




Abstract
The brain is targeted by human immunodeficiency virus type 1 (HIV-1) during the course of untreated infection, leading to cognitive impairment, neurological damage and HIV encephalitis (HIVE). To study early dynamics of HIV entry into the brain, we examined a unique autopsy series of samples obtained from 15 untreated individuals who died in the presymptomatic stages of infection from non-HIV causes. HIV was detected and quantified by limiting dilution PCR and genetically characterized in the V3 region of env. Limiting dilution was shown to be essential for correct estimation of genetic partitioning between brain- and lymphoid-associated HIV populations. While no actively expressing HIV-infected cells were detected by immunohistochemistry, variable and generally extremely low levels of proviral DNA were detected in presymptomatic brain samples. V3 region sequences were frequently genetically distinct from lymphoid-associated HIV variants, with association index (AI) values similar to those observed in cases of HIVE. Infiltration of CD8 lymphocytes in the brain was strongly associated with expression of activation markers (MHCII; R = 0.619; P < 0.05), the presence of HIV-infected cells (proviral load; R = 0.608; P < 0.05) and genetic segregation of brain variants from populations in lymphoid tissue (AI value, R = −0.528; P ≈ 0.05). CD8 lymphocytes may thus limit replication of HIV seeded into the brain in early stages of infection. Neurological complications in AIDS occur when this control breaks down, due to systemic immunosuppression from HIV that destroys CD8 lymphocyte function and/or through the evolution of more aggressive neuropathogenic variants.
Introduction
Infection with human immunodeficiency virus type 1 (HIV-1) is persistent in humans and, when untreated, eventually leads to disease manifestations secondary to the profound immunodeficiency that develops in acquired immunodeficiency syndrome (AIDS). Apart from CD4+ lymphocytes, HIV may contribute to AIDS-related disease through damage to other cells types expressing CD4 and co-receptors such as CCR5. The best documented manifestation of this is HIV encephalitis (HIVE), where infection of microglia leads to severe tissue damage in the central nervous system (CNS), manifested initially as impairment of cognitive function, memory and personality change, and progressing to dementia and neurological symptoms.
The development of HIVE is associated with the immunohistochemical detection of HIV-expressing microglia and infiltrating macrophages, whose fusion produces the syncytia (or giant cells) pathognomic of HIVE. HIVE is also closely associated with increased HIV proviral loads throughout the brain (Pang et al., 1990; Bell et al., 1993, 1996, 1998; Achim et al., 1994; Donaldson et al., 1994b). The timing of entry of HIV into the brain during the course of HIV infection remains uncertain. The relatively impermeable blood–brain barrier (BBB) restricts entry of free HIV virions from the peripheral circulation, but HIV-infected monocyte-derived macrophages and T lymphocytes may migrate through vascular endothelium and initiate infection of resident cell types such as microglia. Similarly, there is considerable interaction between cells in peripheral and CNS compartments in the highly vascular choroid plexus, and this has been considered as a possible major portal of HIV entry into the CNS (Falangola et al., 1995; Petito et al., 1999; Chen et al., 2000).
While lack of detectable HIV-related brain damage or atrophy in presymptomatic individuals provides no evidence for early entry of HIV into the CNS, other findings are supportive of early brain infection, such as the high titre virus detected in the CNS of an individual iatrogenically infected a few days before death (Davis et al., 1992). Virus and locally produced antibody to HIV are also frequently detected in the cerebrospinal fluid in presymptomatic subjects. The low or undetectable proviral loads and the repeated failure to detect virus expression in CNS tissue from presymptomatic individuals may therefore reflect the effectiveness of immune responses during this stage (Bell et al., 1993; Gray et al., 1996; Tomlinson et al., 1999). Evidence for a highly active immune response against replicating HIV is indeed provided by observations of the marked perivascular infiltration of cytotoxic T lymphocytes (Bell et al., 1993; Tomlinson et al., 1999) and upregulation of macrophage markers in the CNS and increased chemokine secretion and expression of activation markers on resident microglia (Pulliam et al., 1991; Genis et al., 1992; Giulian et al., 1993; Gelbard et al., 1994; Bukrinsky et al., 1995; Gray et al., 1996; Griffin, 1997; Heyes et al., 1998; Tomlinson et al., 1999).
The hypothesis for early entry is further supported by the genetic divergence of variants recovered from the brain from those found outside the CNS in the lymphoid tissue. Comparison of p17gag region sequences of CNS and non-CNS virus populations in HIVE cases predicted times of divergence several years before the onset of AIDS-defining illnesses (Hughes et al., 1997). Indeed the almost invariably observed genetic partitioning of CNS-derived sequences (Liu et al., 1990; Epstein et al., 1991; Haggerty and Stevenson, 1991; Li et al., 1991; Pang et al., 1991; Steuler et al., 1992; Keys et al., 1993; Ball et al., 1994; Donaldson et al., 1994a; Korber et al., 1994; Power et al., 1994; Di Stefano et al., 1996; Reddy et al., 1996; Gartner et al., 1997; van't Wout et al., 1998; Gatanaga et al., 1999; Morris et al., 1999) supports further the concept of an early established and autonomous CNS-adapted virus population in the brain.
An alternative interpretation of the genetic isolation of brain populations of HIV is that they represent part of a much more widely disseminated population of infected macrophages in the body. The characteristic sequence differences observed in V3 and elsewhere may therefore represent adaptations for replication in cells of the macrophage lineage rather than specifically for microglia and/or other cell types in the CNS. The observation that virus variants recovered from p24-positive multinucleated tissue macrophages in the lung and gastrointestinal tract showed V3 sequences closely related to those found in the brain (Wang et al., 2001) and bone marrow (Liu et al., 2000) supports this hypothesis. The association of HIVE with perivascular infiltration of activated, frequently productively infected macrophages into the brain (Fischer-Smith et al., 2001, 2004) along with the appearance of increased numbers of activated monocytes in peripheral blood (Pulliam et al., 1997) demonstrates a much more dynamic interaction between CNS and peripheral populations of HIV than entailed in previous models.
Most investigations of HIV infection of the CNS have been necessarily limited to autopsy samples from individuals with late stage HIV-related disease. It has therefore not been possible to examine whether CNS- or macrophage-specific populations of HIV are established early in the course of infection, and whether such early CNS infection is associated with evidence for virus expression and infiltration of specific cell types bringing in or responding to the traffic of infected cells. In this study, we have made use of the unique availability of a large set of archived brain and other tissue samples from untreated study subjects who died for reasons unrelated to HIV in the presymptomatic stage of infection. The results show evidence for the early establishment of genetically distinct populations of HIV in the CNS in several individuals, which is associated with a marked inflammatory process and infiltration of cytotoxic T cells.
Materials and methods
Study subjects
All tissue samples for the study subjects were selected from the Edinburgh MRC HIV Brain and Tissue Bank (Western General Hospital, Edinburgh, UK). Consent for use of these post-mortem samples was obtained according to the Declaration of Helsinki, and ethical approval for research on autopsy tissues carried out in this study was provided by the Lothian Research Ethics Committee (LREC2002/4/36). Tissue samples from several anatomically distinct regions of the brain, as well as lymph node or spleen, were obtained from autopsies carried out on the study subjects NA200, NA218, NA240, NA285, NA273, NA305, NA279, NA159, NA178, NA227, NA234, NA225, NA286, NA274 and NA237. All were HIV-positive and had a CD4 count/μl of ≥200 before death except the subject NA286, who had a CD4 count/μl of 167 (data not available for subjects NA274 and NA237). All study subjects died from HIV-unrelated causes [alcohol/drug overdose (n = 11), cirrhosis (n = 2), suicide (n = 1), bronchopneumonia (n = 1)], with no evidence of HIVE, or other HIV-related neuropathology at autopsy. Similarly, there was no evidence for infection with cytomegalovirus, Pneumocystis carinii, JC virus, toxoplasma or other opportunistic infections or neoplasia associated with immunosuppression. Subjects were therefore all considered presymptomatic (Table 1). The median duration of HIV infection before death was 7 years (data available from 11 of the 15 study subjects). No study subject had received antiretroviral therapy. Control subjects comprised seven individuals with a history of past or current injecting drug use (IDUs), but were HIV-uninfected (mean age 25 years; range 20–36), and five non-IDU, HIV-negative individuals (mean age 31 years; range 21–45).
Clinical backgrounds and virology testing of study subjects
| Study subject | Age (years) | Sexa | Risk group | CD4b | CCR5c | Proviral loadd | Segregation (AI value) | Predicted phenotypee | Percentage staining | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | Lymphoid | Brain | Lymphoid | CD3 | CD8 | CD14 | CD16 | MHC | CD68 | CD20 | |||||||||||||||
| NA218 | 44 | M | IDU | 824 | wt, hmz | 21 | 1050 | 1.313 | All X4 | All X4 | 0.02 | 0.01 | 0.03 | 0.01 | 0.02 | 0.35 | 0.23 | ||||||||
| NA285 | 39 | F | IDU | 630 | wt, hmz | 2 | 14 | 1.112 | All R5 | All R5 | 0.01 | 0.02 | 0.06 | 0.03 | 0.69 | 0.79 | 0.07 | ||||||||
| NA240 | 40 | M | IDU | 496 | wt, hmz | 2 | 39 | 0.805 | All R5 | 2 X4, 6 R5 | 0.03 | 0.02 | 0.11 | 0.40 | 0.99 | 0.50 | 0.13 | ||||||||
| NA200 | 30 | M | IDU | 418 | wt, hmz | 33 | 29 | 0.140 | All R5 | 4 X4, 6 R5 | 0.13 | 0.09 | 0.22 | 0.05 | 4.83 | 0.53 | 0.26 | ||||||||
| NA178 | 30 | M | IDU | 342 | wt, hmz | <1 | 3 | 1.037 | All R5 | All R5 | 0.01 | 0.01 | 2.60 | 4.36 | 2.21 | 2.57 | 0.01 | ||||||||
| NA279 | 37 | M | IDU | 268 | wt, hmz | 1 | 101 | 0.144 | All R5 | All R5 | 0.07 | 0.02 | 0.10 | 0.51 | 0.29 | 0.39 | 0.08 | ||||||||
| NA273 | 31 | M | IDU | 257 | wt, hmz | 1 | 28 | 0.784 | All R5 | All R5 | 0.03 | 0.01 | 0.04 | 0.01 | 0.04 | 0.27 | 0.41 | ||||||||
| NA225 | 31 | F | IDU | 247 | wt, hmz | <1 | 639 | 0.398 | All R5 | All R5 | 0.02 | 0.02 | 0.03 | 0.57 | 0.45 | 0.33 | 0.01 | ||||||||
| NA234 | 23 | F | IDU | 246 | wt, hmz | 11 | 726 | 0.463 | All R5 | All R5 | 0.12 | 0.16 | 0.07 | 0.01 | 4.30 | 0.35 | 0.06 | ||||||||
| NA227 | 29 | F | IDU | 245 | wt, hmz | 58 | 421 | 0.590 | All R5 | All R5 | 0.02 | 0.03 | 0.05 | 0.02 | 1.42 | 0.56 | 0.10 | ||||||||
| NA159 | 32 | F | IDU | 240 | wt, hmz | 16 | 56 | 0.600 | All R5 | All R5 | 0.39 | 0.45 | 0.24 | 1.18 | 10.9 | 1.43 | 0.29 | ||||||||
| NA305 | 34 | M | IDU | 229 | wt, hmz | 3 | 9 | 0.0003 | All R5 | All R5 | 0.02 | 0.04 | 0.09 | 0.02 | 0.64 | 0.32 | 0.13 | ||||||||
| NA286 | 33 | M | IDU | 167 | wt, hmz | <1 | 335 | 0.764 | All R5 | 1 X4, 10 R5 | 0.01 | 0.01 | 0.01 | 1.71 | 1.68 | 0.23 | 0.01 | ||||||||
| NA237 | 36 | M | IDU | n.d. | wt, hmz | 4 | 5244 | 0.605 | All R5 | All R5 | 0.01 | 0.01 | 0.02 | 0.02 | 0.34 | 0.23 | 0.04 | ||||||||
| NA274 | 36 | M | IDU | n.d. | wt, hmz | 4 | 1220 | 0.291 | All R5 | All R5 | 0.04 | 0.08 | 0.05 | 3.17 | 2.91 | 1.05 | 0.01 | ||||||||
| Geometric mean | 3.5 | 125 | 0.33 | 0.031 | 0.029 | 0.074 | 0.128 | 0.823 | 0.502 | 0.065 | |||||||||||||||
| Study subject | Age (years) | Sexa | Risk group | CD4b | CCR5c | Proviral loadd | Segregation (AI value) | Predicted phenotypee | Percentage staining | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | Lymphoid | Brain | Lymphoid | CD3 | CD8 | CD14 | CD16 | MHC | CD68 | CD20 | |||||||||||||||
| NA218 | 44 | M | IDU | 824 | wt, hmz | 21 | 1050 | 1.313 | All X4 | All X4 | 0.02 | 0.01 | 0.03 | 0.01 | 0.02 | 0.35 | 0.23 | ||||||||
| NA285 | 39 | F | IDU | 630 | wt, hmz | 2 | 14 | 1.112 | All R5 | All R5 | 0.01 | 0.02 | 0.06 | 0.03 | 0.69 | 0.79 | 0.07 | ||||||||
| NA240 | 40 | M | IDU | 496 | wt, hmz | 2 | 39 | 0.805 | All R5 | 2 X4, 6 R5 | 0.03 | 0.02 | 0.11 | 0.40 | 0.99 | 0.50 | 0.13 | ||||||||
| NA200 | 30 | M | IDU | 418 | wt, hmz | 33 | 29 | 0.140 | All R5 | 4 X4, 6 R5 | 0.13 | 0.09 | 0.22 | 0.05 | 4.83 | 0.53 | 0.26 | ||||||||
| NA178 | 30 | M | IDU | 342 | wt, hmz | <1 | 3 | 1.037 | All R5 | All R5 | 0.01 | 0.01 | 2.60 | 4.36 | 2.21 | 2.57 | 0.01 | ||||||||
| NA279 | 37 | M | IDU | 268 | wt, hmz | 1 | 101 | 0.144 | All R5 | All R5 | 0.07 | 0.02 | 0.10 | 0.51 | 0.29 | 0.39 | 0.08 | ||||||||
| NA273 | 31 | M | IDU | 257 | wt, hmz | 1 | 28 | 0.784 | All R5 | All R5 | 0.03 | 0.01 | 0.04 | 0.01 | 0.04 | 0.27 | 0.41 | ||||||||
| NA225 | 31 | F | IDU | 247 | wt, hmz | <1 | 639 | 0.398 | All R5 | All R5 | 0.02 | 0.02 | 0.03 | 0.57 | 0.45 | 0.33 | 0.01 | ||||||||
| NA234 | 23 | F | IDU | 246 | wt, hmz | 11 | 726 | 0.463 | All R5 | All R5 | 0.12 | 0.16 | 0.07 | 0.01 | 4.30 | 0.35 | 0.06 | ||||||||
| NA227 | 29 | F | IDU | 245 | wt, hmz | 58 | 421 | 0.590 | All R5 | All R5 | 0.02 | 0.03 | 0.05 | 0.02 | 1.42 | 0.56 | 0.10 | ||||||||
| NA159 | 32 | F | IDU | 240 | wt, hmz | 16 | 56 | 0.600 | All R5 | All R5 | 0.39 | 0.45 | 0.24 | 1.18 | 10.9 | 1.43 | 0.29 | ||||||||
| NA305 | 34 | M | IDU | 229 | wt, hmz | 3 | 9 | 0.0003 | All R5 | All R5 | 0.02 | 0.04 | 0.09 | 0.02 | 0.64 | 0.32 | 0.13 | ||||||||
| NA286 | 33 | M | IDU | 167 | wt, hmz | <1 | 335 | 0.764 | All R5 | 1 X4, 10 R5 | 0.01 | 0.01 | 0.01 | 1.71 | 1.68 | 0.23 | 0.01 | ||||||||
| NA237 | 36 | M | IDU | n.d. | wt, hmz | 4 | 5244 | 0.605 | All R5 | All R5 | 0.01 | 0.01 | 0.02 | 0.02 | 0.34 | 0.23 | 0.04 | ||||||||
| NA274 | 36 | M | IDU | n.d. | wt, hmz | 4 | 1220 | 0.291 | All R5 | All R5 | 0.04 | 0.08 | 0.05 | 3.17 | 2.91 | 1.05 | 0.01 | ||||||||
| Geometric mean | 3.5 | 125 | 0.33 | 0.031 | 0.029 | 0.074 | 0.128 | 0.823 | 0.502 | 0.065 | |||||||||||||||
M = male; F = female.
Median interval between CD4 count and death: 70 days; n.d. = not determined/not known.
wt, wild type; hmz, homozygous.
Proviral copies/106 cells.
Based on charge of V3 loop of env, X4 variants are shown in boldface.
Clinical backgrounds and virology testing of study subjects
| Study subject | Age (years) | Sexa | Risk group | CD4b | CCR5c | Proviral loadd | Segregation (AI value) | Predicted phenotypee | Percentage staining | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | Lymphoid | Brain | Lymphoid | CD3 | CD8 | CD14 | CD16 | MHC | CD68 | CD20 | |||||||||||||||
| NA218 | 44 | M | IDU | 824 | wt, hmz | 21 | 1050 | 1.313 | All X4 | All X4 | 0.02 | 0.01 | 0.03 | 0.01 | 0.02 | 0.35 | 0.23 | ||||||||
| NA285 | 39 | F | IDU | 630 | wt, hmz | 2 | 14 | 1.112 | All R5 | All R5 | 0.01 | 0.02 | 0.06 | 0.03 | 0.69 | 0.79 | 0.07 | ||||||||
| NA240 | 40 | M | IDU | 496 | wt, hmz | 2 | 39 | 0.805 | All R5 | 2 X4, 6 R5 | 0.03 | 0.02 | 0.11 | 0.40 | 0.99 | 0.50 | 0.13 | ||||||||
| NA200 | 30 | M | IDU | 418 | wt, hmz | 33 | 29 | 0.140 | All R5 | 4 X4, 6 R5 | 0.13 | 0.09 | 0.22 | 0.05 | 4.83 | 0.53 | 0.26 | ||||||||
| NA178 | 30 | M | IDU | 342 | wt, hmz | <1 | 3 | 1.037 | All R5 | All R5 | 0.01 | 0.01 | 2.60 | 4.36 | 2.21 | 2.57 | 0.01 | ||||||||
| NA279 | 37 | M | IDU | 268 | wt, hmz | 1 | 101 | 0.144 | All R5 | All R5 | 0.07 | 0.02 | 0.10 | 0.51 | 0.29 | 0.39 | 0.08 | ||||||||
| NA273 | 31 | M | IDU | 257 | wt, hmz | 1 | 28 | 0.784 | All R5 | All R5 | 0.03 | 0.01 | 0.04 | 0.01 | 0.04 | 0.27 | 0.41 | ||||||||
| NA225 | 31 | F | IDU | 247 | wt, hmz | <1 | 639 | 0.398 | All R5 | All R5 | 0.02 | 0.02 | 0.03 | 0.57 | 0.45 | 0.33 | 0.01 | ||||||||
| NA234 | 23 | F | IDU | 246 | wt, hmz | 11 | 726 | 0.463 | All R5 | All R5 | 0.12 | 0.16 | 0.07 | 0.01 | 4.30 | 0.35 | 0.06 | ||||||||
| NA227 | 29 | F | IDU | 245 | wt, hmz | 58 | 421 | 0.590 | All R5 | All R5 | 0.02 | 0.03 | 0.05 | 0.02 | 1.42 | 0.56 | 0.10 | ||||||||
| NA159 | 32 | F | IDU | 240 | wt, hmz | 16 | 56 | 0.600 | All R5 | All R5 | 0.39 | 0.45 | 0.24 | 1.18 | 10.9 | 1.43 | 0.29 | ||||||||
| NA305 | 34 | M | IDU | 229 | wt, hmz | 3 | 9 | 0.0003 | All R5 | All R5 | 0.02 | 0.04 | 0.09 | 0.02 | 0.64 | 0.32 | 0.13 | ||||||||
| NA286 | 33 | M | IDU | 167 | wt, hmz | <1 | 335 | 0.764 | All R5 | 1 X4, 10 R5 | 0.01 | 0.01 | 0.01 | 1.71 | 1.68 | 0.23 | 0.01 | ||||||||
| NA237 | 36 | M | IDU | n.d. | wt, hmz | 4 | 5244 | 0.605 | All R5 | All R5 | 0.01 | 0.01 | 0.02 | 0.02 | 0.34 | 0.23 | 0.04 | ||||||||
| NA274 | 36 | M | IDU | n.d. | wt, hmz | 4 | 1220 | 0.291 | All R5 | All R5 | 0.04 | 0.08 | 0.05 | 3.17 | 2.91 | 1.05 | 0.01 | ||||||||
| Geometric mean | 3.5 | 125 | 0.33 | 0.031 | 0.029 | 0.074 | 0.128 | 0.823 | 0.502 | 0.065 | |||||||||||||||
| Study subject | Age (years) | Sexa | Risk group | CD4b | CCR5c | Proviral loadd | Segregation (AI value) | Predicted phenotypee | Percentage staining | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | Lymphoid | Brain | Lymphoid | CD3 | CD8 | CD14 | CD16 | MHC | CD68 | CD20 | |||||||||||||||
| NA218 | 44 | M | IDU | 824 | wt, hmz | 21 | 1050 | 1.313 | All X4 | All X4 | 0.02 | 0.01 | 0.03 | 0.01 | 0.02 | 0.35 | 0.23 | ||||||||
| NA285 | 39 | F | IDU | 630 | wt, hmz | 2 | 14 | 1.112 | All R5 | All R5 | 0.01 | 0.02 | 0.06 | 0.03 | 0.69 | 0.79 | 0.07 | ||||||||
| NA240 | 40 | M | IDU | 496 | wt, hmz | 2 | 39 | 0.805 | All R5 | 2 X4, 6 R5 | 0.03 | 0.02 | 0.11 | 0.40 | 0.99 | 0.50 | 0.13 | ||||||||
| NA200 | 30 | M | IDU | 418 | wt, hmz | 33 | 29 | 0.140 | All R5 | 4 X4, 6 R5 | 0.13 | 0.09 | 0.22 | 0.05 | 4.83 | 0.53 | 0.26 | ||||||||
| NA178 | 30 | M | IDU | 342 | wt, hmz | <1 | 3 | 1.037 | All R5 | All R5 | 0.01 | 0.01 | 2.60 | 4.36 | 2.21 | 2.57 | 0.01 | ||||||||
| NA279 | 37 | M | IDU | 268 | wt, hmz | 1 | 101 | 0.144 | All R5 | All R5 | 0.07 | 0.02 | 0.10 | 0.51 | 0.29 | 0.39 | 0.08 | ||||||||
| NA273 | 31 | M | IDU | 257 | wt, hmz | 1 | 28 | 0.784 | All R5 | All R5 | 0.03 | 0.01 | 0.04 | 0.01 | 0.04 | 0.27 | 0.41 | ||||||||
| NA225 | 31 | F | IDU | 247 | wt, hmz | <1 | 639 | 0.398 | All R5 | All R5 | 0.02 | 0.02 | 0.03 | 0.57 | 0.45 | 0.33 | 0.01 | ||||||||
| NA234 | 23 | F | IDU | 246 | wt, hmz | 11 | 726 | 0.463 | All R5 | All R5 | 0.12 | 0.16 | 0.07 | 0.01 | 4.30 | 0.35 | 0.06 | ||||||||
| NA227 | 29 | F | IDU | 245 | wt, hmz | 58 | 421 | 0.590 | All R5 | All R5 | 0.02 | 0.03 | 0.05 | 0.02 | 1.42 | 0.56 | 0.10 | ||||||||
| NA159 | 32 | F | IDU | 240 | wt, hmz | 16 | 56 | 0.600 | All R5 | All R5 | 0.39 | 0.45 | 0.24 | 1.18 | 10.9 | 1.43 | 0.29 | ||||||||
| NA305 | 34 | M | IDU | 229 | wt, hmz | 3 | 9 | 0.0003 | All R5 | All R5 | 0.02 | 0.04 | 0.09 | 0.02 | 0.64 | 0.32 | 0.13 | ||||||||
| NA286 | 33 | M | IDU | 167 | wt, hmz | <1 | 335 | 0.764 | All R5 | 1 X4, 10 R5 | 0.01 | 0.01 | 0.01 | 1.71 | 1.68 | 0.23 | 0.01 | ||||||||
| NA237 | 36 | M | IDU | n.d. | wt, hmz | 4 | 5244 | 0.605 | All R5 | All R5 | 0.01 | 0.01 | 0.02 | 0.02 | 0.34 | 0.23 | 0.04 | ||||||||
| NA274 | 36 | M | IDU | n.d. | wt, hmz | 4 | 1220 | 0.291 | All R5 | All R5 | 0.04 | 0.08 | 0.05 | 3.17 | 2.91 | 1.05 | 0.01 | ||||||||
| Geometric mean | 3.5 | 125 | 0.33 | 0.031 | 0.029 | 0.074 | 0.128 | 0.823 | 0.502 | 0.065 | |||||||||||||||
M = male; F = female.
Median interval between CD4 count and death: 70 days; n.d. = not determined/not known.
wt, wild type; hmz, homozygous.
Proviral copies/106 cells.
Based on charge of V3 loop of env, X4 variants are shown in boldface.
Samples of white matter from the occipital region of the cerebral cortex were obtained from each study subject and control. Samples were paraffin embedded for immunohistochemical analysis, and for HIV-infected subjects an additional sample was stored at −80°C for use in subsequent PCR and sequence analyses, along with samples of lymph node or spleen. For two of the HIV-positive individuals, samples from several other regions of the brain (frontal, temporal and parietal lobes, pons, basal ganglia and cerebellum) were also obtained for immunohistochemical analysis.
Immunohistochemical examination
Following formalin fixation, autopsy tissues were processed through a routine 41 h programme in the Vacuum Infiltration Processor (Tissue Tek), followed by paraffin wax embedding using a Tissue Tek embedding console. Sections of 5 μm, formalin-fixed paraffin-embedded tissue, from the occipital lobe of all study subjects were prepared for immunohistochemistry using all markers. For study subjects NA200 and NA218, further 5 μm white matter sections from frontal lobe, temporal lobe, parietal parasagittal region, pons, basal ganglia and cerebellum were also prepared for immunohistochemical examination using all markers. Whenever possible, serial sections were used to allow for a close comparison of different markers. Immunohistochemistry was performed using the standard avidin–biotin complex (ABC) method (DAKO) or the tyramide signal amplification (TSA) technique, as described previously (Strappe et al., 1997).
HIV-infected cells in the brain were detected by detection of the p24 antigen, as previously described (Bell et al., 1993). Infiltrating T lymphocytes were detected by monoclonal antibodies to CD3 and CD8 (cytotoxic T lymphocytes; CTLs). Staining for CD4 in paraffin-embedded tissues with any of the available monoclonal antibodies did not give reproducible results, and their use was discontinued for this study. Macrophages and activated microglia were detected by monoclonal antibodies to CD14, CD16, CD68 and major histocompatibility complex class II (MHCII). B lymphocytes were detected by anti-CD20. Antibodies used for CD14, CD16, CD68 and MHCII, and their pretreatments, were as described previously (Anthony et al., 2005a). Antibodies for CD3 (Dako), CD8 (Novocastra) and CD20 (DAKO) were used at 1:50, 1:40 and 1:500, respectively. Slides for CD3 and CD8 were pre-treated with 0.001 M EDTA (pH 8.0), and microwaved for 15 min. All three were detected using the ABC method. Briefly, tissue sections were de-paraffinized using xylene and endogenous peroxidases, if any, were blocked with 5% H2O2. Sections were then incubated in normal rabbit serum for 10 min before 30 min incubations with both primary and then secondary antibodies. Following 30 min incubation with ABC, diaminobenzidine (Vector) was used as a visualizing agent. All slides were routinely counterstained with haematoxylin.
Tissue sections were scored for immunopositive cells both manually and by use of the Image-Pro Plus image analysis software and a Roper scientific video camera for semi-automated quantitation. Both perivascular and parenchymal stainings (excluding blood vessels) were quantified in white matter areas of the brain sections studied. For the manual counts, 10 fields were counted using a 10× objective, and the mean averages were taken. Using Image-Pro Plus, a number of contiguous separate images were captured and then combined into a large composite, and from this the total area stained (expressed as a percentage) was determined.
DNA extraction and amplification
DNA was extracted from frozen brain, mediastinal or peritoneal lymph nodes and spleen samples by re-suspension of small pieces of tissue in 400 μl lysis buffer [100 mM NaCl, 50 mM Tris (pH 8.0), 1 mM EDTA (pH 8.0) and 0.5% SDS] containing 2 mg/ml of proteinase K. Digestion of the samples was allowed to continue for 2 h at 65°C. Following phenol–chloroform extraction and ethanol precipitation, samples were re-suspended in 50–100 μl of nuclease-free water. Total DNA concentrations were determined by UV absorbance at wavelengths of 260 and 280 nm. Amplifications of proviral DNA were carried out using a nested-primer PCR technique, which uses Taq polymerase (Promega) and primers spanning the hypervariable V3 loop and flanking regions of env, as described previously (Simmonds et al., 1990). The nucleotide sequences of the primers were as follows: V3 a, TACAATGTACACATGGAATT, + (sense), 6958; V3 b, TGGCAGTCTAGCAGAAGAAG, +, 7010; V3 c, CTGGGTCCCCTCCTGAGG, −(antisense), 7332; and V3 d, ATTACAGTAGAAAAATTCCCC, −, 7382, with all positions numbered according to the HXB2 (GenBank accession no. K03455) genome (1026 Myers). Amplification of the V3 loop and flanking region was achieved by using a thermal cycle of 18 s at 94°C, 21 s at 50°C and 90 s at 72°C. Each template strand was subjected to 25 cycles of amplification. To carry out CCR5 genotyping, the following primers were used: R5F, CTTCATCATCCTCCTGACAATCG, +, 590; R5A, AGCCCTGTGCCTCTTCTTC, −, 938, with all positions numbered according to the human CCR5 gene (GenBank accession no. X91492). Amplification was performed using a thermal cycle of 18 s at 94°C, 30 s at 55°C and 90 s at 72°C. Each template strand was subjected to 30 amplification cycles. All reactions were performed with appropriate positive and negative controls.
Isolation and quantitation of provirus
For all study subjects, single copies of proviral DNA from both brain and lymphoid tissue were amplified and quantified using a limiting dilution and nested PCR technique (Simmonds et al., 1990). Briefly, following a ten-fold serial dilution nested PCR amplification reaction, a series of replicate nested PCR reactions were performed using a 1:3 dilution of the last positive in the dilution series. This allows for the isolation of single copies of provirus. The number of proviral copies was estimated by assuming a Poisson distribution for each sample by −log(1 − p)/d (where p = proportion of positive samples and d = dilution). Proviral load was expressed as copies per million cells, based on the DNA composition of human diploid cells of 6.6 pg of DNA.
Sequence analysis
Single molecules of provirus isolated by the limiting dilution and nested PCR technique produced sufficient DNA to allow for subsequent sequencing of the PCR products. For three of the study subjects (NA200, NA218 and NA159), 1 μg aliquots of DNA samples extracted from both brain and lymphoid tissue were amplified by nested PCR and cloned directly into the pGEM-T Easy vector system (Promega). Following transformation of competent bacteria and minipreps of 10 of the resulting colonies for each sample, the presence of inserts was screened for by digestion using appropriate restriction enzymes, and all positive samples were selected for sequencing. All sequences from both the limiting dilution and cloning studies were then sequenced directly using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems). Sequences were aligned and distances calculated using the Simmonic 2005 Sequence Editor package (http://polio.vir.gla.ac.uk/software).
Analysis of phylogenetic groupings
Phylogenetic trees were constructed by the neighbour-joining method using Jukes–Cantor corrected sequence distances with the MEGA program (Kumar et al., 1993). An epidemiologically unlinked sequence (HIV-1YU2) was used as an outgroup. The nucleotide sequences for the V3 and flanking regions from each of the 15 study subjects were compared with each other and with a range of standard HIV-1 variants. Each set of sequences compared was monophyletic and distinct from those of the published sequences of subtype B: HIV-1SF2 (K02007), HIV-1RF (M17451), HIV-1OYI (M26727), HIV-1LAI (K02013), HIV-1JRFL (M74978), HIV-1YU2 (M93258), HIV-1CAMI (D10112), HIV-1NY5CG (M38431), HIV-1HAN (U43141), HIV-1WMJ22 (M12507) and HIV-1SFAAA (M65024). This comparison provided no evidence for co-infection with more than one epidemiologically unrelated HIV strain, or for inter-sample or exogenous laboratory contamination.
The degree of phylogenetic segregation between the sets of sequences was scored using the association index (AI) method (Wang et al., 2001) as implemented in the Simmonic Sequence Editor version 1.4 (www.polio.vir.gla.ac.uk/software). Briefly, calculation of the AI value involves the use of a tree scoring method which analyses trees constructed by the PHYLIP program (Felsenstein, 1989) for incompatibilities between the phylogenetic grouping of the groups of sequences being tested (i.e. brain versus lymphoid). The tree score is calculated for native sequences and compared with the nul expectation (the score of a tree where group membership is randomly reassigned). The influence of tree robustness on the association index value was indicated by performing 100 bootstrap re-samplings using PHYLIP.
Statistical analyses
Quantitative data obtained in the study was analysed using non-parametric tests (Mann–Whitney U-test, Spearman rank correlation) as implemented in the SYSTAT package (version 10.2).
Nucleotide sequence accession numbers
Sequences obtained in this study have been deposited into GenBank and have been assigned the accession numbers DQ288393 to DQ288268.
Results
Immunohistochemistry
To assess the extent of inflammation in the CNS and the frequency of infiltration of specific cell types into the brain following infection with HIV, immunohistochemistry was performed using 5 μm serial sections obtained from autopsy samples of the occipital region of the brain for all presymptomatic study subjects and controls. Immunohistochemical staining for CD3 (to detect all T lymphocytes), CD8 (cytotoxic T cells), CD20 (B lymphocytes), CD14, CD16 and CD68 (monocytes/macrophages), and MHCII (antigen presenting cells, e.g. activated macrophages/microglia) was undertaken, and staining was quantified in areas of white matter by both manual counts (data not shown) and semi-automated quantitation using the Image-Pro Plus software.
All brain sections examined from the presymptomatic individuals were negative for the p24 antigen. However, there was a marked variability between individuals in the frequency of staining for cellular markers identifying different cell types (Figs 1 and 2). For example, many of the HIV-positive study subjects showed only low degrees of perivascular staining for CD8, with no staining in the parenchyma. However, three of the study subjects (NA200, NA234 and NA274) had extensive staining around vessels, and one study subject (NA159) had both perivascular and parenchymal staining, indicating an infiltration of T cells into deeper areas of the brain. Similar variability in staining was found for markers of activated macrophages and/or microglia (CD16 and MHCII). Despite the intra-group variability, staining for CD3, CD8, CD14, CD68 and MHCII was significantly greater among the HIV-positive individuals than controls (Fig. 2) irrespective of drug use. Indeed, for most markers, IDUs and non-IDUs in the control group produced comparable degrees of staining, with only CD14 showing significantly greater staining in IDUs (Fig. 2). The difference in lymphocyte infiltration and activation markers between HIV-positive and -negative individuals was therefore not the result of injecting drug use.
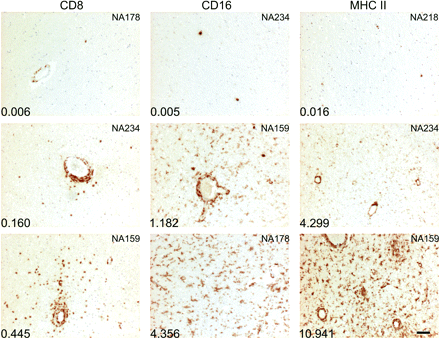
Range of staining intensities of cell type-specific markers. Representative images of staining with monoclonal antibodies to CD8 (T cell), CD16+ (infiltrating macrophages) and MHCII (antigen presenting cells) of sections showing low, intermediate and high frequencies of target cells. The patient identifier is shown in the top right-hand corner of each panel. The total area of the section stained for each marker quantified by automated Image-Pro Plus image analysis is shown in the bottom left-hand corner of each panel. The scale bar represents 50 microns.

Frequency of staining with monoclonal antibodies CD3, CD8, CD20, CD14, CD16, CD68 and MHCII of sections of the occipital region from each study subject and controls. For the control subjects, the following symbols were used: closed circles = HIV-negative IDUs; open circles = HIV-negative, non-IDUs. Staining was quantified as the percentage of total area stained for each marker by automated Image-Pro Plus image analysis (shown as a log scale on the y-axis; median values shown at top of graphs). Staining in study subjects and controls was compared by the Kruskal–Wallace non-parametric test (P-values < 0.05 shown in bold; P-values < 0.01 underlined; n.s. = not significant).
Other regions of the brain were studied in order to determine whether staining levels observed in the occipital region of the brain were representative of the brain as a whole. For these studies, two HIV-infected individuals were chosen who had consistently low or medium to high levels of staining of occipital sections across the panel of markers (NA218 and NA200, respectively; Table 1). Sections of 5 μm of the frontal region, parietal parasagittal region, cerebellum, pons and basal ganglia were used in immunohistochemical studies with antibodies against the range of specific cell-type markers (Fig. 3).
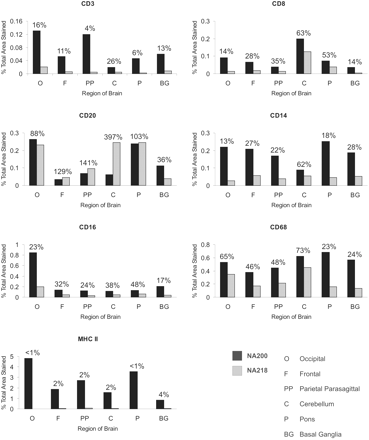
Immunohistochemical quantitation of markers for specific cell types in various regions of the brain in two study subjects showing high and low levels of cell infiltration in the occipital region (NA200 and NA218). Immunostaining in five other areas of the brain for each of the seven cell markers was quantified using the Image-Pro Plus software (percentage area stained shown on y-axis). The ratio of staining in sections from NA218 to that observed in sections from NA200 is shown above each pair of bars.
These comparisons revealed substantial variability in the degree of infiltration between brain regions for most markers. For example, in the occipital region, NA200 and NA218 had 0.091 and 0.013% total CD8 staining, respectively, while in the cerebellum total CD8 staining was 0.199 (NA200) and 0.125% (NA218), greater by 2.2- and 9.6-fold. With CD16, a much higher level of staining was observed in the occipital region than any of the other regions studied for NA200 (0.847% compared with 0.113–0.208%). This variability between regions was observed across the panel of markers for both study subjects. However, in almost all cases the actual difference in staining between NA200 and NA218 observed in the occipital region was reproduced in other brain regions. For CD3, CD8, CD14, CD16, CD68 and MHCII, the level of staining observed in samples from NA218 was consistently lower than the level observed in those from NA200 (NA218 percentages range from <1–73% total staining level of NA200). The ratio in staining levels between the two study subjects varied from marker to marker, but was reproducible in each brain region analysed. Thus the occipital region provides a representative view of the CNS of the relative (but not absolute) degree of lymphocytic and monocytic infiltration and the degree of activation of microglia in the brain.
For the occipital region from the HIV-infected subjects, there was an association between the detection of CD3 with CD8 (Table 2); the quantitative similarity in the extent of staining with the two monoclonal antibodies (Table 1; Fig. 2) provides evidence that the majority of infiltrating T cells were CTLs. There were also associations between the staining for the various macrophage and microglia markers with each other (CD14, CD16 and CD68; data not shown), as would be predicted. Expression of an activation marker, MHCII, correlated with CD8 infiltration of the CNS (R = 0.619; P < 0.05; Table 2). B cell infiltration (CD20) was not associated with any other cell markers (data not shown).
Correlations between cell infiltration, study subject, virology and segregation results in the study group
| Category | R-valuea | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infiltration | CD4 count | Proviral load | ||||||||||||
| CD3 | CD8 | CD16 | MHCII | Brain | Lymph | |||||||||
| Infiltration | ||||||||||||||
| CD3 | ||||||||||||||
| CD8 | 0.641 | |||||||||||||
| CD16 | −0.080 | −0.116 | ||||||||||||
| MHCII | 0.355 | 0.619 | 0.397 | |||||||||||
| CD4 count | −0.006 | −0.228 | −0.159 | −0.319 | ||||||||||
| Proviral load | ||||||||||||||
| Brain | 0.452 | 0.608 | −0.466 | 0.265 | 0.127 | |||||||||
| Lymphoid | 0.200 | 0.009 | −0.118 | −0.107 | −0.082 | 0.333 | ||||||||
| Segregation | ||||||||||||||
| AI value | −0.468 | −0.528 | −0.058 | −0.261 | 0.528 | −0.199 | −0.086 | |||||||
| Category | R-valuea | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infiltration | CD4 count | Proviral load | ||||||||||||
| CD3 | CD8 | CD16 | MHCII | Brain | Lymph | |||||||||
| Infiltration | ||||||||||||||
| CD3 | ||||||||||||||
| CD8 | 0.641 | |||||||||||||
| CD16 | −0.080 | −0.116 | ||||||||||||
| MHCII | 0.355 | 0.619 | 0.397 | |||||||||||
| CD4 count | −0.006 | −0.228 | −0.159 | −0.319 | ||||||||||
| Proviral load | ||||||||||||||
| Brain | 0.452 | 0.608 | −0.466 | 0.265 | 0.127 | |||||||||
| Lymphoid | 0.200 | 0.009 | −0.118 | −0.107 | −0.082 | 0.333 | ||||||||
| Segregation | ||||||||||||||
| AI value | −0.468 | −0.528 | −0.058 | −0.261 | 0.528 | −0.199 | −0.086 | |||||||
Correlation coefficients using Spearman's non-parametric test. Values in boldface: P ≤ 0.05.
Correlations between cell infiltration, study subject, virology and segregation results in the study group
| Category | R-valuea | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infiltration | CD4 count | Proviral load | ||||||||||||
| CD3 | CD8 | CD16 | MHCII | Brain | Lymph | |||||||||
| Infiltration | ||||||||||||||
| CD3 | ||||||||||||||
| CD8 | 0.641 | |||||||||||||
| CD16 | −0.080 | −0.116 | ||||||||||||
| MHCII | 0.355 | 0.619 | 0.397 | |||||||||||
| CD4 count | −0.006 | −0.228 | −0.159 | −0.319 | ||||||||||
| Proviral load | ||||||||||||||
| Brain | 0.452 | 0.608 | −0.466 | 0.265 | 0.127 | |||||||||
| Lymphoid | 0.200 | 0.009 | −0.118 | −0.107 | −0.082 | 0.333 | ||||||||
| Segregation | ||||||||||||||
| AI value | −0.468 | −0.528 | −0.058 | −0.261 | 0.528 | −0.199 | −0.086 | |||||||
| Category | R-valuea | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infiltration | CD4 count | Proviral load | ||||||||||||
| CD3 | CD8 | CD16 | MHCII | Brain | Lymph | |||||||||
| Infiltration | ||||||||||||||
| CD3 | ||||||||||||||
| CD8 | 0.641 | |||||||||||||
| CD16 | −0.080 | −0.116 | ||||||||||||
| MHCII | 0.355 | 0.619 | 0.397 | |||||||||||
| CD4 count | −0.006 | −0.228 | −0.159 | −0.319 | ||||||||||
| Proviral load | ||||||||||||||
| Brain | 0.452 | 0.608 | −0.466 | 0.265 | 0.127 | |||||||||
| Lymphoid | 0.200 | 0.009 | −0.118 | −0.107 | −0.082 | 0.333 | ||||||||
| Segregation | ||||||||||||||
| AI value | −0.468 | −0.528 | −0.058 | −0.261 | 0.528 | −0.199 | −0.086 | |||||||
Correlation coefficients using Spearman's non-parametric test. Values in boldface: P ≤ 0.05.
Quantitation of proviral DNA by limiting dilution
To determine the frequency of infected cells in brain and lymphoid tissue, we quantified proviral DNA using a highly-sensitive nested PCR technique in DNA extracted from samples of frozen occipital region (brain) and lymph node/spleen (lymphoid) tissue from the 15 HIV-infected presymptomatic individuals. PCR amplification reactions were performed at limiting dilution and observed frequencies of positive reactions were converted to proviral concentrations by standard Poisson distribution formulae (Table 1).
Proviral loads in both brain and lymphoid tissue were highly variable from individual to individual, varying from 3 up to 5244 provirus copies/106 cells in the lymphoid tissue. Proviral loads were invariably lower in brain samples than in the lymphoid tissue, ranging from <1 copy of provirus/106 cells to 58 copies/106 cells. To investigate the relationship between the numbers of infiltrating T cells in the brain and the levels of virus present in brain and lymphoid tissue, proviral loads were plotted against the level of CD8 staining in the brain as quantified by semi-automated methods (Fig. 4). There was a clear correlation between the level of infiltration of CD8+ cells into the brain and brain proviral load (Fig. 4A; Table 2), but none with proviral load in the lymphoid tissue (Fig. 4B; Table 2).
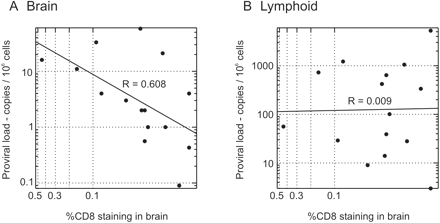
Association between degree of T cell infiltration of the brain with proviral load in (A) brain and (B) lymphoid tissue for each of the 15 study subjects. Proviral loads were calculated by titration to limiting dilution of DNA extracted from tissues amplified by nested PCR using primers from the V3 region.
Proviral loads in the brain were compared with a range of other variables, including the levels of specific cell types in the brain and disease progression. Apart from CD8 infiltration (see above), no significant correlations were found. Importantly, there was no relationship between the frequency of infected cells in the brain and disease progression or with the proviral load in lymphoid tissue (Table 2).
Comparisons of HIV proviral sequences from different tissues
The limiting dilution method developed previously by our group (Simmonds et al., 1990) is very suitable for the isolation of target sequences used in genetic comparisons of HIV populations in different tissues, because it guarantees that individual sequences are not re-sampled from the original populations. This is a frequently observed artefact that arises from cloning of the amplified PCR product (Kwok et al., 1990; Liu et al., 1996; van't Wout et al., 1998). To demonstrate the value of the limiting dilution method for population sampling, we carried out population comparisons of brain- and lymphoid-derived variants obtained at limiting dilution with those derived from cloned sequences (Fig. 5). Nested PCR amplification reactions were performed on brain and lymphoid tissue from study subjects NA200, NA159 and NA218. This was carried out either at limiting dilution to isolate single copies of provirus or by cloning of the resultant heterogeneous population into pGEM-T Easy. With the cloning method, following growth of the vector plus inserts in bacteria, positive colonies were selected for preparation of DNA and subsequent sequencing of the V3 and flanking region of env.
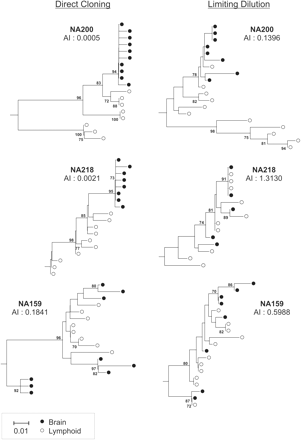
Phylogenetic comparison of brain and lymphoid V3 sequences obtained from study subjects NA200, NA218 and NA159 by two different methods. Trees were constructed from sequences obtained from cloning of amplified V3 sequences directly into pGEM-T Easy and sequencing of individual clones (left column) and from sequences obtained at limiting dilution by V3 nested PCR (right column). The AI value for each method is shown under the patient identifier for each tree. Trees were constructed by the neighbour-joining method using the MEGA 2.1 package; the frequencies of bootstrap replicates supporting individual clades are indicated on branches (only values ≥70% are shown). All trees were plotted to the scale indicated by the bar at the bottom of the figure.
As described previously (Wang et al., 2001), the AI value determines numerically the degree of genetic segregation between virus populations. AI values of ≈0.6 and under show evidence for a substantial degree of genetic segregation between different tissues. AI values were calculated and phylogenetic trees constructed of sequences obtained by both methods. AI values calculated from sequences obtained by conventional cloning methods consistently showed a higher degree of genetic segregation of HIV variants in brain and lymphoid tissue than those obtained through the limiting dilution nested PCR technique (Fig. 5). Study subject NA200 had AI values of 0.0005 and 0.140 when obtained by cloning and limiting dilution, respectively, a difference evident from comparison of phylogenetic trees constructed from the sequences (Fig. 5). Similarly, study subject NA159 had AI values of 0.184 (cloning) and 0.599 (limiting dilution), and study subject NA218 had AI values of 0.002 (cloning) and 1.31 (limiting dilution). The phylogenetic trees constructed following the limiting dilution method showed slightly interspersed (NA159) or very interspersed (NA218) populations of provirus, whereas the trees constructed following the cloning method showed highly segregated brain and lymphoid variants for both. In addition, several brain sequences obtained from NA159 by the cloning method encoded for stop codons, suggesting erroneous amplification of sequences. As discussed previously, template re-sampling in the cloning method was probably the cause of the higher degree of genetic segregation than those obtained by the limiting dilution technique.
To avoid artefactual results arising from re-sampling, the limiting dilution nested PCR technique was used to investigate population differences in HIV variants present in brain and lymphoid tissue, of the remaining study subjects. This analysis was necessarily limited to proviral sequences due to the nature of the autopsy tissue available; it was not possible to amplify viral RNA sequences from the tissues which might have provided a better indication of the actively replicating population of HIV (Simmonds et al., 1991; Ho et al., 1995; Wei et al., 1995). However, irrespective of whether archival populations were present among the proviral sequences, AI values calculated from the brain and lymphoid tissue would still be able to demonstrate whether populations in these tissues were distinct from each other or not.
AI values varied widely between the 15 study subjects (Table 1), ranging from 0.0003 (NA305), indicative of complete genetic segregation between virus populations in brain and lymphoid tissue to values of 1.3 and 1.1 (from NA218 and NA285) demonstrating equivalence of the CNS and lymphoid proviral populations. Since HIV variants in lymph nodes are similar to those found in peripheral blood cells (van't Wout et al., 1998), in these cases, proviral sequences detected in the CNS may well have arisen due to blood contamination of the brain samples. In total, 9 from 15 study subjects showed evidence for genetic segregation between brain and lymphoid populations (AI values ≈0.6 or under). In these individuals, the degree of genetic segregation (geometric mean value of AI values 0.16; range 0.003–0.61) was comparable to those previously observed in HIV variants associated with HIVE (geometric mean value 0.02; range 0.0002–0.52) (Wang et al., 2001).
Comparison of V3 hypervariable region amino acid sequences
To investigate whether variants recovered from brain tissue by limiting dilution nested PCR shared predicted phenotypes with those recovered from lymphoid tissue using the same method, a comparison of the overall charge of the V3 loop of each sequence was undertaken. Sequences with an overall net charge of between +2 and +4 indicate an R5 phenotype, reflecting both acidic and basic residues (Chesebro et al., 1992; de Jong et al., 1992; Fouchier et al., 1992). V3 sequences with an overall net charge of ≥+5 would have an X4 predicted phenotype, possessing a reduced frequency of acidic residues and an increased frequency of basic residues. With one exception, all brain sequences recovered had an R5 predicted phenotype (Table 1). Similarly, the majority of lymphoid sequences recovered had a predicted R5 phenotype, with only a few of the study subjects showing some sequences with a predicted X4 phenotype. NA218 was unusual in that all brain and lymphoid sequences had a predicted X4 phenotype, although in this case, the lack of genetic segregation (AI value 1.3) and the proviral load in the brain (21 proviral copies/106 cells) could indeed be accounted for by peripheral blood contamination of the tissue sample (Donaldson et al., 1994b).
Correlations with genetic segregation
The AI value provides a numerical measure of the degree of distinctness between the brain and lymphoid populations and could therefore be used in non-parametric tests to investigate correlations with other study group variables (Table 2). There were significant associations between genetic segregation and the degree of CD8 infiltration in the brain (R = −0.528; P < 0.05), and with CD4 count (R = 0.528; P < 0.05), indicating greater segregation in individuals with disease progression (Table 2; Fig. 6). These associations were not simply secondary associations arising from an increased likelihood of CNS invasion with HIV progression, as there was no correlation between proviral load in the brain with CD4 count [R = 0.127 (n.s.); Table 2].
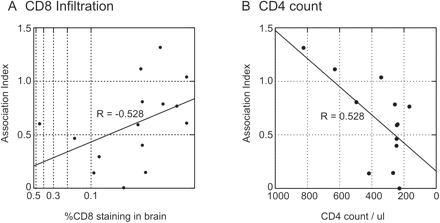
Factors influencing genetic segregation of brain and lymphoid tissue-derived virus populations, as determined by calculation of association indices (y-axis). (A) Semi-quantitation of CD8 lymphocyte infiltration of the brain. (B) CD4 lymphocyte counts in peripheral blood.
In summary, this study group showed widely ranging degrees of inflammatory cell infiltration into the CNS, accompanied by varying but frequently low or undetectable proviral loads. Populations of HIV were usually genetically distinct from those recovered from lymphoid tissue. Uniquely amongst the cell markers tested, CD8 infiltration was linked both with greater cellular activation and with higher levels of genetically distinct HIV populations in the brain. The cytotoxic T cell response therefore appeared to be a key correlate of the early stages of HIV invasion of the CNS.
Discussion
This study examined early pathology of the CNS associated with HIV infection. From a methodological point of view, a highly sensitive and specific nested PCR method was required to recover and accurately quantify the frequently very low numbers of infected cells in brain samples. Limiting dilution (Simmonds et al., 1990) was shown to be essential for correct genetic analysis of virus populations in the CNS, avoiding the phenomenon of template re-sampling that produces spurious estimates of population diversity, and the detection of segregation where none is present (Fig. 5). These methods were therefore key factors in our ability to detect and genetically characterize the virus populations in the CNS in this study.
Inflammatory changes in the CNS of presymptomatic individuals
We obtained evidence for varying and frequently extensive inflammatory changes occurring in the CNS in the presymptomatic study group, consistent with an infection process. High level expression of MHCII was found in the brains of the majority of subjects, demonstrating the levels of cellular activation approaching that observed in cases of AIDS (Table 1; Fig. 2) (Anthony et al., 2005a). Immunocytochemistry quantitation also revealed increased expression of CD14 and CD16, known to be upregulated on microglia/infiltrating macrophages in association with trauma and inflammatory processes (Ulvestad et al., 1994; Becher et al., 1996; Nadeau and Rivest, 2000; Beschorner et al., 2002). Increased staining for CD3, CD8 and CD20 compared with seronegative controls showed substantial infiltration of T and B lymphocytes.
Despite this, generally very low levels or undetectable frequencies of HIV-infected cells were found in the CNS by PCR. Furthermore, no actively virus-expressing cells were detected using immunohistochemistry for p24 antigen. However, genetic analysis revealed the frequent presence of a separate, brain-associated virus population in the majority of study subjects; brain autopsy samples frequently contained sequences in the V3 region that were largely or entirely genetically distinct from those recovered from infected cells in the lymph node or spleen. Indeed, in the study subjects with AI values of ≈0.6 or under, the distinct nature of the brain-derived sequences, particularly the absence of viruses with a predicted X4 phenotype (despite their detection in lymphoid tissue), was more consistent with the partitioning of HIV variants found in HIVE (Liu et al., 1990; Epstein et al., 1991; Haggerty and Stevenson, 1991; Li et al., 1991; Pang et al., 1991; Steuler et al., 1992; Keys et al., 1993; Ball et al., 1994; Donaldson et al., 1994a; Korber et al., 1994; Power et al., 1994; Di Stefano et al., 1996; Reddy et al., 1996; Gartner et al., 1997; van't Wout et al., 1998; Gatanaga et al., 1999; Morris et al., 1999).
Despite the evident restriction in virus replication, findings of frequent genetic partitioning argue for early and continuing trafficking of HIV into the CNS, potentially triggering the inflammatory responses observed. The absence of detectable virus expression may indeed reflect the effectiveness of CTLs in pre-AIDS in destroying antigen-expressing cells, and reduce their frequencies to levels undetectable by immunocytochemistry.
CD8 lymphocyte infiltration of the CNS
While infiltration of CD8 lymphocytes into the CNS of presymptomatic individuals has been documented previously (Bell et al., 1993), the current study was additionally able to clearly link this process to infection of the CNS by HIV. As described, there were correlations between CD8 infiltration with both HIV proviral load in the brain and degree of genetic segregation of CNS and lymphoid-derived variants, as well as with markers of cellular activation (Table 2). Importantly, these changes were not simply a reflection of disease progression, as CD8 infiltration in the brain was not associated with the peripheral CD4 count.
There was no detectable association between infiltration of the CNS with CD14+/CD16+ macrophages or activation of resident microglia (both putative target cells for HIV infection) with the degree of virus infection of the CNS, a finding seemingly inconsistent with higher detection frequencies of these cell types in cases of HIVE (Fischer-Smith et al., 2004). However, as the frequencies of HIV-infected cells in presymptomatic stages of infection were generally so low, it is unlikely that they would have been detected in a background of more general inflammatory processes occurring in the pre-AIDS CNS.
Although macrophage trafficking is widely accepted as the source of CNS infection of the CNS in HIVE, it is possible that different processes are responsible for CNS invasion in pre-AIDS. As described, it was not possible to localize and identify morphologically any p24 antigen-expressing cells in the section, and cell-sorting methods could not be applied to previously frozen brain samples. Amongst other candidate infected cell types, the observed B cell infiltration would be unlikely to be associated with CNS invasion, as this cell type is uninfected in vivo. For other lymphocyte subsets, it was unfortunately not possible to determine the extent of CD4 infiltration in the study subjects, as monoclonal antibodies to this marker are unreliable for paraffin-embedded tissue. However, the similarity in the degree of CD3 and CD8 staining of sections (Table 1; Fig. 2) provided evidence that CD4 lymphocytes were at most a minor component of the lymphocytic infiltration of the brain. Secondly, as CD4 lymphocyte is the predominant cell type infected in lymphoid tissue, the often highly genetically distinct brain- and lymphoid tissue-associated virus populations would not have been observed.
Infected CD8 lymphocytes are a more plausible source of HIV detected in the brain, particularly as their degree of infiltration was directly correlated with proviral load and genetic segregation in the brain (Table 1). CD8 lymphocytes in peripheral blood are detectably infected throughout the course of HIV infection (Livingstone et al., 1996; Cochrane et al., 2004), at frequencies of between 1 and 300 copies/106 cells (median values 4 and 8 copies/106 cells in the two studies). However, these frequencies of infected CD8 lymphocytes, combined with their low overall proportion of cells in brain tissue (<1%), are clearly unable to account for the brain proviral loads observed in the current study. The marked genetic segregation, particularly the absence of X4 viruses observed in brain-associated populations is similarly inconsistent with the CD8 viral origin hypothesis.
Together, the findings in this study support a scenario in which a functioning immune system plays a pivotal role in controlling infection of the CNS. From a wealth of immunopathology, virology and genetic information it has become established that infected macrophages disseminate HIV infection throughout the body, carrying variants of HIV distinct from those in lymphocytes, in particular in their restriction to variants using the CCR5 receptor for entry. The lymphocytosis and cellular activation observed in the CNS of pre-AIDS individuals is therefore the outward manifestation of the control of this process exerted by a still functioning immune system. Thus CD8 infiltration is a more active process in individuals with greater degrees of CNS invasion (as observed in this study), even though the process is efficient enough to prevent the detection of actively infected cells by immunohistochemistry.
In this model, the eventual development of HIVE reflects a breakdown of this control as a result of more generalized immunosuppression. Measures that restore immune function, such as antiretroviral therapy (ART), would therefore be predicted to prevent the progression of the infection process to a productive phase in the brain, irrespective of whether therapeutic levels of antiviral drugs are able to penetrate the BBB or not. The evidence in this study for the control of CNS infection, even in individuals with CD4 counts ∼200 copies/μl (Table 1), suggests that even modest improvements in frequencies of circulating CD4 lymphocytes by therapy would restore control of infection with HIV and potentially of opportunistic infections in the brain. As suggested by this model, while proviral loads and the incidence of HIVE and other HIV-associated neuropathology would be expected to decline with effective ART, inflammatory processes and microglial activation associated with CTL-mediated containment of infection would continue. This prediction is indeed borne out by our recent study of an autopsy series of individuals with HIV infections controlled by highly active antiretroviral therapy (HAART) (Anthony et al., 2005b); these individuals showed similar MHCII expression and CD8 infiltration in the brain to that found in AIDS or HIVE cases (and the presymptomatic individuals studies here), despite the clearance or containment of active HIV infection.
The model developed here therefore does not entail the existence of an autonomous, potentially neuroadapted or neuropathogenic population of HIV in the CNS. The sequence differences observed between CNS and lymphoid tissue-derived viruses likely reflect deeper divisions between HIV variants adapted for replication in lymphocytes and macrophages, a conclusion borne out by phenotypic analysis (Gorry et al., 2001, 2002) and by the observation of CNS-related populations of HIV in other macrophage-infiltrated tissues such as the lung and bone marrow (Wang et al., 2001). However, the ‘immune-control’ model for HIV CNS infection described here still does not directly account for the varying incidences of HIVE in different individuals and clinical backgrounds. Indeed, innate variation in the effectiveness of immune control, other factors that damage the CNS independently (such as injecting drug-induced activation of microglia) and the evolution of specifically neuropathogenic variants of HIV are potentially further factors in the eventual development of HIVE that can be incorporated into this model.
The authors are grateful to Stephen Ramage for the assistance with immunohistochemistry. This study was funded by NIH grants 5R21MH65860-03, 1RO1DA131127 and 1RO1DA13840 using samples archived in the Medical Research Council HIV Brain Bank G9708080.
References
Achim CL, Wang R, Miners DK, Wiley CA. Brain viral burden in HIV infection.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection?
Anthony IC, Ramage SN, Carnie FW, Simrnonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation.
Ball JK, Holmes EC, Whitwell H, Desselberger U. Genomic variation of human immunodeficiency virus type 1 (HIV-1)—molecular analyses of HIV-1 in sequential blood samples and various organs obtained at autopsy.
Becher B, Fedorowicz V, Antel JP. Regulation of CD14 expression on human adult central nervous system-derived microglia.
Bell JE, Busuttil A, Ironside JW, Rebus S, Donaldson YK, Simmonds P, et al., Human immunodeficiency virus and the brain—investigation of virus load and neuropathologic changes in pre-AIDS subjects.
Bell JE, Donaldson YK, Lowrie S, McKenzie CA, Elton RA, Chiswick A, et al. Influence of risk group and zidovudine therapy on the development of HIV encephalitis and cognitive impairment in AIDS patients.
Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement.
Beschorner R, Schluesener HJ, Gozalan F, Meyermann R, Schwab JM. Infiltrating CD14+ monocytes and expression of CD14 by activated parenchymal microglia/macrophages contribute to the pool of CD14+ cells in ischemic brain lesions.
Bukrinsky MI, Nottet HSLM, Schmidtmayerova H, Dubrovsky L, Flanagan CR, Mullins ME, et al. Regulation of nitric oxide synthase activity in human immunodeficiency virus type 1 (HIV-1)-infected monocytes: implications for HIV-associated neurological disease.
Chen H, Wood C, Petito CK. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: Potential role of choroid plexus in the pathogenesis of HIV encephalitis.
Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparsion with T cell-tropic isolates: definition of critical amino acids involved in cell tropism.
Cochrane A, Imlach S, Leen C, Scott G, Kennedy D, Simmonds P. High levels of human immunodeficiency virus infection of CD8 lymphocytes expressing CD4 in vivo.
Davis LE, Hjelle BL, Miller VE, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection.
de Jong JJ, de Ronde A, Keulen W, Tersmette M, Goudsmit J. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution.
Di Stefano M, Wilt S, Gray F, DuboisDalcq M, Chiodi F. HIV type 1 V3 sequences and the development of dementia during AIDS.
Donaldson YK, Bell JE, Holmes EC, Hughes ES, Brown HK, Simmonds P. In vivo distribution and cytopathology of variants of human immunodeficiency virus type 1 showing restricted sequence variability in the V3 loop.
Donaldson YK, Bell JE, Ironside JW, Brette RP, Robertson JR, Busuttil A, et al. Redistribution of HIV outside the lymphoid system with onset of AIDS.
Epstein LG, Kuiken C, Blumberg BM, Hartman S, Sharer LR, Clement M, et al. HIV-1 V3 domain variation in brain and spleen of children with AIDS: tissue-specific evolution within host-determined quasispecies.
Falangola MF, Hanly A, Galvao-Castro B, Petito CK. HIV infection of human choroid plexus: a possible mechanism of viral entry into the CNS.
Fischer-Smith T, Croul S, Sverstiuk AE Capini C, L'Heureux D, Regulier EG, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection.
Fischer-Smith T, Croul S, Adeniyi A, Rybicka K, Morgello S, Khalili K, et al. Macrophage/microglial accumulation and proliferating cell nuclear antigen expression in the central nervous system in human immunodeficiency virus encephalopathy.
Fouchier RAM, Groenink M, Kootstra NA, Tersmette MH, Huisman G, Miedema F, et al. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule.
Gartner S, McDonald RA, Hunter EA, Bouwman F, Liu Y, Popovic M. Gp120 sequence variation in brain and in T-lymphocyte human immunodeficiency virus type 1 primary isolates.
Gatanaga H, Oka S, Ida S, Wakabayashi T, Shioda T, Iwamoto A. Active HIV-1 redistribution and replication in the brain with HIV encephalitis.
Gelbard HA, Nottet HSLM, Swindells S, Jett M, Dzenko KA, Genis P, et al. Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin.
Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, et al. Cytokines and arachidonic acid metabolites produced during HIV-infected macrophage-astroglial interactions: implications for the neuropathogenesis of HIV disease.
Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of human immunodeficiency virus type-1 stimulates release of neurotoxins from monocytes.
Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity.
Gorry PR, Taylor J, Holm GH, Mehle A, Morgan T, Cayabyab M, et al. Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate.
Gray F, Scaravilli F, Everall I, et al. Neuropathology of early HIV-1 infection.
Griffin DE. Cytokines in the brain during viral infection: clues to HIV- associated dementia.
Haggerty S, Stevenson M. Predominance of distinct viral genotypes in brain and lymph node compartments of HIV-infected individuals.
Heyes MP, Saito K, Lackner A, Wiley CA, Achim CL, Markey SP. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques.
Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection.
Hughes ES, Bell JE, Simmonds P. Investigation of the dynamics of the spread of human immunodeficiency virus to brain and other tissues by evolutionary analysis of sequences from the p17(gag) and env genes.
Keys B, Karis J, Fadeel B, Valentin A, Norkrans G, Hagberg L, et al. V3 sequences of paired HIV-1 isolates from blood and cerebrospinal fluid cluster according to host and show variation related to the clinical stage of disease.
Korber BT, Kunstman KJ, Patterson BK, Furtado M, McEvilly MM, Levy R, et al. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences.
Kwok S, Kellogg DE, Mckinney N, Spasic D, Goda L, Levenson C, et al. Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies.
Li YX, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes.
Liu ZQ, Wood C, Levy JA, Cheng Mayer C. The viral envelope gene is involved in macrophage tropism of a human immunodeficiency virus type 1 strain isolated from brain tissue.
Liu SL, Rodrigo AG, Shankarappa R, Learn GH. HIV quasispecies and resampling.
Liu Y, Tang XP, Mcarthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain.
Livingstone WJ, Moore M, Innes D, Bell JE, Simmonds P. Frequent infection of peripheral blood CD8-positive T-lymphocytes with HIV-1.
Morris A, Marsden M, Halcrow K, Hughes ES, Brettle RP, Bell JE, et al. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations.
Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia.
Pang S, Koyanagi Y, Miles S, Wiley C, Vinters HV, Chen IS. High levels of unintegrated HIV-1 DNA in brain tissue of AIDS dementia patients.
Pang S, Vinters HV, Akashi T, OBrien WA, Chen IS. HIV-1 env sequence variation in brain tissue of patients with AIDS-related neurologic disease.
Petito CK, Chen H, Mastri AR, Torres-Munoz J, Roberts B, Wood C. HIV infection of choroid plexus in AIDS and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection.
Power C, Mcarthur JC, Johnson RT, Griffin DE, Glass JD, Perryman S, et al. Demented and nondemented patients with AIDS differ in brain-derived human immunodeficiency virus type 1 envelope sequences.
Pulliam L, Herndier BG, Tang NM, McGrath MS. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains.
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia.
Reddy RT, Achim CL, Sirko DA, Tehranchi S, Kraus FG, Wong-Staal F, et al. Sequence analysis of the V3 loop in brain and spleen of patients with HIV encephalitis.
Simmonds P, Balfe P, Peutherer JF, Ludlam CA, Bishop JO, Leigh Brown AJ. Human immunodeficiency virus-infected individuals contain provirus in small numbers of peripheral mononuclear cells and at low copy numbers.
Simmonds P, Zhang LQ, McOmish F, Balfe P, Ludlam CA, Leigh Brown AJ. Discontinuous sequence change of human immunodeficiency virus (HIV) type 1 env sequences in plasma viral and lymphocyte-associated proviral populations in vivo: implications for models of HIV pathogenesis.
Steuler H, Storch Hagenlocher B, Wildemann B. Distinct populations of human immunodeficiency virus type 1 in blood and cerebrospinal fluid.
Strappe PM, Wang TH, McKenzie CA, Lowrie S, Simmonds P, Bell JE. Enhancement of immunohistochemical detection of HIV-1 p24 antigen in brain by tyramide signal amplification.
Tomlinson GS, Simmonds P, Busuttil A, Chiswick A, Bell JE. Upregulation of microglia in drug users with and without pre-symptomatic HIV infection.
Ulvestad E, Williams K, Mork S, Antel J, Nyland H. Phenotypic differences between human monocytes/macrophages and microglial cells studied in situ and in vitro.
van't Wout AB, Ran LJ, Kuiken CL, Kootstra NA, Pals ST, Schuitemaker H. Analysis of the temporal relationship between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy.
Wang TH, Donaldson YK, Brettle RP, Bell JE, Simmonds P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system.
Author notes
1Centre for Infectious Diseases, 2Department of Pathology, Western General Hospital and 3Regional Infectious Diseases Unit, Western General Hospital, University of Edinburgh, Edinburgh, UK


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}