




Abstract
MicroRNAs are non-coding small RNAs that regulate gene expression by Watson–Crick base pairing to target messenger RNA (mRNA). They are involved in most biological and pathological processes, including tumorigenesis. The binding of microRNA to mRNA is critical for regulating the mRNA level and protein expression. However, this binding can be affected by single-nucleotide polymorphisms that can reside in the microRNA target site, which can either abolish existing binding sites or create illegitimate binding sites. Therefore, polymorphisms in microRNA can have a differing effect on gene and protein expression and represent another type of genetic variability that can influence the risk of certain human diseases. Different approaches have been used to predict and identify functional polymorphisms within microRNA-binding sites. The biological relevance of these polymorphisms in predicted microRNA-binding sites is beginning to be examined in large case–control studies.
Introduction
Recently, Stefano Landi et al. ( 1 ), who performed a case–control study that examined the association of eight polymorphisms within microRNA-binding sites with the risk of sporadic colorectal cancer, found that the variant alleles of CD86 and insulin receptor ( INSR ) genes were statistically significantly associated with the risk of colorectal cancer. This was the first epidemiological study showing an association between microRNA-binding single-nucleotide polymorphism (SNP) sequences and cancer risk.
However, an association between microRNA-binding SNPs and the risk of common diseases was indicated in three earlier studies. In a landmark study, Abelson et al. ( 2 ) showed that a 3′ untranslated region (UTR) SNP in the human SLITRK1 gene strengthens an existing miR-189 target site, thereby amplifying the downregulation of SLITRK1 , which is implicated in Tourette syndrome. Another study demonstrated that a 3′ UTR SNP in the sheep myostatin ( Gdf8 ) gene creates a new illegitimate microRNA target site, which leads to the significant downregulation of Gdf8 and contributes to the development of muscular hypertrophy ( 3 ). In the third study, two groups almost simultaneously investigated the 3′ UTR of the human AGTR1 gene that contains the SNP rs5186 and showed that hsa-miR-155 specifically downregulates the expression of only the 1166A, and not the 1166C, allele of rs5186 ( 4 , 5 ). Both groups concluded that, by abrogating the regulation by hsa-miR-155, the 1166C allele may be functionally associated with hypertension and cardiovascular disease.
All the studies mentioned above were uniformly founded on the assumption that microRNAs regulate a variety of biological and pathological processes, such as cell growth, differentiation, apoptosis and tumorigenesis ( 6–15 ). From this, it is apparent that genetic polymorphisms that reside on microRNA-binding sites can alter the microRNA–target interaction and result in the difference of target gene expression, which can in turn influence the individual risk of common diseases. Although so far there are only a few isolated examples of associations between microRNA variation and diseases, with the increasing trend toward genome-wide association studies, this situation is highly probably to change in the near future. Perhaps the most widespread manifestation of microRNA variations in diseases will be shown by genetic association studies of common complex diseases since the potential number of target sites where a variation might occur is very high and the impact of these variations fits in well with the subtle and complex phenotypes of many common diseases. In this short review, we summarize the history of microRNAs and what is known about their biogenesis. We also describe the mechanism by which microRNAs bind to their target messenger RNAs (mRNAs). In addition, we discuss the potential effect of polymorphisms within microRNA-binding sites in the development of common diseases, and cancer in particular.
History of microRNAs
MicroRNAs are evolutionarily conserved, endogenous, single-stranded, non-coding RNA molecules of ∼22 nt in length that function as posttranscriptional gene regulators by pairing to the mRNA of protein-coding genes, which has several possible consequences: it can result in the cleavage of mRNA ( 16 ) or the repression of productive translation ( 17 , 18 ) or even the destabilization and reduction in the mRNA concentration by accelerating poly (A) tail removal ( 19 , 20 ). Recently, a deviation from the above understanding of microRNA function was brought to light by the finding that miR-369-3 can upregulate the translation of the tumor necrosis factor alpha ( 21 ), suggesting that microRNA function has an added level of complexity.
Lin-4 , identified in 1993 in Caenorhabditis elegans , is the first microRNA ever discovered ( 22 ). It does not code for a protein, however, but rather is transcribed into a 22 nt RNA molecule, which can repress the expression of the lin-14 mRNA by directly interacting with its 3′ UTR. Although this discovery generated intense interest at the time, lin-4 was only then regarded as an on–off oddity with no other homologous family members in C.elegans. Major breakthroughs in our understanding of microRNA occurred 7 years later with the discovery of two key characteristics of let-7 and small interfering RNA (siRNAs). The first discovery was that the sequence and temporal expression pattern of let-7 was largely conserved in a variety of organisms, which supported the important and conserved roles these small RNAs might play in gene regulation ( 23 ). The second discovery was that the regulation mechanisms are similar to those of siRNA ( 24 ). These findings sparked the rapid progression of research in this area such that now hundreds of microRNAs have been identified in different species ( 25–28 ). In humans, up to 500 microRNAs have been identified so far ( 29 ), and computational analyses have predicted that up to 1000 microRNAs exist in the genome ( 30 ). Of further note, since a single microRNA can bind to >100 different target transcripts ( 31 ), it has been estimated that microRNAs may be able to regulate up to 30% of the protein-coding genes in the human genome ( 32 ). This further highlights their importance as global regulators of gene expression.
Biogenesis of microRNAs
MicroRNAs are transcribed from different genomic locations ( 33 ): they are embedded in either independent non-coding RNAs or in the introns of protein-coding genes. Additionally, some microRNAs are clustered in polycistronic transcripts ( 34 , 35 ). Several microRNAs are expressed in a tissue-specific and developmental stage-specific manner ( 36 , 37 ).
In brief, the synthesis and maturation of microRNAs are made up of the following steps ( 38–41 ). Initially, the microRNAs are transcribed from microRNA genes as long primary RNAs (pri-microRNAs). Since pri-microRNAs usually contain the cap structure and the poly (A) tail, it has been suggested that the transcription of microRNAs is carried out by RNA polymerase II ( 42 ). In the nucleus, the pri-microRNAs are processed by Drosha, a member of the RNase III enzyme family, into precursors (pre-microRNAs) of ∼70 nt in length with a stem-loop structure, in conjunction with the double-stranded RNA-binding protein DGCR8/Pasha. Pre-microRNAs are exported from the nucleus to the cytoplasm by exportin-5 in a Ran-guanosine triphosphate (GTP)-dependent manner, where they are processed by another RNase III enzyme, Dicer. This causes the release of a ∼22 nt double-stranded RNA duplex that is incorporated into a RNA-induced silencing complex in a manner analogous to that observed for siRNA ( 43 ). In this complex, one strand is retained as the mature microRNA, whereas the other strand is generally degraded. This complex is now capable of regulating its target genes.
MicroRNA-binding target
The microRNA within the RNA-induced silencing complex acts by binding to the 3′ UTRs of target mRNAs ( 44 ), which inhibits their translation. The critical region for microRNA binding is nucleotides 2–8 from the 5′ end of the microRNA, called the ‘seed region’, which binds to its target site on a given mRNA by Watson–Crick complementarity. Asymmetry is the general rule for matches between a microRNA and its target, in that the 5′ end of the microRNA tends to have more bases complementary to the target than the 3′ end does. In a series of experiments done to determine the minimal requirements for a functional microRNA target site, Cohen et al. ( 45 ) concluded that the complementarity of seven or more bases to the 5′ end microRNA (miRNA) is sufficient to confer regulation and that sites with weaker 5′ complementarity require compensatory pairing to the 3′ end, but that only extensive pairing to the 3′ end of the microRNA is not sufficient to confer regulation. Additional Watson–Crick pairing to four contiguous nucleotides at nucleotides 12–17 enhances microRNA targeting, especially nucleotides 13–16 ( 46 ).
In truth, the existence of a seed region is not a generally reliable predictor of a real miRNA target ( 47 ). Because it is an energy-consuming process to free the base pair within mRNA in order to make the target accessible for microRNA binding, secondary structures are also required for target recognition to occur. In this regard, Zhao et al. ( 48 ) demonstrated that a common feature of most validated targets is that microRNAs preferentially target 3′ UTR sites that do not have complex secondary structures and are located in accessible regions of the RNA based on favorable thermodynamics. Kertesz et al. ( 49 ) systematically investigated the role of target site accessibility in microRNA target recognition and experimentally showed that mutations diminishing target accessibility substantially reduce microRNA-mediated translational repression. Since target accessibility may be a critical feature of miRNA target recognition, it has been suggested that the free energy (ΔG) of the 70 nucleotides flanking the 5′ and 3′ sides of the predicted miRNA-binding sites be determined using the mFold web server ( 50 ). Judging by the resultant ΔG calculated using the nucleotide sequence surrounding the miRNA-binding sites, those sites with a ΔG higher than randomly expected may not be accessible to miRNAs. However, a model based on the binding energy of the microRNA–target duplex often correlates poorly with the observed degree of repression. To overcome this problem, Kertesz et al. ( 49 ) devised an energy-based score, ΔΔG, for measuring microRNA–target interactions; this score represents the difference between the free energy gained by the binding of the microRNA to the target and the free energy lost by unpairing the target-site nucleotides. In the new model, ΔΔG strongly correlates ( r = 0.7, P < 4 × 10 −4 ) with the measured degree of repression.
Current consensus is that microRNA target specificity is truly determined by both sequence matching and target accessibility. This is because even sequences with high complementarity might not be the real targets of a microRNA ( 47 ). In addition, the same site might be rendered accessible under certain cellular conditions that would promote the unfolding of stable secondary structures ( 51 ). This might add yet another layer of regulation to microRNA target selection, one that is controlled by cellular events corresponding to various stresses or that is regulated in a tissue-specific manner. Further, microRNAs could function co-operatively to bind to one accessible site ( 52 ), affecting the neighboring regions, which would alter the secondary structure and enable or inhibit the binding of other microRNAs.
There are a number of algorithms that have been used to identify putative microRNA-binding sites. PicTar computes a maximum likelihood score that indicates to what extent a given mRNA 3′ UTR is targeted by a fixed set of miRNA ( 53 ). DIANA-MicroT finds miRNA–target duplexes that are conserved in humans and mice with the minimum free energy ( 54 ). miRBase includes the miRNA gene nomenclature, miRNA sequence data and annotation and predicted miRNA target genes ( 29 ). miRanda considers the sequence complementarity of the mature miRNA and the target site, the binding energy of the miRNA–target duplex and the evolutionary conservation of the target position in aligned UTRs of homologous genes ( 55 ). TargetScan searches the 3′ UTRs for segments of perfect Watson–Crick complementarity to bases 2–8 of the miRNA numbered from the 5′ end and assigns a free energy to the miRNA–target site interaction ( 31 ). MicroInspector generates a list of possible target sites, which are then sorted by free-energy values ( 56 ). PupaSuite selects SNPs with potential phenotypic effect and is specifically oriented to help in the design of large-scale genotyping projects ( 57 ). PolymiRTS database is a collection of naturally occurring DNA variations in putative microRNA target sites and integrates sequence polymorphism, phenotype and expression microarray data ( 58 ). Patrocles database lists putative polymorphic microRNA–target interactions as a novel source of phenotypic variation ( 59 ).
When utilizing these algorithms for target prediction, it is important to note that multiple algorithms should be utilized because these programs often predict distinct miRNA-binding sites. For example, when the AGTR1 gene was analyzed for putative miRNA-binding sites, miRanda predicted 27 miRNA-binding sites, whereas TargetScan predicted 37 sites with overlap of only 8 of the predicted miRNA-binding sites from that predicted by miRanda algorithms. In addition, when the AGTR1 gene was analyzed by the PicTar algorithm, no miRNA-binding site was predicted because of the rigid requirement for sequence conservation of binding sites across different species (i.e. human, chimp, mouse, rat and dog). Therefore, it is advisable to use multiple algorithms to predict miRNA binding for genotyping studies ( 60 ).
Further insights into the microRNA–mRNA interaction mechanism are needed to make target prediction much more accurate and efficient, knowledge that is critical to understanding microRNA biology ( 61 ). Recently, four criteria were recommended for validating the authenticity of a functional microRNA target: (i) the microRNA–target interaction must be verified, (ii) the predicted microRNA and mRNA target gene must be coexpressed, (iii) a given microRNA must have a predictable effect on target protein expression and (iv) microRNA-mediated regulation of target gene expression should equate with altered biological function ( 60 ). Although all these criteria might not be met under all conditions, it is advisable that as many as possible be met.
Polymorphisms within microRNA-binding sites
Since the sequence complementarity and thermodynamics of binding play an essential role in the interaction of microRNA with its target mRNA, sequence variations such as SNPs in the microRNA-binding seed region should disrupt the microRNA–mRNA interaction and affect the expression of microRNA targets ( 49 , 62 ). Simply put, an SNP may either abolish or weaken a microRNA target or create a perfect sequence match to the seed of a microRNA that otherwise was not associated with the given mRNA ( 63 , 64 ). The increase or decrease in microRNA binding caused by the SNP variation would probably lead to a corresponding decrease or increase in protein translation ( 63 , 65 , 66 ). This would make it possible to predict the effect of this type of SNP. For example, SNP-associated deregulation of the expression of an oncogene or tumor suppressor might contribute to tumorigenesis ( 67–69 ).
In their study examining the genetics of papillary thyroid carcinoma, He et al. ( 70 ) sequenced the regions harboring two microRNA-binding sites in the KIT gene from papillary thyroid carcinoma patients. A polymorphism was found in each binding site. The G>A SNP (rs17084733) is located within the KIT 3′ UTR complementary to the seed region of miR-221/222. The GA heterozygosity of this SNP was found to lead to a notable increase in ΔG. The synonymous G>C SNP (rs3733542) in exon 18 is located within the site complementary to the seed region of miR-146a/146b. This genotype leads to changes in the microRNA–mRNA duplex interaction and results in hybridization with a different region. Both SNPs have been associated with deregulated expression of the KIT protein and contribute to papillary thyroid carcinoma pathogenesis; possible mechanisms are shown in Figure 1 .
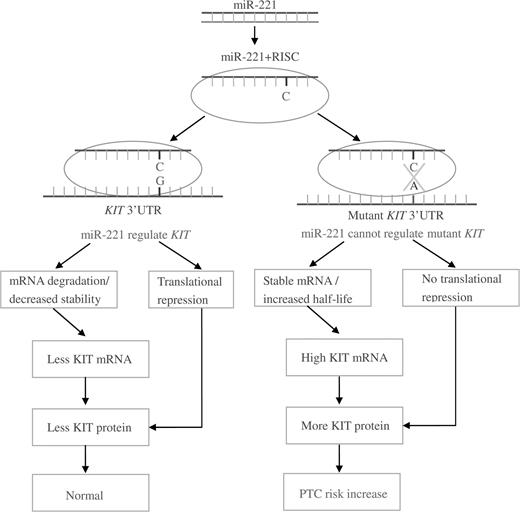
Possible mechanism for SNP within KIT 3′ UTR and papillary thyroid carcinoma risk.
Almost all microRNAs have multiple targets and regulate the expression of hundreds of genes. Some of these genes have important cellular functions. Therefore, in principle, a variation in microRNAs themselves may have an extensive and often deleterious impact ( 71 ), and the cell with the variant microRNA is vulnerable to negative selection ( 72 ). Consistently, germ line alterations in the primary transcript of miR-16 were found in chronic lymphocytic leukemia, both in humans ( 73 ) and in a mouse model of this disease ( 74 ). Moreover, because microRNA is very short and its sequence highly conserved, the frequency of polymorphisms/mutations within microRNA is expected to be low. Indeed, one study has indicated that the likelihood of an SNP occurring in a microRNA seed region is <1% ( 75 ). The significance of such SNPs/mutations from the standpoint of population genetics still has to be determined in large sets of cancer patients. In contrast to the microRNA seed region, the effect of polymorphisms within target sequences is limited only to the target mRNA, but considering the large amount of target sequences and the less conserved non-coding 3′ UTR, the frequency of this kind of SNP should be much higher, and such SNPs are more important from an epidemiological standpoint.
The recent development of methods to identify microRNAs and their binding sites, along with the availability of comprehensive genomic databases of SNPs, is now providing an unprecedented opportunity to explore human evolution at the microRNA target level ( 29 , 76–81 ). Already, a bioinformatic genome-wide survey of human SNPs has revealed an appreciable level of variation within predicted or experimentally verified microRNA target sites ( 75 ). The investigators in this study speculated that some of these variations may have biological relevance and are worth further investigation in case–control studies examining their association with certain biological or pathological events. The issue now is how to determine which of the numerous SNPs within target sites are potentially related to certain biological or pathological events of concern to epidemiologists?
Two groups have focused on identifying SNPs of relevance to cancer. Yu’s group ( 82 ) comprehensively defined the frequencies of microRNA-binding SNPs in cancers versus normal tissues through the mining of EST databases. Interestingly, they found that the allele frequencies of some microRNA-binding SNPs differed significantly between the human cancer EST libraries and the dbSNP database. More importantly, in the case–control association studies, they found 12 microRNA-binding SNPs that displayed an aberrant allele frequency in human cancers. Landi’ s group ( 83 ) selected the 3′ UTRs of 129 genes involved in pathways commonly recognized as important to cancer and identified putative microRNA-binding sites using specialized algorithms. Then they investigated the ability of 79 SNPs within the putative binding sites to affect or impair the binding of the SNP with the microRNA by assessing the ΔΔG through comparisons of the wild-type alleles and their corresponding variant alleles. By considering the validation status of the SNPs and their frequencies, they identified 15 candidate polymorphisms of potential relevance to cancer. The SNPs with a relatively higher population frequency in most ethnic groups from the two studies are summarized in Table I .
Candidate polymorphisms of biological relevance within miRNA target sites
| Target gene | SNP | Alleles | Frequency (A/AA/E/SSA) a | MiRNA |
| RYR3 | rs1044129 | A/G | -/-/0.680/0.275 | mir-153 |
| DAG1 | rs12583 | G/T | -/-/0.302/- | mir-184 |
| SETD8 | rs16917496 | C/T | 0.22/-/-/- | mir-502 |
| IQGAP1 | rs1042538 | A/T | 0.438/0.043/0.100/0.093 | mir-124 |
| ACAA2 | rs7233791 | C/G | 0.114/0.159/0.050/0.186 | mir-124 |
| MTPN | rs17168525 | C/T | 0.083/0.000/0.008/0.000 | let7/mir-98 |
| ALOX15 | rs916055 | T/C | 0.333/0.217/0.295/0.142 | mir-588/183 |
| CD4 | rs3213428 | A/G | 0.458/0.333/0.159/0.292 | mir-518 |
| CD86 | rs17281995 | G/C | 0.027/0.071/0.109/0.102 | mir-337/184/200a/212/582 |
| CDKN2A | rs11515 | C/G | 0.011/0.121/0.125/0.142 | mir-601 |
| IL12B | rs1368439 | A/C | 0.022/0.032/0.217/0.025 | mir-513/210 |
| IL16 | rs1131445 | T/C | 0.211/0.250/0.358/0.317 | mir-135a/135b |
| IL18 | rs360727 | G/A | -/0.152/0.167/- | mir-197/361 |
| INSR | rs1051690 | G/A | 0.033/-/0.276/0.271 | mir-612/618 |
| NOD2 | rs3135500 | G/A | 0.289/0.604/0.397/0.568 | mir-158/215 |
| PLA2G2A | rs11677 | C/T | 0.174/0.022/0.125/0.000 | mir-187 |
| PTGER4 | rs16870224 | G/A | 0.396/0.022/0.104/0.000 | mir-9/30a-3p |
| RAF1 | rs1051208 | C/T | 0.062/0.364/0.146/0.316 | mir-213 |
| Target gene | SNP | Alleles | Frequency (A/AA/E/SSA) a | MiRNA |
| RYR3 | rs1044129 | A/G | -/-/0.680/0.275 | mir-153 |
| DAG1 | rs12583 | G/T | -/-/0.302/- | mir-184 |
| SETD8 | rs16917496 | C/T | 0.22/-/-/- | mir-502 |
| IQGAP1 | rs1042538 | A/T | 0.438/0.043/0.100/0.093 | mir-124 |
| ACAA2 | rs7233791 | C/G | 0.114/0.159/0.050/0.186 | mir-124 |
| MTPN | rs17168525 | C/T | 0.083/0.000/0.008/0.000 | let7/mir-98 |
| ALOX15 | rs916055 | T/C | 0.333/0.217/0.295/0.142 | mir-588/183 |
| CD4 | rs3213428 | A/G | 0.458/0.333/0.159/0.292 | mir-518 |
| CD86 | rs17281995 | G/C | 0.027/0.071/0.109/0.102 | mir-337/184/200a/212/582 |
| CDKN2A | rs11515 | C/G | 0.011/0.121/0.125/0.142 | mir-601 |
| IL12B | rs1368439 | A/C | 0.022/0.032/0.217/0.025 | mir-513/210 |
| IL16 | rs1131445 | T/C | 0.211/0.250/0.358/0.317 | mir-135a/135b |
| IL18 | rs360727 | G/A | -/0.152/0.167/- | mir-197/361 |
| INSR | rs1051690 | G/A | 0.033/-/0.276/0.271 | mir-612/618 |
| NOD2 | rs3135500 | G/A | 0.289/0.604/0.397/0.568 | mir-158/215 |
| PLA2G2A | rs11677 | C/T | 0.174/0.022/0.125/0.000 | mir-187 |
| PTGER4 | rs16870224 | G/A | 0.396/0.022/0.104/0.000 | mir-9/30a-3p |
| RAF1 | rs1051208 | C/T | 0.062/0.364/0.146/0.316 | mir-213 |
A, Asian; AA, African-American; E, European; SSA, sub-Saharan African.
Candidate polymorphisms of biological relevance within miRNA target sites
| Target gene | SNP | Alleles | Frequency (A/AA/E/SSA) a | MiRNA |
| RYR3 | rs1044129 | A/G | -/-/0.680/0.275 | mir-153 |
| DAG1 | rs12583 | G/T | -/-/0.302/- | mir-184 |
| SETD8 | rs16917496 | C/T | 0.22/-/-/- | mir-502 |
| IQGAP1 | rs1042538 | A/T | 0.438/0.043/0.100/0.093 | mir-124 |
| ACAA2 | rs7233791 | C/G | 0.114/0.159/0.050/0.186 | mir-124 |
| MTPN | rs17168525 | C/T | 0.083/0.000/0.008/0.000 | let7/mir-98 |
| ALOX15 | rs916055 | T/C | 0.333/0.217/0.295/0.142 | mir-588/183 |
| CD4 | rs3213428 | A/G | 0.458/0.333/0.159/0.292 | mir-518 |
| CD86 | rs17281995 | G/C | 0.027/0.071/0.109/0.102 | mir-337/184/200a/212/582 |
| CDKN2A | rs11515 | C/G | 0.011/0.121/0.125/0.142 | mir-601 |
| IL12B | rs1368439 | A/C | 0.022/0.032/0.217/0.025 | mir-513/210 |
| IL16 | rs1131445 | T/C | 0.211/0.250/0.358/0.317 | mir-135a/135b |
| IL18 | rs360727 | G/A | -/0.152/0.167/- | mir-197/361 |
| INSR | rs1051690 | G/A | 0.033/-/0.276/0.271 | mir-612/618 |
| NOD2 | rs3135500 | G/A | 0.289/0.604/0.397/0.568 | mir-158/215 |
| PLA2G2A | rs11677 | C/T | 0.174/0.022/0.125/0.000 | mir-187 |
| PTGER4 | rs16870224 | G/A | 0.396/0.022/0.104/0.000 | mir-9/30a-3p |
| RAF1 | rs1051208 | C/T | 0.062/0.364/0.146/0.316 | mir-213 |
| Target gene | SNP | Alleles | Frequency (A/AA/E/SSA) a | MiRNA |
| RYR3 | rs1044129 | A/G | -/-/0.680/0.275 | mir-153 |
| DAG1 | rs12583 | G/T | -/-/0.302/- | mir-184 |
| SETD8 | rs16917496 | C/T | 0.22/-/-/- | mir-502 |
| IQGAP1 | rs1042538 | A/T | 0.438/0.043/0.100/0.093 | mir-124 |
| ACAA2 | rs7233791 | C/G | 0.114/0.159/0.050/0.186 | mir-124 |
| MTPN | rs17168525 | C/T | 0.083/0.000/0.008/0.000 | let7/mir-98 |
| ALOX15 | rs916055 | T/C | 0.333/0.217/0.295/0.142 | mir-588/183 |
| CD4 | rs3213428 | A/G | 0.458/0.333/0.159/0.292 | mir-518 |
| CD86 | rs17281995 | G/C | 0.027/0.071/0.109/0.102 | mir-337/184/200a/212/582 |
| CDKN2A | rs11515 | C/G | 0.011/0.121/0.125/0.142 | mir-601 |
| IL12B | rs1368439 | A/C | 0.022/0.032/0.217/0.025 | mir-513/210 |
| IL16 | rs1131445 | T/C | 0.211/0.250/0.358/0.317 | mir-135a/135b |
| IL18 | rs360727 | G/A | -/0.152/0.167/- | mir-197/361 |
| INSR | rs1051690 | G/A | 0.033/-/0.276/0.271 | mir-612/618 |
| NOD2 | rs3135500 | G/A | 0.289/0.604/0.397/0.568 | mir-158/215 |
| PLA2G2A | rs11677 | C/T | 0.174/0.022/0.125/0.000 | mir-187 |
| PTGER4 | rs16870224 | G/A | 0.396/0.022/0.104/0.000 | mir-9/30a-3p |
| RAF1 | rs1051208 | C/T | 0.062/0.364/0.146/0.316 | mir-213 |
A, Asian; AA, African-American; E, European; SSA, sub-Saharan African.
In general, at least three factors should be considered in the design of a case–control study: the target genes should be related directly or indirectly to the disease under study; the polymorphisms should be within the seed region in most cases and there should be definite data regarding the population frequency of the SNPs in the ethnic group being studied and a frequency of no <5% for the variant allele is preferable.
Perspectives for epidemiologists
Researchers have tried different bioinformatics methods to select functional candidate polymorphisms in microRNA target sites, but different methods have identified different sets of SNPs, indicating that each study provided only a partial picture that may not reflect the real situation. Nevertheless, some of the candidate SNPs in the microRNA-binding targets have been investigated in case–control studies, which have revealed promising results. In particular, these early studies have shown that microRNAs are a new class of genetic factors with a role in human diseases. Now, computational predictions in combination with experimental verifications will reveal better SNP targets in microRNA-binding sites for case–control studies, and case–control studies will in turn validate the effectiveness of the predictions and the importance of the target SNPs in human diseases. In this respect, case–control studies will serve both as a bridge between computational algorithms and biological analyses and as a sieve that filters out from the relatively large number of candidate SNPs the ones with potential functional significance for in-depth functional characterization.
The genomics revolution continues to transform epidemiology studies. The resultant new genomic knowledge is being increasingly integrated into epidemiological research, and microRNA is no exception. Indeed, recent studies have provided solid evidence that microRNA is closely involved at the genetic level in the susceptibility to, progression and prognosis of and response to therapy of many complex diseases, including cancer ( 70 , 73 , 84–86 ). Therefore, it is expected that molecular epidemiologists will conduct extensive investigations of microRNA as an etiological factor for diseases in the coming years. A better understanding of distinct polymorphisms in microRNA-related gene and protein expression regulation in different diseases at the population level will be a critical step toward the clinical utilization of this new subclass of genetic variations in health management. Large and well-characterized population-based, case–control studies will play a central role in the future implementation of microRNA target SNP-based gene analysis. A new gold mine and a new gold rush are in the offing.
Abbreviations
- mRNA
messenger RNA
- miRNA
microRNA
- SNP
single-nucleotide polymorphism
- UTR
untranslated region
We would like to thank Beth Notzon in the Department of Scientific Publication of the M. D. Anderson Cancer Center for her editorial assistance.
Conflict of Interest Statement: None declared.
References
Author notes
Both are first authors

{kind=link}