




Abstract
DEP-1/HPTPη , a receptor-type protein tyrosine phosphatase, is a candidate tumor suppressor gene because its expression was blocked in rat and human thyroid transformed cells, and its restoration reverted their neoplastic phenotype. In addition, loss of DEP-1/HPTPη heterozygosity has been described in mammary, lung and colon primary tumors. We now show that DEP-1/HPTPη is drastically reduced in several cell lines originating from human epithelial pancreatic carcinomas compared with normal pancreatic tissue. We also show that the infection of AsPC1 and PSN1 cells with a recombinant adenovirus carrying r-PTPη cDNA (the rat homolog of DEP-1/HPTPη ) inhibits their proliferation. Flow cytometric analysis of the infected cells demonstrated that restoration of r-PTPη activity disrupts their cell cycle and leads to apoptosis. Finally, the growth of PSN1 xenograft tumors was blocked by the intratumoral injection of a recombinant adeno-associated virus carrying r-PTPη . The data suggest that restoration of DEP-1/HPTPη expression could be a useful tool for the gene therapy of human pancreatic cancers.
Introduction
Duct cell adenocarcinoma, the most common type of pancreatic cancer, is the fifth leading cause of cancer death in the US. Each year, almost 30 000 cases are diagnosed in the US and in most cases the disease is fatal ( 1 ). The incidence of duct cell adenocarcinoma increases steadily with age and ∼80% of the cancers occur within the seventh and eighth decades of life ( 2 ). Pancreatic cancer is one of the most difficult malignancies to treat. Currently, radiation and chemotherapy are not effective and complete surgical resection remains the only therapeutic option. However, only 5% of patients are resectable at diagnosis, and for all stages of surgically resectable disease combined, the overall 5-year survival is <15% ( 3 , 4 ).
Several genetic lesions have been identified in pancreatic cancers ( 5 , 6 ). Mutations of the K- ras gene occur in over 90% of cases ( 7 ), and the protooncogenes Akt2 and Myb are found amplified in 10–20 and 10%, respectively ( 8 , 9 ).
Tumor suppressor genes are also involved in the generation of human pancreatic carcinomas. In fact, loss of heterozygosity (LOH) is found in >40% of chromosomal arms in pancreatic cancer ( 10 ). The tumor suppressor genes most commonly inactivated in this neoplasia are p16, p53 and DPC4, which are genetically inactivated in 50% or more of cases ( 11 – 16 ). Lastly, alterations of FHIT have also been identified in pancreatic cancers ( 17 ).
Very little is known about the role of protein tyrosine phosphatases in exocrine pancreatic tumorigenesis, even though they might inhibit pancreatic malignant cell growth ( 18 , 19 ). Interestingly, the dual-specificity protein phosphatase PTEN, which is a key factor in human tumors ( 20 , 21 ), is unchanged in pancreatic tumors ( 22 , 23 ).
The aim of this study was to decipher the role of the receptor-type tyrosine phosphatase DEP-1/HPTPη in human pancreatic carcinoma. DEP-1/HPTPη encodes a receptor-type protein tyrosine phosphatase that contains an extracellular region, a short transmembrane segment and a unique intracellular catalytic domain ( 24 , 25 ). This protein, also known as CD148 and PTPRJ, is physiologically involved in such cellular processes as contact inhibition ( 24 ). It is present in all hematopoietic lineages and is involved in signal transduction with lymphocytes ( 26 ), and in the regulation of T-cell activation ( 27 ); CD148 was also found expressed in functional splenic memory B cells ( 28 ). Finally, CD148 was reported to be a negative regulator of T-cell activation by interfering with the phosphorylation state and function of PLCγ1 and LAT ( 29 ).
We previously showed that the r-PTPη gene is drastically reduced in rat thyroid cells transformed by acute murine retroviruses and in human thyroid carcinomas ( 30 ). The re-expression of the r-PTPη gene in highly malignant rat thyroid cells transformed by retroviruses carrying the v-mos and v-ras-Ki oncogenes suppresses their malignant phenotype. Protein levels of p27 Kip1 , a crucial cyclin-dependent kinase inhibitor ( 31 ), was increased in the r-PTPη -transfected cells consequent to the reduced proteasome-dependent degradation rate ( 30 ).
DEP-1/HPTPη is relevant in the development of several human cancers. It is associated with a frequent allelic deletion and with a missense mutation that generates an A1176C substitution which, in turn, might result in conformational and functional changes. Furthermore, Ptprj is the candidate for the mouse colon cancer susceptibility locus Scc1 ( 32 ).
The levels of DEP-1/HPTPη were very low in most of the pancreatic carcinoma cell lines analyzed in our study compared with normal pancreatic tissue. Two pancreatic carcinoma cell lines, AsPC1 and PSN1, infected with the adenovirus Ad r-PTPη carrying the r-PTPη gene in a sense orientation, underwent apoptosis. In contrast, infection with the same adenoviral vector carrying only the GFP gene (control) had no effect. Lastly, a recombinant adeno-associated virus (AAV) carrying r-PTPη injected into pre-existing PSN1 xenograft tumors arrested tumor growth.
Materials and methods
Cell lines
Human pancreatic cancer cell lines AsPC1, BxPC3, PANC1, Hs766T, Capan2, MIAPACA, CFPAC and Su8686 were obtained from American Type Culture Collection (ATCC, Manassas, VA). PSN1 cells were obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK). ARO cells originate from a human thyroid anaplastic tumor ( 33 ). Cells were maintained at 37°C in a humidified atmosphere of 5% CO 2 and grown in RPMI containing 10% fetal calf serum. The HEK293 cell line, obtained from American Type Culture Collection, was used for packaging both adenoviruses and AAVs and for amplifying the adenoviral vectors; HEK293 cells were grown in DMEM containing 10% fetal calf serum.
RNA extraction and northern blot analysis
Total RNA was extracted by using Trireagent (Molecular Research Center, Cincinnati, OH) following the manufacturer's procedure. For northern blot analysis, 20 μg of total RNA per lane were separated in 1% agarose/2.2 M formaldehyde gels and blotted onto nylon membranes (Hybond-Amersham, Piscataway, NJ). DEP-1/HPTPη was detected by using a 728 bp Xho I– Xho I fragment from the r-PTPη cDNA. A specific GAPDH probe was used to verify an equally loaded RNA. The filters were hybridized overnight with radiolabeled probes, washed at high stringency and finally exposed (Kodak films).
Protein extraction and western blot analysis
Total proteins were extracted as described previously ( 30 ). Western blot analyses were performed with conventional methods and an enhanced chemiluminescence detection kit (Amersham, Piscataway, NJ), as described previously ( 34 ). Filters were reacted with polyclonal antibodies raised against r-PTPη ( 30 ), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
DNA extraction and sequence analysis
Genomic DNA was extracted from pancreatic cells by using the QIAamp DNA kit (Qiagen, Valencia, CA) following the manufacturer's instructions. DEP-1/HPTPη exons 5, 6 and 7 were amplified by PCR, verified by 2% agarose gel and sequenced in both strands with the same primers used for the amplification on the Applied Biosystem Prism 377 DNA sequencing system (Perkin-Elmer, Foster City, CA). The following oligonucleotides have been used: (i) exon 5 forward: 5′-gAA ggT gAC TgC ATA TAT CT-3′; (ii) exon 5 reverse: 5′-AgA ACA TTT AgT TAC TgA AAg-3′; (iii) exon 6 forward: 5′-CCT CTT gTg gTT gAT gTC TT-3′; (iv) exon 6 reverse: 5′-CCC CTA TTA TCA AAg TTC AAC-3′; (v) exon 13 forward: 5′-CTg CCA TCA CTT TCT TAT gAT-3′; (vi) exon 13 reverse: 5′-CCC AAA gAg TAA gAA CCA gA-3′. Genomic samples of 200 ng were subjected to PCR in 25 µl aliquots containing 25 pmol of each primer, 1× PCR buffer, 2.5 mM MgCl 2 , 400 µM dNTPs and 2 U of Taq Gold DNA polymerase (Perkin-Elmer). PCR cycles included one cycle of 95°C for 5 min followed by 40 cycles each of 94°C for 30 s, 55°C for 30 s and 72°C for 30 s and a final extension step of 72°C for 7 min.
Plasmids, DNA transfection and colony assay
Wild-type r-PTPη and mutant r-PTPηC → S cDNAs were cloned in a sense orientation in the eukaryotic expression vector pMV-7, as described previously ( 30 ). The pMV-7 vector has a neomycin gene that confers G418 resistance for transfectant selection. Fugene reagent (Boehringer/Roche, Indianapolis, IN) was used to transfect AsPC1 and PSN1 cells with 5 µg of pMV-7 empty vector and with an equimolar amount of either wild-type or mutated r-PTPη according to the manufacturer's instructions. G418 was added to the medium at a final concentration of 400 µg/ml. Two weeks later, the cells were stained with 500 µg of crystal violet/ml in 20% methanol and the colonies were counted.
Generation of the r-PTPη viral vectors
A recombinant adenovirus carrying the r-PTPη coding sequence under the transcriptional control of a CMV promoter was generated as described previously ( 34 ). This expression cassette carries also GFP cDNA under the control of a second CMV promoter. An adenovirus carrying GFP under a CMV promoter (Ad5.CMV-GFP; Qbiogene, Carlsbad, CA) served as a control.
To generate a recombinant r-PTPη transfer vector for the AAV preparation, we used the original vector pAM/EF-EGFP-SV40 late pA ( 35 ) in a multi-step cloning procedure. pAM/EF-EGFP-SV40 late pA was digested with Eco RV and Eco RI to remove the GFP cDNA. These two ends were used for the directional insertion of a polyC adapter to stabilize the r-PTPη mRNA ( 36 ). The primers sequences were: 5′-AAT TCA AgC TTC CCA ACg ggC CCT CCT CCC C-3′ and 5′-ggg gAg gAg ggC CCg TTg ggA AgC TTg-3′. We inserted a Hin dIII restriction site immediately downstream from the Eco RI end. We digested intermediate vector with Kpn I and Hin dIII to remove the EF promoter so that we could replace it with a shorter CMV promoter. We digested the transfer vector pAM/CMV polyC SV40 late pA so obtained with Hin dIII, and used Klenow to generate blunt ends for ligating the r-PTPη cDNA. To eliminate the r-PTPη 3′ untranslated sequences, we replaced the Hin cII– Hin dIII fragment from the pGEM-3Z/ r-PTPη (vector with a shorter Hin cII– Hin dIII PCR fragment containing the stop codon immediately before the Hin dIII site). We used the following primers in the PCR amplification procedure described above: forward 5′-gAC CTC ATC TAT CAg AAC AC-3′ and reverse 5′-TgC TAA gCT TCT Agg CgA TgT AAC CAT TAg-3′. The r-PTPη cDNA was excised from pGEM-3Z/ r-PTPη by digesting it with Pau I (10 bp upstream from the ATG) and Hin dIII. The insert was blunted with Klenow and sub-cloned into pAM/CMV poly(C) SV40 late pA prepared as reported above. We sequenced the vector to check for both correct orientation and sequence of the fragments used in this multi-step cloning. The recombinant AAV r-PTPη was packaged as described previously ( 35 ). A recombinant AAV carrying the reporter gene GFP under the control of a CMV promoter served as a control.
In-vitro transduction
Cell lines were transduced with adenoviral vectors by directly applying the diluted viruses into the growth medium at multiplicity of infection (MOI) 50. Direct visualization of Ad GFP and Ad r-PTPη viruses in transduced cell populations by fluorescent cell microscopy was used to verify the proportion of GFP-expressing cells after infection to determine their transduction efficiency. Cell morphology was assessed through fluorescence microscopy after nuclei staining with Hoechst 33258 (Sigma, St Louis, MO).
RT–PCR analysis
RNAs from wild-type and virally transduced AsPC1 and PSN1 cells were extracted by using Trireagent (Molecular Research Center, Cincinnati, OH). Total RNA was also extracted from xenograft tumors originating from PSN1 cells injected in nude mice in experiments carried out with AAV. Two micrograms of total RNA were reverse transcribed and amplified with r-PTPη -specific primers by means of the RT–PCR Accession kit (Promega, Madison, WI) used according to the manufacturer's instructions. Thirty-five cycles per amplification were performed. The r-PTPη primers were: (i) forward primer: 5′-TCg gTg TgA CAC Agg CTC TT-3′; (ii) reverse primer: 5′-CTg TgA CCC ATA AgT CAT gC-3′. RT–PCR samples were loaded on a 1.0% agarose gel for electrophoresis and stained with ethidium bromide. A specific human GAPDH fragment was amplified to normalize the samples (BD Biosciences Clontech, Palo Alto, CA).
Assessment of in-vitro growth curves
Cells were seeded at 5 × 10 4 cells per 60-mm diameter dish and infected with Ad GFP or Ad r-PTPη at MOI 50. The cells were monitored and counted at 24-h intervals after infection.
Flow cytometry
Wild-type, GFP and r-PTPη virally transduced pancreatic cells were collected and washed in PBS solution. DNA was stained with propidium iodide (50 µg/ml) and analyzed with a FACScan flow cytometer (Becton Dickinson, San Jose, CA) interfaced with a Hewlett-Packard (Palo Alto, CA) computer. Cell cycle data were analyzed with the CELL-FIT program (Becton Dickinson, San Jose, CA).
TUNEL assay
Wild-type, Ad GFP- and Ad r-PTPη -infected cells were assessed for the induction single strand breaks (indicative of apoptosis) by the terminal deoxynucleotidyl transferase mediated X-dUTP nick end labeling (TUNEL) assay using the in situ cell death detection kit (Boehringer/Roche, Indianapolis, IN) according to the manufacturer's recommendations.
Animal studies
Nude mice were obtained from Charles River (Cambridge, MA). Animal experiments were conducted under institutional guidelines established for the Animal Facility at the Kimmel Cancer Center. 1 × 10 6 of PSN1 cells were subcutaneously injected into the right flank of the nude mice; after the tumors appeared (10 days), PBS, AAV GFP and AAV r-PTPη [1 ×10 10 viral particles (VP) per tumor] were intratumorally injected and the mice were killed 12 days after infection and the tumors were extracted. The volume of tumors was calculated with the formula: tumor volume = length × (width) 2 /2.
Results
DEP-1/HPTPη gene expression in human pancreatic cancer cell lines
DEP-1/HPTPη protein expression in the pancreas was assessed previously by immunohistochemistry on frozen sections of normal tissue where it was basically detectable in all components of pancreatic tissue, including acini, ducts and Langerhans islets ( 37 ). Here, we first analyzed the DEP-1/HPTPη gene expression in a panel of nine pancreatic carcinoma cell lines by northern blot analysis using a fragment of the rat r-PTPη corresponding to the catalytic domain of the gene sharing a 100% homology with the human DEP-1/HPTPη gene. DEP-1/HPTPη expression was significantly reduced or suppressed compared with normal pancreas; in fact, DEP-1/HPTPη was barely detectable in PSN1 and MIAPACA cells, and absent in BxPC3, AsPC1 and PANC1 cells; intermediate expression levels were demonstrated in Capan2, CFPAC1, Su8686 and HS766T cells ( Figure 1A ). These results were paralleled by the DEP-1/HPTPη protein expression levels assessment through western blot using a polyclonal antibody raised against the catalytic domain of the molecule ( 30 ). Normal thyroid cells and the ARO cell line, derived from a human thyroid anaplastic tumor ( 33 ), served as positive and negative controls, respectively ( Figure 1B ).
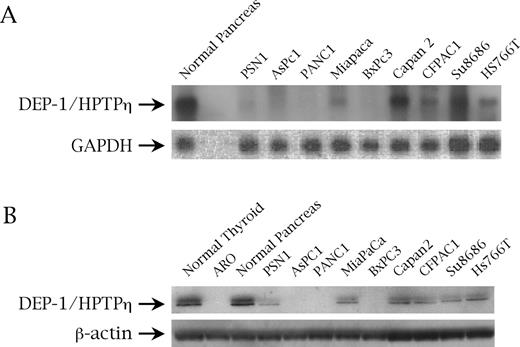
( A ) DEP-1/HPTPη gene expression assessed by northern blot analysis on normal pancreas and pancreatic cancer cell lines. GAPDH was used as a control for RNA loading. ( B ) DEP-1/HPTPη protein expression assessed on normal pancreatic tissue and pancreatic malignant cells by western blot with an anti-r-PTPη antibody. β-Actin is to demonstrate an equal protein loading.
We further analyzed the sequences of exons 5, 6 and 13 of DEP-1/HPTPη in DNA samples from pancreatic cells. These exons harbor three nucleotide polymorphisms, A1176C, G1326A and C2965G, which result in the coding polymorphisms Gln276Pro, Arg326Gln and Asp872Glu, respectively. Gln276Pro has been proposed as implicated in susceptibility to human cancer ( 32 ). The results of such an analysis are reported in Table I .
Genotypic profile of DEP-1 / HPTPη / PTPRJ in human pancreatic carcinoma cell lines
| Cell line | 276 genotype | 326 genotype | 872 genotype |
|---|---|---|---|
| PSN1 | Gln/Pro | Arg/Gln | Glu/Glu |
| AsPC1 | Gln/Gln | Arg/Arg | Glu/Glu |
| PANC1 | Gln/Gln | Arg/Arg | Asp/Asp |
| MIAPACA2 | Gln/Pro | Arg/Gln | Asp/Asp |
| BxPC3 | Gln/Gln | Arg/Arg | Asp/Glu |
| Capan2 | Gln/Gln | Arg/Arg | Asp/Glu |
| CFPAC | Gln/Pro | Arg/Gln | Glu/Glu |
| Su8686 | Gln/Pro | Arg/Gln | Asp/Glu |
| Hs766T | Gln/Pro | Arg/Gln | Asp/Asp |
| Cell line | 276 genotype | 326 genotype | 872 genotype |
|---|---|---|---|
| PSN1 | Gln/Pro | Arg/Gln | Glu/Glu |
| AsPC1 | Gln/Gln | Arg/Arg | Glu/Glu |
| PANC1 | Gln/Gln | Arg/Arg | Asp/Asp |
| MIAPACA2 | Gln/Pro | Arg/Gln | Asp/Asp |
| BxPC3 | Gln/Gln | Arg/Arg | Asp/Glu |
| Capan2 | Gln/Gln | Arg/Arg | Asp/Glu |
| CFPAC | Gln/Pro | Arg/Gln | Glu/Glu |
| Su8686 | Gln/Pro | Arg/Gln | Asp/Glu |
| Hs766T | Gln/Pro | Arg/Gln | Asp/Asp |
Genotypic profile of DEP-1 / HPTPη / PTPRJ in human pancreatic carcinoma cell lines
| Cell line | 276 genotype | 326 genotype | 872 genotype |
|---|---|---|---|
| PSN1 | Gln/Pro | Arg/Gln | Glu/Glu |
| AsPC1 | Gln/Gln | Arg/Arg | Glu/Glu |
| PANC1 | Gln/Gln | Arg/Arg | Asp/Asp |
| MIAPACA2 | Gln/Pro | Arg/Gln | Asp/Asp |
| BxPC3 | Gln/Gln | Arg/Arg | Asp/Glu |
| Capan2 | Gln/Gln | Arg/Arg | Asp/Glu |
| CFPAC | Gln/Pro | Arg/Gln | Glu/Glu |
| Su8686 | Gln/Pro | Arg/Gln | Asp/Glu |
| Hs766T | Gln/Pro | Arg/Gln | Asp/Asp |
| Cell line | 276 genotype | 326 genotype | 872 genotype |
|---|---|---|---|
| PSN1 | Gln/Pro | Arg/Gln | Glu/Glu |
| AsPC1 | Gln/Gln | Arg/Arg | Glu/Glu |
| PANC1 | Gln/Gln | Arg/Arg | Asp/Asp |
| MIAPACA2 | Gln/Pro | Arg/Gln | Asp/Asp |
| BxPC3 | Gln/Gln | Arg/Arg | Asp/Glu |
| Capan2 | Gln/Gln | Arg/Arg | Asp/Glu |
| CFPAC | Gln/Pro | Arg/Gln | Glu/Glu |
| Su8686 | Gln/Pro | Arg/Gln | Asp/Glu |
| Hs766T | Gln/Pro | Arg/Gln | Asp/Asp |
r-PTPη gene restoration reduces colony formation by AsPC1 and PSN1 cells
To ascertain the potential growth-suppressor effect of r-PTPη on pancreatic cancer cells, we evaluated colony formation by transfecting r-PTPη cDNA and its corresponding empty vector, pMV-7, into AsPC1 and PSN1 cells. In both cell lines, the number of colonies formed after transfection with r-PTPη was much lower (at least 20-fold) compared with the number of colonies after transfection with the empty vector. The r-PTPη catalytic activity was responsible for this effect since the transfection of both pancreatic cancer cell lines with a mutant r-PTPη ( 30 ) bearing a point mutation critical for the abolishment of its enzymatic activity (r-PTPη C → S ) showed no reduction of the colony number compared with the backbone vector ( Table II ).
Colony assay of AsPC1 and PSN1 pancreatic cancer cells with r-PTPη
| Number of colonies | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| pMV-7 | 197 ± 18 | 165 ± 11 | |
| pMV/ r-PTPη | 12 ± 2 | 9 ± 3 | |
| pMV/ r-PTPη C→S | 190 ± 12 | 157 ± 10 | |
| Number of colonies | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| pMV-7 | 197 ± 18 | 165 ± 11 | |
| pMV/ r-PTPη | 12 ± 2 | 9 ± 3 | |
| pMV/ r-PTPη C→S | 190 ± 12 | 157 ± 10 | |
Data represents mean ± SD values obtained from three different experiments.
Colony assay of AsPC1 and PSN1 pancreatic cancer cells with r-PTPη
| Number of colonies | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| pMV-7 | 197 ± 18 | 165 ± 11 | |
| pMV/ r-PTPη | 12 ± 2 | 9 ± 3 | |
| pMV/ r-PTPη C→S | 190 ± 12 | 157 ± 10 | |
| Number of colonies | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| pMV-7 | 197 ± 18 | 165 ± 11 | |
| pMV/ r-PTPη | 12 ± 2 | 9 ± 3 | |
| pMV/ r-PTPη C→S | 190 ± 12 | 157 ± 10 | |
Data represents mean ± SD values obtained from three different experiments.
A recombinant adenovirus carrying the r-PTPη gene induces morphological changes and reduces the growth rate of AsPC1 and PSN1 cells
To evaluate the r-PTPη gene as a therapeutic agent, we generated a replication-defective adenovirus carrying r-PTPη cDNA. The cDNA was cloned into the transfer vector pQBI AdCMV5-GFP under the transcriptional control of a CMV promoter. This vector also possesses the GFP reporter gene whose expression is controlled by another independent CMV promoter ( 34 ). AsPC1 and PSN1 cells were infected with either Ad GFP or Ad r-PTPη at MOI 50.
We assessed the susceptibility of these cell lines to adenovirus infection by observing the fluorescence of GFP proteins, carried by the Ad GFP and Ad r-PTPη viruses, with confocal microscopy 24 h after infection. As described previously ( 38 ), AsPC1 were almost 100% GFP-positive, whereas only ∼30% of PSN1 was transduced with Ad GFP and Ad r-PTPη (data not shown). To determine the expression of the exogenous r-PTPη gene in the infected cells, we performed an RT–PCR analysis using specific primers for r-PTPη selected from a non-homolog sequence between the human and rat transcript to avoid amplification of the endogenous messenger. As shown in Figure 2A , both AsPC1 and PSN1 Ad r-PTPη -infected cells expressed the r-PTPη gene; conversely, no amplification was observed in wild-type or Ad GFP-infected cells. Finally, no bands appeared after amplification of non-retrotranscribed RNAs (data not shown).
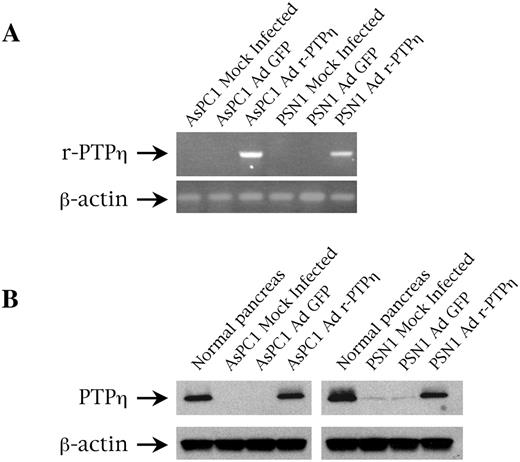
( A ) r-PTPη expression determined by RT–PCR in AsPC1 and PSN1 pancreatic malignant cells transduced with Ad r-PTPη . Expression was analyzed 24 h after infection with Ad r-PTPη at MOI 50. Ethidium bromide staining of RT–PCR amplification performed with oligonucleotides specific for the rat sequence of the exogenous r-PTPη cDNA. Human GAPDH was amplified to assess the quality of the RNA used. ( B ) r-PTPη protein expression assessed on normal pancreas and AsPC1 and PSN1 pancreatic malignant cells infected with Ad r-PTPη by western blot with an anti-r-PTPη antibody. β-Actin is to demonstrate an equal protein loading.
r-PTPη protein expression in Ad r-PTPη -transduced cells was also assessed by western blot with a rabbit polyclonal antibody raised against the catalytic domain ( 30 ); as shown in Figure 2B , r-PTPη expression in AsPC1 and PSN1 Ad r-PTPη -transduced cells was comparable or lower than normal pancreatic tissue, respectively.
After infection with the Ad r-PTPη virus, dramatic morphological changes appeared in both pancreatic carcinoma cell lines. In fact, as assessed by fluorescence microscopy, AsPC1- and PSN1-Ad r-PTPη -transduced cells acquired a flattened appearance and were much larger than cells infected with Ad GFP as a control ( Figure 3 ).
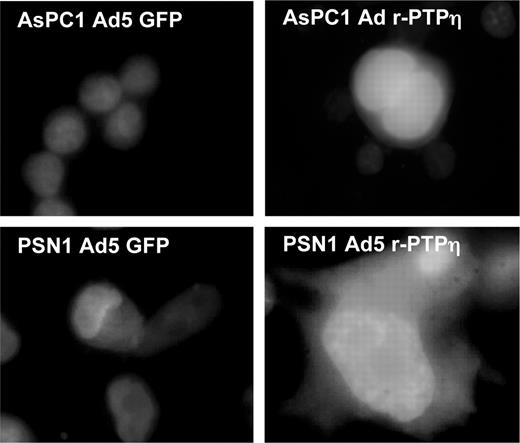
Fluorescent micrograph (original magnification, ×100) showing the morphology of AsPC1 and PSN1 pancreatic cells infected with Ad GFP (left panels) and Ad r-PTPη (right panels) 24 h after infection at MOI 50. Counterstaining with Hoechst dye was used to facilitate nuclei localization. See online supplementary material for a colour version of this figure.
To study the effect of Ad r-PTPη on pancreatic carcinoma cell growth we plated the AsPC1 and PSN1 carcinoma cells (50 000/well), and the next day exposed them to MOI 50 of either Ad r-PTPη or Ad GFP. Cells were harvested daily for cell counting. The infection of the pancreatic carcinoma cells with the Ad r-PTPη virus significantly reduced cell number over the 4 days of the experiment. Growth arrest was much more pronounced in AsPC1 than in PSN1 because the former are more susceptible to adenoviral infection. Cell growth was unchanged in the AsPC1 and PSN1 cells infected with the Ad GFP control virus compared with their mock-infected counterparts ( Figure 4 ).
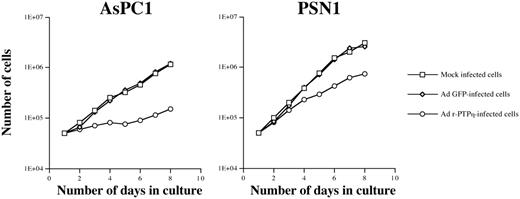
In vitro growth curve of AsPC1 and PSN1 pancreatic cancer cell lines. Cell number was determined by counting the cells after Trypan blue exclusion at 24-h intervals. Cells were transduced with either Ad GFP or Ad r-PTPη at MOI 50.
Ad r-PTPη induces apoptosis in AsPC1 and PSN1 cells
We used FACS analysis to investigate cell cycle changes in AsPC1 and PSN1 cells virally transduced with Ad r-PTPη . There were dramatic changes in the profile of the AsPC1 cell cycle already 48 h after infection with Ad r-PTPη . In fact, AsPC1 cells infected with Ad r-PTPη had a large population of sub-G 1 cells, a strongly reduced percentage of G 0 /G 1 cells, and, finally, an increased percentage of G 2 /M cells, whereas there were no significant changes in the number of S-phase cells. These changes were even more evident 96 h after infection. There were no significant differences in the cell cycle between mock-infected and Ad GFP-infected AsPC1 cells ( Figure 5 ).
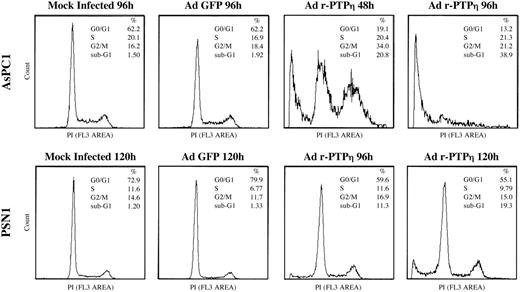
Flow cytometry of AsPC1 and PSN1 pancreatic malignant cells infected with Ad r-PTPη . Flow cytometry analysis of AsPC1 48 and 72 h (top row) and PSN1 96 and 120 h after infection with Ad r-PTPη (bottom row). Both cell lines were transduced at MOI 50. Two other experiments performed in parallel gave the same results. The percentages of cell cycle distribution and apoptotic cells (i.e. cells sub-G 1 DNA content) are shown on each histogram.
At 96 h post-infection, changes appeared in the cycle of PSN1 cells infected with Ad r-PTPη as witnessed by a sub-G 1 peak and a reduced percentage of PSN1 Ad r-PTPη -infected cells in G 0 /G 1 stage versus mock-transduced PSN1 cells. These alterations were more pronounced 120 h after infection. There were no differences in the cell cycle profile of uninfected and Ad GFP-infected PSN1 cells ( Figure 5 ).
The large number of Ad r-PTPη sub-G 1 cells phase suggested apoptosis. In fact, a TUNEL assay showed that most of the sub-G 1 population of AsPC1 and PSN1 cells infected with r-PTPη was predominantly composed of apoptotic cells. The results of the TUNEL assay are summarized in Table III . This set of experiments show that virus-mediated over-expression of r-PTPη induces apoptosis of malignant pancreatic cells.
TUNEL assay of AsPC1 and PSN1 pancreatic cancer cells infected with Ad r-PTPη
| Apoptosis (%) | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| Mock infected | 1.5 ± 0.3 | 0.3 ± 0.1 | |
| Ad GFP | 2.8 ± 0.7 | 0.5 ± 0.1 | |
| Ad r-PTPη | 25.1 ± 3.8 | 12 ± 1.3 | |
| Apoptosis (%) | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| Mock infected | 1.5 ± 0.3 | 0.3 ± 0.1 | |
| Ad GFP | 2.8 ± 0.7 | 0.5 ± 0.1 | |
| Ad r-PTPη | 25.1 ± 3.8 | 12 ± 1.3 | |
The percentages represent mean ± SD of the values of three different experiments.
TUNEL assay of AsPC1 and PSN1 pancreatic cancer cells infected with Ad r-PTPη
| Apoptosis (%) | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| Mock infected | 1.5 ± 0.3 | 0.3 ± 0.1 | |
| Ad GFP | 2.8 ± 0.7 | 0.5 ± 0.1 | |
| Ad r-PTPη | 25.1 ± 3.8 | 12 ± 1.3 | |
| Apoptosis (%) | |||
|---|---|---|---|
| AsPC1 | PSN1 | ||
| Mock infected | 1.5 ± 0.3 | 0.3 ± 0.1 | |
| Ad GFP | 2.8 ± 0.7 | 0.5 ± 0.1 | |
| Ad r-PTPη | 25.1 ± 3.8 | 12 ± 1.3 | |
The percentages represent mean ± SD of the values of three different experiments.
Suppression of pancreatic cell lines tumorigenicity by the virally transduced r-PTPη
AAVs are promising candidates for cancer gene therapy ( 39 ). In this context, we generated a recombinant AAV r-PTPη vector as described in the ‘Materials and methods’ ( Figure 6A ). For the in vivo experiments with AAV r-PTPη , 1 × 10 6 PSN1 cells were injected into the right flank of nude mice. Ten days after injection, small tumors (∼8 mm in diameter) appeared in all mice. One group (each group consisted of six mice) was injected with PBS, a second group was injected with 1 × 10 10 VP of AAV GFP, and the third group was injected with 1 × 10 10 VP of AAV r-PTPη . Five days after injection, one mouse per group was killed and r-PTPη transgene expression was evaluated in the tumors by RT–PCR ( Figure 6B ). The remaining mice were monitored for 12 days after infection, killed and the tumors measured. The tumors in the mice treated with AAV r-PTPη were ∼2.5-fold smaller than those detected in untreated and AAV GFP-injected mice ( Figure 6C ).
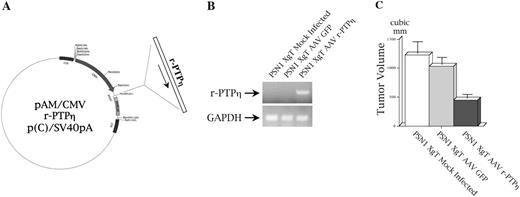
In-vivo transduction effects of r-PTPη using recombinant AAV r-PTPη . ( A ) Preparation of the transfer vector pAM/CMV r-PTPη p(C)/SV40pA. The full-length cDNA of r-PTPη was cloned under the transcriptional control of a CMV promoter to generate the plasmid pAM/CMV r-PTPη p(C)/SV40pA; this vector was used to prepare a recombinant AAV r-PTPη (for details, see ‘Materials and methods’). ( B ) Intratumoral expression of r-PTPη . PSN1 cells were injected into nude mice; 10 days later the tumors were injected with 1 × 10 10 VP of either AAV GFP or AAV r-PTPη . Five days after infection, one mouse per group was killed and total RNA was extracted from tumors. RT–PCR analysis was performed with oligonucleotides specific for r-PTPη . ( C ) PSN1 xenograft tumor (XgT) volume evaluation after AAV r-PTPηin vivo treatment. Twelve days after intratumoral viral administration, mice were killed and tumor volume was assessed with the formula: tumor volume = length × (width) 2 /2. Each bar represents the mean volume ± SD of tumors from five mice injected with PSN1 cells.
Discussion
Pancreatic carcinoma is one of the most aggressive tumors in humans. Because diagnosis is usually made at an advanced stage of the disease and there is no effective treatment, the mortality rate approximates the number of diagnosed cases. Consequently, there is an urgent need for new therapeutic approaches ( 40 ). Gene therapy could fulfil this need. Many proto-oncogenes and anti-oncogenes have been implicated in pancreatic tumors, and some have proved to be effective both in vitro and in vivo , i.e. p53 ( 41 , 42 ), p16INK4a ( 42 , 43 ), p21WAF1 ( 44 ), a dominant negative H-ras mutant ( 45 ) and FHIT ( 38 ).
In this study, we investigated the expression of a receptor-type protein tyrosine phosphatase, DEP-1/HPTPη , and its therapeutic use in nine human pancreatic cancer cell lines. DEP-1/HPTPη expression was very low in most cell lines examined. Although the polymorphism Gln276Pro has been proposed as a candidate for cancer susceptibility in humans ( 32 ), no specific association among this polymorphism and malignancy in pancreatic cancer cells was established.
Consequent to our finding that colony formation was dramatically lower in AsPC1 and PSN1 cells transfected with an r-PTPη expression vector ( 30 ) versus cells transfected with an empty vector, we used a recombinant adenovirus carrying the r-PTPη gene in our experiments. Fluorescence microscopy showed that the AsPC1 and PSN1 cells differed in transduction efficiency: transduction by Ad GFP cells at MOI 50 was almost total in AsPC1 cells, and merely 25–30% in PSN1 cells. This difference could reflect their diverse expression of the Coxsackie adenovirus receptor (CAR), the surface molecule governing entry of the adenovirus into the cell ( 46 , 47 ). This might represent a major limit for pancreatic cancer gene therapy and modification of the CAR-specificity of the fiber knob protein of the adenovirus vector has attracted interest as a promising way to combine high transduction efficiency of the viral vector and targetability in vivo ( 48 , 49 ). Very promising results on PSN1 cells have been obtained by using a chimeric Ad5-35 vector carrying the GFP reporter protein, where the efficiency of infection was extended to nearly all treated cells (Trapasso, unpublished results). This vector could represent one more possibility for the treatment of both pancreatic primary tumors and their metastatic disease.
Expression of the transgene in AsPC1 and PSN1 caused the cells to become flat and significantly reduced their proliferation rate. The cell cycle analysis of AsPC1 cells transduced with Ad r-PTPη revealed a massive peak of sub-G 1 cells and accumulation of cells at the G 2 /M phase. Less dramatically, there was only a small sub-G 1 peak in PSN1 r-PTPη -transduced cells, and the percentage of G 0 /G 1 cells was much lower compared with PSN1 cells transduced with Ad GFP. TUNEL assay showed that the sub-G 1 population peak consisted of apoptotic cells.
AAV is a promising tool for cancer gene therapy ( 39 ). Here, we found that tumors were 3-fold smaller in mice injected with PSN1 cells and subsequently intratumorally injected with AAV r-PTPη compared with tumors from controls. These results suggest the fascinating prospect of sequential gene therapy using the r-PTPη gene transduced by both adenoviruses and AAV in patients with pancreatic cancer.
Our findings expand the role of the DEP-1/HPTPη gene as a candidate tumor suppressor also to pancreatic cancer, after its involvement was already demonstrated in human mammary, thyroid, lung and colon carcinomas ( 50 , 30 , 32 ). Until now, the mechanism mainly invoked for DEP-1/HPTPη aberration in cancer is LOH in its locus ( 32 ); however, epigenetic silencing of DEP-1/HPTPη in human carcinogenesis cannot be excluded and further studies will be required to investigate the possibility of the DEP-1/HPTPη down-regulation through CpG island hypermethylation.
In conclusion, DEP-1/HPTPη is a promising candidate for the treatment of human pancreatic cancer, also in combination with other conventional therapies.
Supplementary material
Supplementary material can be found at: http://www.carcin.oupjournals.org/
This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), by the Programma Italia-USA sulla Terapia dei Tumori coordinated by Prof. Cesare Peschle, Progetto Finalizzato ‘Biotecnologie’ of Consiglio Nazionale delle Ricerche, Progetto ‘5%’ of the Consiglio Nazionale delle Ricerche, and the MURST Project ‘Terapie antineoplastiche innovative’. We are grateful to Jean Ann Gilder (Scientific Communication) for editing the manuscript and to David Fineman for his friendly support to our group.
References
Parker,S.L., Tong,T., Bolden,S. and Wingo,P.A. (
Gold,E.B. (
Warshaw,A. and Fernandez-del Castillo,C. (
Ahrendt,S.A. and Pitt,H.A. (
Hilgers,W. and Kern,S.E. (
Bardeesy,N. and DePinho,R.A. (
Almoguera,C., Shibata,D., Forrester,K., Martin,J., Arnheim,N. and Perucho,M. (
Cheng,J., Ruggeri,B., Klein,W.M., Sonoda,G., Altomare,D.A., Watson,D.K. and Testa,J.R. (
Wallrapp,C., Muller-Pillasch,F., Solinas-Toldo,S., Lichter,P., Friess,H., Buchler,M., Fink,T., Adler,G. and Gress,T.M. (
Hahn,S.A., Seymour,A.B., Hoque,A.T. et al . (
Caldas,C., Hahn,S.A., da Costa,L.T., Redston,M.S., Schutte,M., Seymour,A.B., Weinstein,C.L., Hruban,R.H., Yeo,C.J. and Kern,S.E. (
Pellegata,N.S., Sessa,F., Renault,B., Bonato,M., Leone,B.E., Solcia,E. and Ranzani,G.N. (
Redston,M.S., Caldas,C., Seymour,A.B., Hruban,R.H., da Costa,L., Yeo,C.J. and Kern,S.E. (
Hahn,S.A., Schutte,M., Hoque,A.T. et al . (
Schutte,M., Hruban,R.H., Hedrick,L. et al . (
Rozenblum,E., Schutte,M., Goggins,M. et al . (
Sorio,C., Baron,A., Orlandini,S., Zamboni,G., Pederzoli,P., Huebner,K. and Scarpa,A. (
Liebow,C., Reilly,C., Serrano,M. and Schally,A.V. (
Douziech,N., Calvo,E., Coulombe,Z., Muradia,G., Bastien,J., Aubin,R.A., Lajas,A. and Morisset,J. (
Li,J., Yen,C., Liaw,D. et al . (
Steck,P.A., Pershouse,M.A., Jasser,S.A. et al . (
Sakurada,A., Suzuki,A., Sato,M., Yamakawa,H., Orikasa,K., Uyeno,S., Ono,T., Ohuchi,N., Fujimura,S. and Horii,A. (
Okami,K., Wu,L., Riggins,G. et al . (
Ostman,A., Yang,Q. and Tonks,N.K. (
Honda,H., Inazawa,J., Nishida,J., Yazaki,Y. and Hirai,H. (
de la Fuente-Garcia,M., Nicolas,J.M., Freed,J.H., Palou,E., Thomas,A.P., Vilella,R., Vives,J. and Gaya,A. (
Tangye,S.G., Phillips,J.H., Lanier,L.L., de Vries,J.E. and Aversa,G. (
Tangye,S.G., Liu,Y.J., Aversa,G., Phillips,J.H. and de Vries,J.E. (
Baker,J.E., Majeti,R., Tangye,S.G. and Weiss,A. (
Trapasso,F., Iuliano,R., Boccia,A., Stella,A., Visconti,R., Bruni,P., Baldassarre,G., Santoro,M., Viglietto,G. and Fusco,A. (
Viglietto,G. and Fusco,A. (
Ruivenkamp,C.A., van Wezel,T., Zanon,C. et al . (
Scala,S., Portella,G., Fedele,M., Chiappetta,G. and Fusco,A. (
Iuliano,R., Trapasso,F., Le Pera,I. et al . (
Luo,J., Kaplitt,M.G., Fitzsimons,H.L., Zuzga,D.S., Liu,Y., Oshinsky,M.L. and During,M.J. (
Holcik,M. and Liebhaber,S.A. (
Autschbach,F., Palou,E., Mechtersheimer,G., Rohr,C., Pirotto,F., Gassler,N., Otto,H.F., Schraven,B. and Gaya,A. (
Dumon,K.R., Ishii,H., Vecchione,A. et al . (
Ponnazhagan,S., Curiel,D.T., Shaw,D.R., Alvarez,R.D. and Siegal,G.P. (
Rosenberg,L. (
Bouvet,M., Bold,R.J., Lee,J. et al . (
Ghaneh,P., Greenhalf,W., Humphreys,M., Wilson,D., Zumstein,L., Lemoine,N.R. and Neoptolemos,J.P. (
Kobayashi,S., Shirasawa,H., Sashiyama,H., Kawahira,H., Kaneko,K., Asano,T. and Ochiai,T. (
Joshi,U.S., Dergham,S.T., Chen,Y.Q., Dugan,M.C., Crissman,J.D., Vaitkevicius,V.K. and Sarkar,F.H. (
Takeuchi,M., Shichinohe,T., Senmaru,N., Miyamoto,M., Fujita,H., Takimoto,M., Kondo,S., Katoh,H. and Kuzumaki,N. (
Pearson,A.S., Koch,P.E., Atkinson,N., Xiong,M., Finberg,R.W., Roth,J.A. and Fang,B. (
Wesseling,J., Bosma,P.J., Krasnykh,V. et al . (
Shayakhmetov,D.M. and Lieber,A. (
Belousova,N., Krendelchtchikova,V., Curiel,D.T. and Krasnykh,V. (
Author notes
1Kimmel Cancer Institute, Thomas Jefferson University, 233S 10th Street, Philadelphia, PA 19107, USA, 2Dipartimento di Medicina Sperimentale e Clinica, Università ‘Magna Græcia’, via T. Campanella, 5, I-88100 Catanzaro, Italy, 3Department of Neurosurgery, CNS Gene Therapy Center, Thomas Jefferson University, 1025 Walnut Street, Philadelphia, PA 19107, USA and 4Centro di Endocrinologia ed Oncologia Sperimentale del C.N.R. c/o Dipartimento di Biologia e Patologia Cellulare e Molecolare, Università di Napoli ‘Federico II’, via Pansini, 5, I-80131 Naples, Italy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}