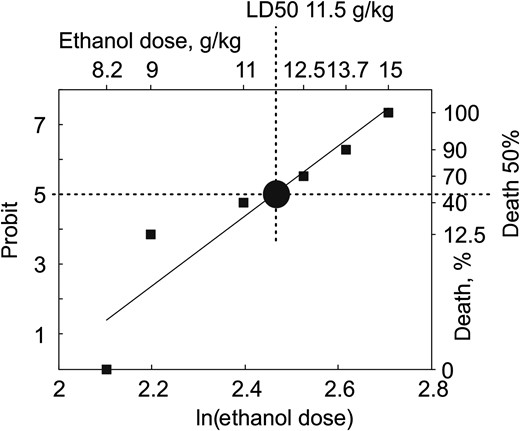
{kind=link}
Toxicology of ethanol in neonatal rat pups. The plot shows the ethanol dose-dependent mortality rate in P4–7 rats. Ethanol was dissolved in normal saline at 20% and administered intraperitoneally at doses of 2.5; 3.7; 6; 6.7; 7.4; 8.2; 9; 11; 12.5; 13.7; and 15 g/kg (total 74 rats). Animals were monitored for 6 h after ethanol administration. The semilethal (LD50) dose of ethanol was 11.5 g/kg.
Blood Alcohol Concentration
Blood alcohol concentrations (BAC) were determined in 25 rat pups of postnatal age P5–7. Alcohol was administered intraperitoneally (20% in normal saline) at doses of 1 g/kg, 1 + 3 g/kg with a 1 h interval, and 6 g/kg (n = 8 animals per dose). Control animals were administered with an equal volume of normal saline. Each group was divided into 2 subgroups of 4 rats, from which blood was sampled intracardially 1 and 4 h after alcohol administration. Blood samples were transferred into vacuum tubes and frozen. Measurements of BAC levels were performed at the Bureau of Criminal and Medical Expertise of the Ministry of Health of the Republic of Tatarstan using a Cristallux-4000 M gas chromatograph (Yoshkar-Ola, Russia).
Apoptosis
Cleaved Caspase-3 immunohistochemistry was used to characterize the neuroapoptotic actions of alcohol in the S1 cortex of rats through postnatal development. Eight hours after ethanol administration (20% solution in normal saline) rats were deeply anesthetized with urethane (2 g/kg, intraperitoneally) and perfused through the left cardiac ventricle with 4% paraformaldehyde. The brains were removed and left for fixation in 4% paraformaldehyde at room temperature. Then the brains were rinsed in PBS (3 × 30 min) and cut in agar blocks into 50-μm-thick coronal sections using a Vibratome (Leica, Germany). After 1–2 h incubation in blocking solution (PBS, 0,3% Triton, 5% Normal Goat Serum), sections were incubated at 4°С with primary antibodies against Cleaved Caspase-3(ASP 175) (Cell Signalling Technology, Beverly, MA) diluted at 1:1000 in a solution containing PBS, 0,3% Triton, 1% Normal Goat Serum. Then sections were rinsed in PBS (4 × 5 min) and incubated for 1 h at room temperature with the secondary fluorescent antibodies Cy3 AffiniPure Donkey Anti-Rabbit IgG (H + L) diluted at 1:1000 in PBS. Then sections were rinsed in water and mounted with a DAPI containing Fluoromount.
Statistics
Statistical analysis was performed using Matlab Statistics toolbox. The Kolmogorov–Smirnov test was used to determine whether datasets were normally distributed. The 2-side Wilcoxon rank sum test was performed on abnormally distributed data with a level of significance kept at P < 0.05.
Results
In the present study we explored and compared the effects of alcohol on (1) neuroapoptosis using active caspase-3 immunochemistry and (2) spontaneous/sensory-evoked cortical activity using multisite silicone probe recordings of the LFP and MUA from the limbs and whisker representation in the primary somatosensory (S1) cortex of P1–23 rats.
Age- and Dose-dependence of Ethanol-induced Neuroapoptosis in the Rat S1 Cortex
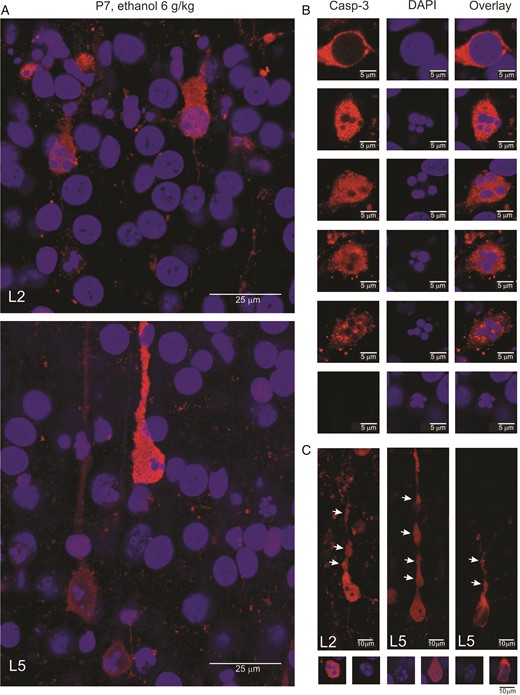
{kind=link}
Morphological features of the ethanol-induced apoptosis in the neonatal rat barrel cortex. (A) Confocal images of the L2 (top) and L5 (bottom) of the barrel cortex of a P7 rat treated with 6 g/kg ethanol (20% wv, i.p.). The sections were stained with antibodies against activated caspase-3 (red) and DAPI staining of the chromatin (blue). Note DAPI-stained apoptotic bodies in the active caspase-3 stained neurons. (B) Examples of the bodies of apoptotic cells showing progression of apoptosis starting with caspase-3 activation without visible changes in the nucleus followed by chromatin condensation into apoptotic bodies with a reduction in caspase-3 signal and morphological degradation. Note that apoptotic bodies are also seen as shadows devoid of caspase-3 staining. (C) Examples of the dendritic degradation in the active caspase-3 stained neurons manifested by formation of thinnings and varicosities (arrows). Below are shown corresponding images of DAPI and overlay of DAPI with active caspase-3 stains. (A–C) Examples from P5–7 rat barrel cortex. Brains were fixed 8 h after ethanol administration.
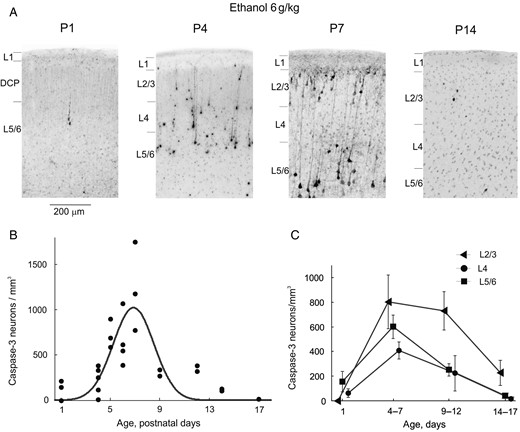
{kind=link}
Age-dependence of ethanol-induced apoptosis in the somatosensory cortex of developing rats. (A) Histological sections from the S1 cortex of rats at different postnatal ages 8 h after treatment with ethanol (6 g/kg, 20% wv, i.p.). Sections have been stained with antibodies to activated caspase-3. (B,C) Age dependency of the total (B) and cross-layer (C) density of activated caspase-3 stained neurons in the S1 barrel cortex from the rats treated with ethanol (6 g/kg, 20% wv, i.p.). Each point on panel B corresponds to an individual animal (total, n = 32 P1–17 rats). The Gaussian fit reveals a peak of ethanol-induced apoptosis in the S1 rat cortex at around P7.
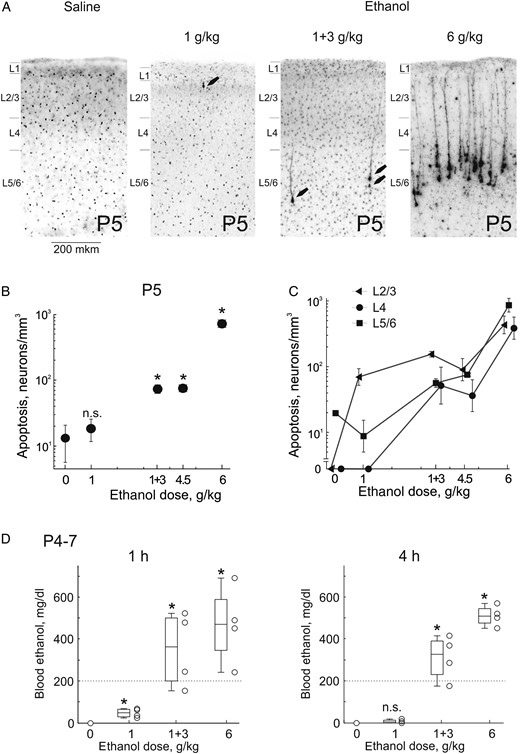
{kind=link}
Dose-dependence of ethanol-induced apoptosis in the somatosensory S1 cortex of neonatal rats. (A) Histological sections from S1 cortex of P5 rats 8 h after treatment with ethanol at different dosage regimens (normal saline, 1 g/kg, 1 and 3 g/kg with a 1 h interval, and 6 g/kg, 20% wv, i.p.). Sections have been stained with antibodies to activated caspase-3. At doses of 1 g/kg and 1 + 3 g/kg, apoptotic neurons are marked by arrows. (B,C) Dependence of the total (B) and cross-layer (C) density of caspase-3 stained neurons on the ethanol dose. Note a logarithmic scale for the caspase-3 stained neuronal density. Pooled data from n = 15 P5 rats. (D) Ethanol blood levels one and 4 h after administration of ethanol at different doses. Each point corresponds to an individual animal. Horizontal bars show median values and boxes indicate 25–75 percentile range. Pooled data obtained from n = 16 P4–7 rats.
Effects of Ethanol on Spontaneous Activity in the S1 Cortex of Neonatal Rats
{kind=link}
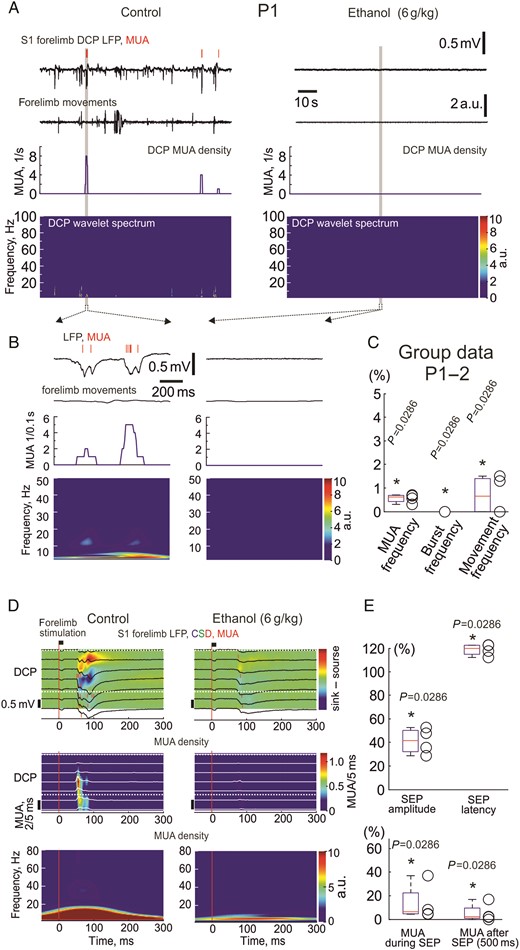
Effects of ethanol on spontaneous and sensory-evoked activity in the primary somatosensory cortex of P1–2 neonatal rats. (A) Example traces of spontaneous electrical activity in the inner part of the dense cortical plate of the forelimb representation in the S1 cortex of a P1 rat (LFP-black traces; MUA-red bars above) in control conditions and 10 min after intraperitoneal injection of 20% ethanol at 6 g/kg. Below are shown corresponding wavelet spectrograms and MUA frequency plots. Note that ethanol almost completely suppresses ongoing LFP and MUA activity. (B) An individual oscillatory burst in control conditions and a typical episode of silence outlined by gray boxes on panel (A) are shown on an expanded time scale. (C) Group data on the effects of ethanol (6 g/kg) on MUA and bursts frequency in the S1 cortex of P1–2 rats (n = 4). (D) Sensory responses evoked by forelimb mechanical stimulation at different cortical depths in a P1 rat S1 forelimb representation (LFP-black traces; MUA-red bars overlaid on a color coded CSD plot in control conditions (left) and 1 h after ethanol injection (6 g/kg, right). Stimulus onset is indicated by a vertical red line. The inner part of the dense cortical plate is indicated by white dashed lines. Below, corresponding stimulus-triggered average (n = 40) for MUA across layers. (E) Group data on the effects of ethanol (6 g/kg) on the parameters of sensory-evoked responses in P1–2 neonatal rat somatosensory cortex (n = 4 rats).
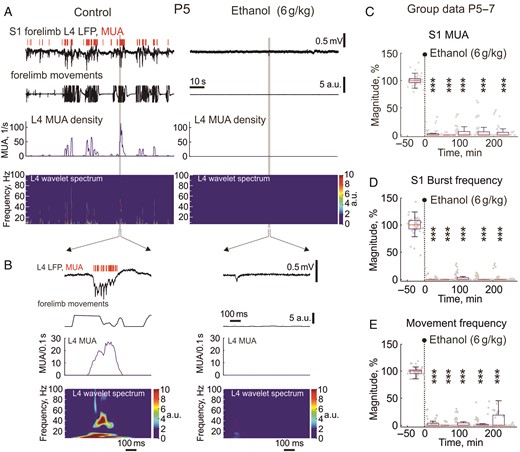
Effects of ethanol on spontaneous activity in the primary somatosensory cortex of neonatal rats. (A) Example traces of spontaneous electrical activity in L4 of the forelimb representation in the S1 cortex of a P5 rat (LFP-black traces; MUA-red bars above) in control conditions and 10 min after intraperitoneal injection of 20% ethanol at 6 g/kg. Below are shown corresponding wavelet spectrograms and MUA frequency plots. Note that ethanol almost completely suppresses ongoing LFP and MUA activity. (B) An individual oscillatory burst in control conditions and a sharp potential outlined by gray boxes on panel (A) are shown on an expanded time scale. (C–E) Group data (P5–7) on the time course of the inhibitory effect of ethanol (6 g/kg, i.p.) on spontaneous MUA frequency (A) and burst frequency (B) in the S1 cortex of neonatal rats and body/limbs movement frequency (C). Each gray dot represents individual response and boxes indicate median and interquartile range during approximately 15 min recording periods for all animals normalized to the control values prior to ethanol injection. Note that a single injection of ethanol (indicated by vertical dashed line) rapidly and profoundly inhibits spontaneous activity and movements during the entire period of recordings (4 h). Pooled data from L2/3, L4, L5/6 of whisker and limbs S1 cortex in ten P5–7 rats, *** = P < 0.001.
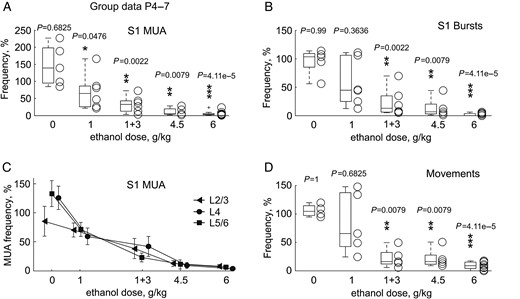
Dose-dependence in the effects of ethanol on electrical activity in the S1 cortex and limb movements in P4–7 rats. (A–D) MUA frequency (A), burst frequency (B), MUA frequency across layers (C), and limb movement frequency (D) 1 h after administration of ethanol at different doses. Each open circle corresponds to an individual animal and boxes indicate median ± error during the 15 min recording period normalized to the control values prior to ethanol injection. Pooled data from 27 P4–7 rats.
Dose-dependence in the inhibitory effects of ethanol on S1 activity was explored in P4–7 rats, using the same dosage regimens as used to study dose-dependence of ethanol-induced apoptosis. At a dosage of 1 g/kg (n = 6 rats), ethanol exerted nearly 2-fold suppressive effects on S1 MUA and bursts, with a similar inhibition of the occurrence of spindle bursts and gamma bursts (to 58 [49–79] % and 56 [34–90]%, respectively, without any change in the oscillation power), and on the frequency of movements (Fig. 7). Ethanol at higher doses, 1 + 3 g/kg with a 1 h interval and 4.5 g/kg produced strong suppression of S1 activity including a decrease in MUA to 18 [4–27]%, bursts frequency to 8 [5–29]% with a similar suppression of spindle-burst occurrence to 8 [2–23]% and gamma-bursts to 7 [5–20]%, a decrease in corresponding alpha–beta and gamma oscillation power to 9 [7–22]% and 33 [25–54]%, respectively, and movements frequency to 16 [12–29]% (pooled data for 1 + 3 and 4.5 g/kg; n = 11 rats). The most robust effect was suppression of activity by ethanol at the highest dose used in the present study, 6 g/kg, where suppression of MUA attained 2.8 [2.3–6.1]%, bursts frequency attained 0.01 [0.0–3.6]% and movement frequency attained 8.5 [4.0–15.3]% of control levels (n = 9 rats). Neuronal activity was similarly suppressed by different doses of ethanol across cortical layers (Fig. 7C). Thus, the dose-dependence of the inhibitory effects of ethanol on cortical activity correlated well with the neuroapoptotic effects described above.
In the group of P4–7 animals we also explored the effects of ethanol on sensory-evoked responses in the S1 cortex. In control conditions, a brief (10 ms) mechanical stimulation of the topographic spots at limbs or a deflection of the principal whisker evoked complex S1 responses consisting of the initial SEP followed by gamma- and spindle-burst oscillations (Supplementary Fig. 1A). In keeping with the results of previous studies, both SEP and the following sensory-evoked activity bursts were associated with maximal LFP signals and MUA in L4. Ethanol (6 g/kg) affected neither SEP amplitude (control, 536 [437–879] μV; ethanol, 434 [350–724] μV; P = 0.22; Supplementary Fig. 1C) or SEP onset latency (control, 36 [34–41] ms; ethanol, 41 [34–45] ms; P = 0.22; n = 9; P4–7; Supplementary Fig. 1D). Although MUA during SEP showed a tendency to decrease, from 5.2 [2.0–8.0] spikes/20 ms to 3.1 [1.2–8.0] spikes/20 ms after ethanol administration, however, this was not significant (P = 0.81). Yet, ethanol strongly suppressed neuronal firing during sensory-evoked activity bursts from 13.7 [6.8–42.8] to 4.0 [1.7–17.4] spikes/500 ms (P = 0.0019). Even stronger, almost complete suppression of sensory-evoked cortical bursts has been observed at P1–2 (Fig. 5D,E).
Developmental Changes in the Effects of Ethanol on Cortical Activity
{kind=link}
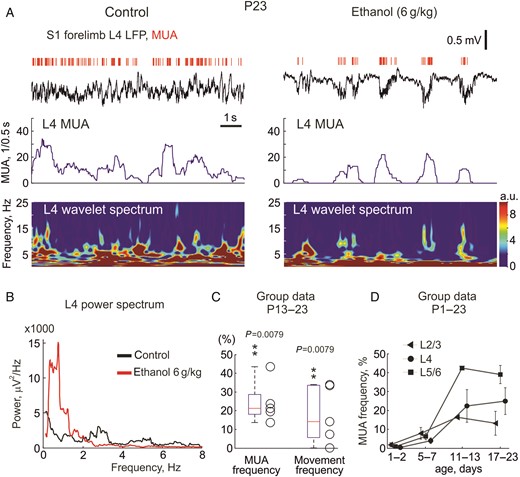
Effects of ethanol on spontaneous activity in the primary somatosensory cortex of a P23 rat. (A) Example traces of spontaneous electrical activity in L4 of the forelimb representation in the S1 cortex of a P23 rat (LFP-black traces; MUA-red bars above) in control conditions and 10 min after intraperitoneal injection of 20% ethanol at 6 g/kg. Below are shown corresponding wavelet spectrograms and MUA frequency plots. Note that ethanol slows down ongoing LFP activity and reduces MUA. (B) The parts of recordings outlined by gray boxes on panel (A) are shown on expanded time scale. (C) Group data on the effects of ethanol (6 g/kg, i.p.) on S1 MUA and limb movement frequency in 5 P13–23 rats. (D) Age-dependence in the effects of ethanol (6 g/kg, 20% wv, i.p.) on MUA frequency across layers.
Discussion
In the present study, we addressed the effects of ethanol on spontaneous and sensory-evoked activity in the S1 cortex of P1 to P23 rats using intracortical recordings of EEG and neuronal firing, and compared these effects with the age- and dose-dependence of the neuroapoptotic actions of ethanol. Our main finding is that during the postnatal period from P4 to P7, when the peak of alcohol-induced apoptosis is observed, ethanol strongly suppresses spontaneous cortical activity and inhibits sensory-evoked oscillatory bursts in a dose-dependent manner, which also correlates with the dose-dependence of the ethanol-induced apoptosis. These findings point to a particular vulnerability of the immature brain activity to ethanol and provide support to the hypothesis that the adverse effects of ethanol involve an inhibitory action of ethanol on brain activity.
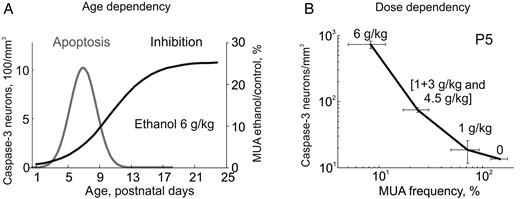
{kind=link}
Summary of the age and dose dependency of the ethanol-induced neuroapoptosis and inhibition of S1 cortical activity in developing rats. (A) Developmental profiles of the ethanol-induced neuroapoptosis (Gaussian fit from the Fig. 3B) and inhibitory effects of ethanol on neuronal firing expressed as a Boltzman fit of the age-dependence of MUA inhibition in the developing rat S1 cortex. Note that ethanol almost completely suppresses neuronal activity during the first postnatal week, when the neuroapoptotic actions of ethanol attain maximal values. However, neurotoxic actions of ethanol only occur within a critical developmental window and inhibition per se is not sufficient to trigger massive apoptosis at P1–2, where the inhibition of cortical activity is most robust. (B) The correlation between the ethanol-induced apoptosis and suppression of S1 activity (MUA frequency after ethanol administration normalized to control values) at different doses of ethanol. Note high correlation levels between the dose-dependent ethanol-induced cortical inhibition and apoptosis.
Our finding of suppression of early cortical activity stays in contrast to the observations made in hippocampal slices of neonatal rats in vitro, where ethanol stimulated neuronal activity through an increase in the frequency of the GDPs (Galindo et al. 2005) that was also observed in our laboratory (Latfullina and Khazipov, unpublished observation). The reasons for this sharp discrepancy in ethanol actions are unclear. This may involve a difference between the brain structures (neocortex vs. hippocampus), and a difference in the generative mechanisms of the in vivo and in vitro activity patterns. One of the plausible explanations is that GABAergic interneurons exert mainly inhibitory network actions during generation of the oscillatory neocortical activity bursts in vivo, evidenced by (1) the effects of GABA(A) receptor antagonists, which eliminate feedforward inhibition, suppress gamma oscillations, and expand the cortical territories activated during spindle-bursts, and (2) inhibitory effects of the drugs potentiating GABA(A) receptors, which reduce burst occurrence and the size of activated cortical areas (Minlebaev et al. 2007; Minlebaev and Khazipov 2011; Minlebaev et al. 2011; Kirmse et al. 2015). This is in contrast to well documented excitatory GABA actions in vitro, both in the neocortex and hippocampus (Ben Ari et al. 2007; Rheims et al. 2008; Khalilov et al. 2015).
Why is the early cortical activity in vivo particularly vulnerable to ethanol? Previous studies have revealed several generative mechanisms of the early cortical activity patterns that are potential targets for the suppressive effects of ethanol. Firstly, the main physiological drive for the early oscillatory bursts in the somatosensory cortex has been shown to involve sensory feedback resulting from spontaneous motor activity including myoclonic twitching (Khazipov et al. 2004; Mohns and Blumberg 2010; Tiriac et al. 2012). We found that ethanol profoundly inhibits spontaneous motor activity in neonatal rat pups thus eliminating the physiological trigger for the cortical activity bursts. However, the nearly complete suppression of the cortical activity cannot be entirely explained by the loss of motor activity as the frequency of spindle-bursts after deafferentation only reduces to about a third of the control values (Khazipov et al. 2004; Yang et al. 2013). In addition, ethanol also reduced neuronal excitation during bursts evoked by direct sensory stimulation. This indicates that ethanol also exerts central actions in the thalamocortical circuits generating the early activity bursts. Previous intracellular and pharmacological studies have shown that generation of the thalamocortical early gamma oscillations and spindle-bursts during the first postnatal week primarily involves activation of the AMPA/kainate and NMDA receptors at glutamatergic synapses and that blockade of cortical NMDA receptors reduces the excitatory currents during spindle-bursts by half (Minlebaev et al. 2007; Minlebaev et al. 2009; Minlebaev et al. 2011). GABAergic interneurons support the early oscillations in a developmental manner and compartmentalize the activated cortical areas via surround inhibition, and blockade of cortical GABA(A) receptors expands the cortical territories activated during spindle-bursts and increases their duration, as well as eliminates the feedforward inhibition and gamma oscillations in >P4 rats, whereas the positive allosteric GABA(A) receptor modulators reduce burst frequency and the size of activated cortical areas (Minlebaev et al. 2007; Minlebaev et al. 2011; Kirmse et al. 2015). Ethanol is a noncompetitive inhibitor of NMDA receptors affecting ion channel gating (Lovinger et al. 1989; Wright et al. 1996). Importantly, ethanol preferentially inhibits NMDA receptors containing the NR2B subunit (Masood et al. 1994), which is preferentially expressed and is responsible for the long decay of NMDA receptor mediated synaptic currents in cortical neurons during the first postnatal week (Carmignoto and Vicini 1992; Monyer et al. 1994; Khazipov et al. 1995). In addition, ethanol also inhibits the kainate type of glutamate receptors prominently expressed in the cortex during the early postnatal period (Dildymayfield and Harris 1995; Kidd and Isaac 1999). On the other hand, ethanol potentiates GABA(A) receptors with a particularly strong action on δ subunit containing GABA(A) receptors, which mediate tonic current (Glykys et al. 2007; Mody et al. 2007) and which are expressed in the cortex shortly after birth (Laurie et al. 1992). Thus, the inhibitory effects of ethanol on cortical activity in the neonatal rats likely involves: (1) peripheral inhibitory actions of ethanol on spinal cord-generated movements, which normally trigger S1 bursts via sensory feedback and (2) central inhibitory actions of ethanol on NMDA and kainate receptors and enhancement of the GABA(A) receptor functions that inhibit early oscillatory thalamocortical activity patterns. The latter mechanism is supported by our recent findings on nearly complete inhibition of S1 activity in rat pups by combined administration of the NMDA receptor antagonist ketamine and the positive GABA(A) receptor allosteric modulator midazolam (Lebedeva, unpublished observation). Thus, mimicking the effects of ethanol on its main presumable targets—suppression of NMDA receptors and potentiation of GABA(A) receptors—produces inhibition of the cortical activity which is very similar to the ethanol-induced inhibition, and, as shown previously, stimulates apoptosis in the neonatal rat pups (Young et al. 2005).
Profound and long lasting suppression of the cortical activity by ethanol in neonatal rat pups may contribute to adverse neurological and behavioral consequences through 2 potential mechanisms: (1) stimulation of programmed cell death and (2) disruption in the activity-dependent formation of the cortical circuits. The neuroapoptotic effects of ethanol during the postnatal period are well documented (Ikonomidou et al. 2000; Olney, Tenkova, et al. 2002; Olney, Wozniak, et al. 2002) and they likely involve suppression of neuronal activity, directly as supported by the results of the present study. Indeed, considerable evidence indicates that physiological activity in developing networks prevents developmental apoptosis and that synaptic activity-dependent neuronal survival is induced by calcium entry through synaptic NMDA receptors and voltage-gated calcium channels, and activation of survival-promoting genes that enable neurons to build up a neuroprotective shield (Ikonomidou et al. 1999; Mennerick and Zorumski 2000; Heck et al. 2008; Leveille et al. 2010; Hagenston and Bading 2011). Suppression of phosphorylation of extracellular signal-regulated kinases (ERK), which is a signaling system that regulates cell survival, appears to play the key role in the apoptogenic effects of ethanol, as lithium, which counteracts the suppressant action of ethanol on pERK efficiently protects against apoptogenic injury induced by ethanol in the immature brain (Zhong et al. 2006; Young et al. 2008). Our finding that ethanol rapidly and almost completely inhibits neuronal activity during the first postnatal week, with the inhibitory effects lasting for several hours provides strong support to the hypothesis that the apoptogenic effects of this drug in the immature brain are caused by suppression of brain activity (Creeley and Olney 2013; Olney 2014). A similar mechanism operates during neuroapoptotic actions exerted by other drugs and pathological factors. For example, in a recent study it has been demonstrated that lipopolysaccharide-induced inflammation causes rapid alterations in the pattern of spontaneous burst activities and subsequently leads to an increase in apoptosis in the neonatal rat cortex (Nimmervoll et al. 2013). This is also in keeping with the apoptogenic (Jevtovic-Todorovic et al. 2003) and powerful inhibitory effects of the general anesthetic isoflurane on cortical activity specifically during the first postnatal week, when this agent totally suppresses spontaneous activity bursts and neuronal firing and inhibits sensory-evoked bursts, similarly to ethanol (Sitdikova et al. 2014). Interestingly, the inhibitory effects of isoflurane on cortical activity are observed during exposure to isoflurane and they rapidly wane after isoflurane withdrawal (Sitdikova et al. 2014) while the apoptogenic effects of isoflurane require long lasting, not <4–6 h, exposure to the drug (Jevtovic-Todorovic et al. 2003). This is in keeping with the in vitro studies, where promotion of apoptosis was observed after 6 h of the activity blockade (Heck et al. 2008). In the present study, we found that a single administration of ethanol at the doses evoking cortical neuroapoptosis produces rapid suppression of cortical activity lasting for more than 4 h. Taken together, these findings suggest that the minimal period of the activity blockade required for stimulation of apoptosis is in the range of several hours, which is achieved in the case of isoflurane by continuous exposure to the drug and in the case of a single administration of alcohol by elevated ethanol levels lasting for several hours after injection (see also Ikonomidou et al. 2000).
Another mechanism through which suppression of cortical activity by ethanol may contribute to the adverse neurobehavioral outcome involves disruption of the activity-dependent process of the development of cortical circuits. There is considerable evidence indicating that the activity patterns expressed in the developing circuits enable correlated neuronal firing, support synaptic plasticity and participate in the formation of the neuronal circuits (Katz and Shatz 1996; Khazipov and Luhmann 2006; Blankenship and Feller 2010; An et al. 2012; Khazipov et al. 2013; Benders et al. 2015). This has been particularly well demonstrated in the somatosensory cortex, where the critical period of the activity-dependent formation of the somatosensory cortical maps occurs during the first postnatal week (Erzurumlu and Gaspar 2012; Mitrukhina et al. 2015). It could be suggested that a long-lasting, for more than several hours, pause in this activity-dependent construction of neuronal circuits after administration of ethanol may disturb this developmental process, contributing to malformations in the cortical networks.
The first postnatal week in rodents is an early phase of the “brain growth spurt,” when neurons show their highest rate of morphological and functional differentiation and formation of synaptic connections (Ben Ari 2001). This period roughly corresponds to a period from 20 to 30 gestation weeks in the human fetus and it is characterized by transient activity patterns homologous to the early activity patterns seen in the neonatal rodents (Khazipov and Luhmann 2006; Milh et al. 2007; Colonnese et al. 2010; Colonnese and Khazipov 2012). Similarly to neonatal rodents, the fetal primate brain is particularly sensitive to the apoptogenic actions of alcohol during this period (Farber et al. 2010; Creeley and Olney 2013; Olney 2014). Reported here neuroapoptotic BAC levels in the range from 200 to 500 mg/dL are attained in human according to Widmark's formula after ingestion from approximately 7 to 16 standard (17 g) alcohol drinks (60 kg woman, 2 h drinking period). Alcohol consumed by a pregnant woman easily crosses the placenta and one to 2 h after maternal ingestion, fetal BAC reaches levels nearly equivalent to maternal levels with reported values at delivery of up to 800 mg/dL and exposure of the fetus to ethanol is aggravated by impaired ethanol elimination by the fetus because of reduced metabolic capacity and prolonged fetal exposure time owing to the reuptake of amniotic-fluid containing ethanol by the fetus (Burd et al. 2012). Alcohol easily reaches the brain through lipid-mediated blood–brain barrier penetrability and equilibrates with the BAC as evidenced by comparisons of CSF and BAC levels (Viamontes et al. 1972; Cornford et al. 1982). In keeping with these observations, the results of our study predict that alcohol should inhibit fetal brain and that the basis of the particular susceptibility of the fetal brain to alcohol may involve particularly high sensitivity of the early brain activity to this agent.
Supplementary Material
Supplementary material can be found at http://www.cercor.oxfordjournals.org/online.
Funding
This work was supported through Fondation pour la Recherche Medicale (DEQ20110421301 to R.K.), Government of the Russian Federation (11.G34.31.0075 to R.K.), Ministry of Education and Research, the subsidy allocated for the state assignment in the sphere of scientific activities and the Program of Competitive growth of Kazan Federal University.
Notes
We thank Drs John Olney and Heiko Luhmann for their careful and critical reading of the manuscript. Conflict of Interest: None declared.
References
Author notes
These authors contributed equally to this study.