





Abstract
Motivation: Amyloids play a role in the degradation of β-cells in diabetes patients. In particular, short amyloid oligomers inject themselves into the membranes of these cells and create pores that disrupt the strictly controlled flow of ions through the membranes. This leads to cell death. Getting rid of the short oligomers either by a deconstruction process or by elongating them into longer fibrils will reduce this toxicity and allow the β-cells to live longer.
Results: We develop a computational method to probe the binding affinity of amyloid structures and produce an amylin analog that binds to oligomers and extends their length. The binding and extension lower toxicity and β-cell death. The amylin analog is designed through a parsimonious selection of mutations and is to be administered with the pramlintide drug, but not to interact with it. The mutations (T9K L12K S28H T30K) produce a stable native structure, strong binding affinity to oligomers, and long fibrils. We present an extended mathematical model for the insulin–glucose relationship and demonstrate how affecting the concentration of oligomers with such analog is strictly coupled with insulin release and β-cell fitness.
Availability and implementation: SEMBA, the tool to probe the binding affinity of amyloid proteins and generate the binding affinity scoring matrices and R-scores is available at: http://amyloid.cs.mcgill.ca
Contact: jeromew@cs.mcgill.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Insulin and amylin molecules are co-secreted from β-cells in the pancreas to lower the concentration of glucose in the blood. Some of the amylin misfold into amyloids and do not continue to carry on their normal functions in the body. In type II diabetes, these amylin amyloids are observed to form in the extracellular space surrounding β-cells and build up into different sizes, some small (oligomers) and others large (fibrils; MacArthur etal., 1999). The exact effect of the very large fibril deposits on the progression of diabetes and the inflammation of β-cells is not really known. However, amyloid oligomers have been clearly observed to be toxic, while the longer fibrils are non-toxic (Meier etal., 2006). These aggregates inject themselves into cell membranes and create weakly selective pores that introduce an uncontrolled influx of ions into and out of the cell (Lashuel et al., 2002; Mirzabekov etal., 1996). Their short length allows for ions to pass through them easily, unlike fibrils. The influx of ions is mainly due to the high concentration gradient of calcium molecules separating the cytoplasm and extracellular space. This influx disrupts cell coupling, impairs insulin secretion, depletes ATP, depolarizes the membrane and weakens cells inducing apoptosis (Ritzel etal., 2007). Upon cell breakdown, the oligomers find other live cells to target (Meier etal., 2006). Figure 1 shows an illustration of amylin oligomers permeating a cell membrane.
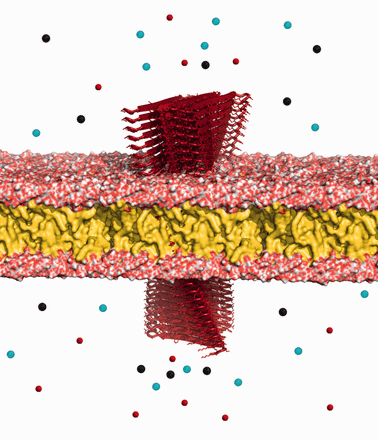
Oligomer toxicity. Amylin amyloid oligomers (red) permeating the cell membrane and disrupting the tightly controlled flow of ions through the membrane. Figure created with PyMol (Schrodinger, 2010)
When too many amylin molecules misfold into amyloids, due to insulin resistance and extreme pressure on the pancreas to secrete insulin, the required levels of amylin in the body to modulate gastric emptying and prevent postprandial spikes in glucose levels are not met. This further complicates the conditions of type II diabetes patients. Several injectable amylin analogs have been created in an effort to find a suitable replacement for this lack of the necessary levels of amylin. The most promising one, pramlintide, is an analog containing 3 proline substitutions at positions 25, 28 and 29, and reveals a weak tendency to aggregate (Cort etal., 2009). Pramlintide is an effective amylin replacement agent that acts as a synergetic partner to insulin (Riddle etal., 2007). Pramlintide does not enter the cells of the pancreas; however, it moves through the circulatory system to reach necessary organs and compensates for the lack of sufficient amylin levels.
Although pramlintide has a weaker tendency to aggregate, and compensates for amylin’s lost functions, it does not solve the problem of amyloid oligomer toxicity. The inhibition of the amyloid production has been studied as a way to elongate cell life in diabetes. Molecules like rifampicin have been designed to prevent fibril formation (Tomiyama etal., 1997), but failed to remove the short toxic oligomers that speed up the death of cells (Meier etal., 2006). Other attempts at designing inhibiting molecules include the red wine compound resveratrol (Mishra etal., 2009), Vanadium complexes (He etal., 2014), Polyphenols (Porat etal., 2004, 2006), selenium-containing phycocyanin molecules (Li etal., 2014) and N-methylation of amylin (Tatarek-Nossol etal., 2005). Many of these molecules presented strong inhibiting results that lower the rate of oligomerization in vitro and are currently undergoing more experimentation. However, none of them are at a stage of being produced as a therapeutic agent.
In this work we explore a different technique to reduce the toxicity of oligomers. Instead of inhibiting fibril extension, we promote it. At a higher rate of amyloid aggregation, longer structures are created. The higher the rate, the lower the concentration of oligomers and the higher the concentration of fibrils. We aim to achieve this high aggregation rate by constructing a molecule that when injected into the human body, binds to these short oligomers and extends their length, promoting aggregation and forcing them into long non-toxic fibrils. A circumspect approach to create such molecule that does not perform unknown and undesirable interactions in the body is to engineer the molecule as an amylin analog with a parsimonious selection of mutations to perform the task. We construct this molecule as an analog of high amyloidogenicity, to have very strong binding affinity to the amyloids, and to have an extremely low dissociation rate. The low dissociation rate is essential in preventing the emergence of more short oligomers through fibrils breaking up. This analog is designed to be administered with the pramlintide drug in a native, non-amyloid form. The analog is engineered to have a low binding affinity to pramlintide to prevent inducing pramlintide to misfold. To conserve structure, the analog is designed through a parsimonious selection of mutations. Moreover, we compare the aggregation potential and stability of our top findings with state-of-the-art tools [FoldX (Schymkowitz etal., 2005), Zyggregator (Tartaglia etal., 2008), PASTA (Walsh etal., 2014) and TANGO (Fernandez-Escamilla etal., 2004)] to increase confidence in our selection.
2 Approach
Ridding the pancreas from toxic, short amyloid oligomers will reduce β-cell death. In theory, we could remove these oligomers in three ways:
Altering the environment around amyloids to reduce the rate of nucleation and inhibit oligomerization.
Designing molecules that bind to the ends of oligomers to stop the flow of ions through pores and to inhibit amyloid extension.
Promoting amyloid extension and fibril growth to reduce oligomer toxicity.
In this work, we develop a novel method to assist researchers with solutions 2 and 3. Although we do not focus on exploring molecules to inhibit amyloid growth, our work will show how the method could be used to assess the binding preferences and affinity of amyloids to other molecules. In particular, we use the method to improve the binding of amylin amyloids to other amyloids, stimulating growth and extension. Promoting amyloid extension forces oligomers to grow into fibrils and decreases their toxicity.
Amyloids inherently bind to each other. They have a strong affinity to aggregate. Introducing the right amino acid mutations into amyloids can increase their binding affinity and likelihood to aggregate. This increase results in longer structures, forcing short oligomers to grow into fibrils. The binding affinity scoring matrix (BASM) method we introduce in this article explores the affinity of amyloid residues to all possible amino acid bindings. We explore the effect of pairing a single amino acid with an amyloid structure at a specific residue position. For each of the residue positions of an amyloid, we generate 20 binding affinity readings, one for every position and amino acid pair, as illustrated in Figure 2. The affinity measures the Lennard-Jones potential between the amino acid and the amyloid structure, the Coulomb interactions, and the Solvation energy of the system. We organize the readings into a n × 20 matrix. The first dimension of the matrix comprises 20 amino acids, the second dimension is the protein residue positions, and each record is what we call an R-score. The lower the R-scores, the stronger the binding affinity between an amino acid and the amyloid structure at the specific residue position.
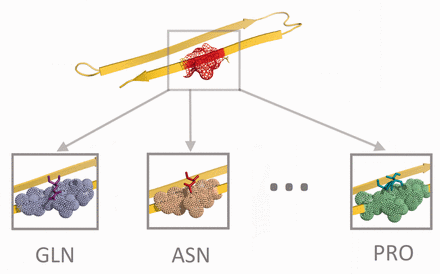
Binding affinity method. Twenty amino acids are placed on top of a specific residue position on the amylin protein, one at a time, to compute the R-scores in Equation (4)
The goal behind the BASM method is to identify bindings that increase affinity. The best structure bindings could be used to engineer an amylin analog that produces amyloids that bind better to the ones in the pancreas. If designed with the proper amyloidogenicity, the engineered analog will not only bind well to oligomers, but will also promote amyloid extension. This extension reduces oligomer toxicity and β-cell death.
The analog can be administered in native, non-amyloid form with pramlintide injections. In its native form, it will carry out the normal functions of amylin and pramlintide. Once it reaches the pancreas (through the bloodstream) and makes contact with the amyloids, it should misfold and aggregate with the surrounding amyloids and oligomers due to its high amyloidogenicity. To prevent any potential aggregation with pramlintide, the analog is designed with lowest affinity to pramlintide. In the Methods section we explain in detail how we develop this analog by generating the BASM R-scores for amylin and pramlintide amyloids.
3 Methods
3.1 BASM R-scores
3.2 BASM matrix
3.2.1 Binding affinity function
3.2.2 Finding best fit to amylin and worst to pramlintide
If we let be the sequence binding affinity function for , we can construct S* for this system by using Equations (12)–(14). S* might contain 1 to n differences in amino acids compared to amylin’s sequence, S1. However, too many differences in amino acids might create a structure that differs from amylin and does not bind to the amyloids. To guarantee that we have a sequence that represents a structurally conserved, folded amylin analog, we will restrict the number of possible differences between S* and S1. Using S*, we will construct a parsimonious sequence SP.
3.3 Amyloidogenicity
3.4 Oligomer concentrations
Out of the set of solutions for Equation (18) we choose the parsimonious solution that has the lowest dissociation potential, producing the lowest number of oligomers. For each solution, we measure the concentration of oligomers they might create. We can quantitatively express these concentrations by computing the free energies of the oligomers. The expression for the concentration of these structures in terms of the chemical potential of the system μ, the free energies of oligomers Fk, and the number of monomers k at each state can be computed by the law of mass action.
Let’s denote by N the initial number of proteins in the solution, V the volume of the system, Nk the number of oligomers of type k. Let a3 be the volume of a protein. The initial volume fraction of the proteins is defined by where is the initial concentration of proteins and the volume fraction of the oligomers of type k by . Note that all and must be positive and smaller than 1.
3.5 Molecular dynamics (MD) and energy minimization (EM)
We used the GROMACS 4.5 (Pronk etal., 2013) molecular simulation package to run MD and EM simulations. Our mutant molecules were solvated in a cubic box (with a minimum distance of 35 Å from any edge of the box to any atom) and neutralized with chloride ions and modeled using the GROMOS96 53a6 force field along with the SPC water model. We used a cutoff of 10 Åfor van der Waals and short range electrostatic interactions, and calculated long range electrostatic interactions using a particle mesh Ewald sum (Darden etal., 1993; Essmann etal., 1995). Simulations were prepared for a full MD run in both isothermal-isobaric (100 ps) and canonical equilibration (100 ps) ensembles. Temperature and pressure were controlled at 300 K and 1 bar using the velocity rescaling thermostat and the Parrinello-Rahman barostat, respectively. A linear constraint solver was used to keep all bonds at their equilibrium length. One million time steps were used with an integration time step of 2 fs. The system’s coordinates were saved every 10 ps for further analysis.
3.6 Dipolar water solvent
We resort to our previous work in (Smaoui etal., 2013) to calculate the Free energy of protein aggregates and plot Figure 3. In particular, we compute a Lennard-Jones, Coulomb and Solvation energy terms. The Solvation term is computed using a fast and detailed dipolar water model that solves the dipolar nonlinear Poisson–Boltzmann–Langevin equation. The three energy terms are used to describe the stability of molecular forces in molecules. Solvation energy was precisely calculated using the AQUASOL routine (Koehl and Delarue, 2010) with the following setup: atomic charges and radii assigned with PDB2PQR using CHARMM force field at neutral pH. A grid or 257 points per edge spaced by 1 Å, a temperature of 300 K, and a solvent accessible surface with an Rprobe of 1.4 Å. All hydrogen-bonds were optimized. We used a trilinear interpolation protocol for projection of fixed charges on the grid, a lattice grid size for the solvent: a = 2.8 Å, solvent made of dipoles of moment p0 = 3.00D at a concentration of Cdip = 55 M. No salt was added to the solution and small ions were used to equilibrate the system when needed. The electrostatic potential was set to zero at the boundaries, and the stopping criteria for residual was sent to: (when possible).
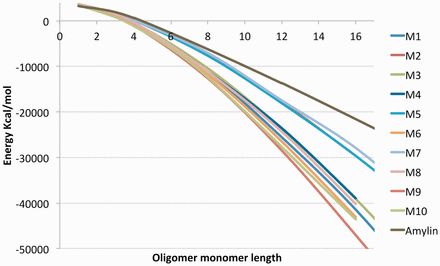
Energies for top 10 amylin analog oligomers
3.7 Building oligomer structures
The 3D structures (PDB files) are required in the process of assessing the concentration of oligomers produced by a particular analog. To create the 3D oligomers for amylin, we start with the amylin PDB structure solved by Wiltzius et. al (2008) and use SCWRL (Wang etal., 2008) to perform the necessary mutations introduced in each analog. The mutated structure is then used by the CreateFibril tool (Smaoui etal., 2013) to construct the oligomers with the following parameters: 8° rotation angle, 5.0 Å between monomers, and a fibril packing distance of 3 units.
3.8 The effect of amyloid oligomers on the insulin–glucose system
The model is analyzed further in the Section 5 and provides a foundation for exploring the relationship between oligomers and β-cell fitness.
4 Results
We introduced the method to compute R-scores of BASM in the Methods section and described how they can be used to engineer an injectable amylin analog that binds well to amyloids in the pancreas while preventing pramlintide from misfolding. Supplementary Table S1 shows the R-scores that we generated for all the residue positions of amylin amyloid, and Supplementary Table S2 shows the R-scores for pramlintide’s amyloid residues. In Table 1, we solve for the sequence S* with optimal binding affinity to BASM for various α and β values. Setting a fixed β = 1 value and increasing α doesn’t drastically change the sequence S*, suggesting that the residues of S* possess a strong binding affinity to amylin. We proceed with sequence S* generated by α = 2 and β = 1. This sequence encompasses a binding affinity to amylin that is twice as large as the repelling forces to pramlintide.
Sequences S* for various α and β binding affinity values
| α | β | Sequence S* | F(S*) |
|---|---|---|---|
| 1 | 0 | KKKKMKKQPKKKKMKNNYHKKYFHNKPLKKNMKNKQK | -5484 |
| 0 | 1 | ETIWDGESDDYRPLTTTQKTFTTFWDFEESKRPEYET | -3944 |
| 1 | 1 | KTKKKKKKKKKKKEKIKKKKFTKPNKHESKKVNKHKK | -3162 |
| 2 | 1 | KKKKMKKQKKKKKMKNEYYKKYFHNKKHKKKVKQKFK | -7913 |
| 4 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKKHKKNWKNKFK | -18737 |
| 10 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKPLKKNMKNKFK | -51585 |
| 1 | 2 | ETIWDGEKDIYRWLTTTQKTFTKRFDHESSKRPEHET | -5720 |
| α | β | Sequence S* | F(S*) |
|---|---|---|---|
| 1 | 0 | KKKKMKKQPKKKKMKNNYHKKYFHNKPLKKNMKNKQK | -5484 |
| 0 | 1 | ETIWDGESDDYRPLTTTQKTFTTFWDFEESKRPEYET | -3944 |
| 1 | 1 | KTKKKKKKKKKKKEKIKKKKFTKPNKHESKKVNKHKK | -3162 |
| 2 | 1 | KKKKMKKQKKKKKMKNEYYKKYFHNKKHKKKVKQKFK | -7913 |
| 4 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKKHKKNWKNKFK | -18737 |
| 10 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKPLKKNMKNKFK | -51585 |
| 1 | 2 | ETIWDGEKDIYRWLTTTQKTFTKRFDHESSKRPEHET | -5720 |
results in a sequence S* that has a binding affinity to amylin amyloids that is equal to its repelling affinity towards pramlintide. A results in a sequence that binds better to amylin amyloids and repels pramlintide less. F(S*) values are in kcal/mol.
Sequences S* for various α and β binding affinity values
| α | β | Sequence S* | F(S*) |
|---|---|---|---|
| 1 | 0 | KKKKMKKQPKKKKMKNNYHKKYFHNKPLKKNMKNKQK | -5484 |
| 0 | 1 | ETIWDGESDDYRPLTTTQKTFTTFWDFEESKRPEYET | -3944 |
| 1 | 1 | KTKKKKKKKKKKKEKIKKKKFTKPNKHESKKVNKHKK | -3162 |
| 2 | 1 | KKKKMKKQKKKKKMKNEYYKKYFHNKKHKKKVKQKFK | -7913 |
| 4 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKKHKKNWKNKFK | -18737 |
| 10 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKPLKKNMKNKFK | -51585 |
| 1 | 2 | ETIWDGEKDIYRWLTTTQKTFTKRFDHESSKRPEHET | -5720 |
| α | β | Sequence S* | F(S*) |
|---|---|---|---|
| 1 | 0 | KKKKMKKQPKKKKMKNNYHKKYFHNKPLKKNMKNKQK | -5484 |
| 0 | 1 | ETIWDGESDDYRPLTTTQKTFTTFWDFEESKRPEYET | -3944 |
| 1 | 1 | KTKKKKKKKKKKKEKIKKKKFTKPNKHESKKVNKHKK | -3162 |
| 2 | 1 | KKKKMKKQKKKKKMKNEYYKKYFHNKKHKKKVKQKFK | -7913 |
| 4 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKKHKKNWKNKFK | -18737 |
| 10 | 1 | KKKKMKKQPKKKKMKNNYYKKYFHNKPLKKNMKNKFK | -51585 |
| 1 | 2 | ETIWDGEKDIYRWLTTTQKTFTKRFDHESSKRPEHET | -5720 |
results in a sequence S* that has a binding affinity to amylin amyloids that is equal to its repelling affinity towards pramlintide. A results in a sequence that binds better to amylin amyloids and repels pramlintide less. F(S*) values are in kcal/mol.
Top results solving the four-residue parsimonious sequences for with α = 2 and β = 1
| Rank | 1 ––––––– 9 –––––––– 18 ––––––– 27 –––––––– 37 | ΔG | Mutations | |
|---|---|---|---|---|
| KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | −1650.0 | 167.6 | NA | |
| 1 | ––––––––––– K ––––––––––––– K–H–K ––––––– | −3013.5 | 125.8 | L12K I26K S28H T30K |
| 2 | ––––––––––– K –––––––––––– N––H–K ––––––– | −3007.5 | 178.0 | L12K A25N S28H T30K |
| 3 | ––––––––––– K ––––––– K ––––––– H–K ––––––– | −3005.4 | 185.9 | L12K S20K S28H T30K |
| 4 | –––K ––––––– K ––––––––––––––– H–K ––––––– | −2993.3 | 246.9 | T4K L12K S28H T30K |
| 5 | ––––––––––– K ––––––––––– H ––– H–K ––––––– | −2992.3 | 210.5 | L12K G24H S28H T30K |
| 6 | ––––––––––– K –––––––––––– NK ––– K ––––––– | −2965.3 | 115.0 | L12K A25N I26K T30K |
| 7 | ––––––– Q ––– K ––––––––––––––– H–K ––––––– | −2957.7 | 191.3 | A8Q L12K S28H T30K |
| 8 | ––––––––––– K ––––––– K –––– N––––K ––––––– | −2957.2 | 122.4 | L12K S20K A25N T30K |
| 9 | –––––––– K––K ––––––––––––––– H–K ––––––– | −2954.9 | 167.7 | T9K L12K S28H T30K |
| 10 | ––––––––––– K ––––––––––– H–K–––K ––––––– | −2950.1 | 129.2 | L12K G24H I26K T30K |
| Rank | 1 ––––––– 9 –––––––– 18 ––––––– 27 –––––––– 37 | ΔG | Mutations | |
|---|---|---|---|---|
| KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | −1650.0 | 167.6 | NA | |
| 1 | ––––––––––– K ––––––––––––– K–H–K ––––––– | −3013.5 | 125.8 | L12K I26K S28H T30K |
| 2 | ––––––––––– K –––––––––––– N––H–K ––––––– | −3007.5 | 178.0 | L12K A25N S28H T30K |
| 3 | ––––––––––– K ––––––– K ––––––– H–K ––––––– | −3005.4 | 185.9 | L12K S20K S28H T30K |
| 4 | –––K ––––––– K ––––––––––––––– H–K ––––––– | −2993.3 | 246.9 | T4K L12K S28H T30K |
| 5 | ––––––––––– K ––––––––––– H ––– H–K ––––––– | −2992.3 | 210.5 | L12K G24H S28H T30K |
| 6 | ––––––––––– K –––––––––––– NK ––– K ––––––– | −2965.3 | 115.0 | L12K A25N I26K T30K |
| 7 | ––––––– Q ––– K ––––––––––––––– H–K ––––––– | −2957.7 | 191.3 | A8Q L12K S28H T30K |
| 8 | ––––––––––– K ––––––– K –––– N––––K ––––––– | −2957.2 | 122.4 | L12K S20K A25N T30K |
| 9 | –––––––– K––K ––––––––––––––– H–K ––––––– | −2954.9 | 167.7 | T9K L12K S28H T30K |
| 10 | ––––––––––– K ––––––––––– H–K–––K ––––––– | −2950.1 | 129.2 | L12K G24H I26K T30K |
values are in kcal/mol. The complete list of all 4-residue parsimonious sequences is given in Supplementary Table S4.
Top results solving the four-residue parsimonious sequences for with α = 2 and β = 1
| Rank | 1 ––––––– 9 –––––––– 18 ––––––– 27 –––––––– 37 | ΔG | Mutations | |
|---|---|---|---|---|
| KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | −1650.0 | 167.6 | NA | |
| 1 | ––––––––––– K ––––––––––––– K–H–K ––––––– | −3013.5 | 125.8 | L12K I26K S28H T30K |
| 2 | ––––––––––– K –––––––––––– N––H–K ––––––– | −3007.5 | 178.0 | L12K A25N S28H T30K |
| 3 | ––––––––––– K ––––––– K ––––––– H–K ––––––– | −3005.4 | 185.9 | L12K S20K S28H T30K |
| 4 | –––K ––––––– K ––––––––––––––– H–K ––––––– | −2993.3 | 246.9 | T4K L12K S28H T30K |
| 5 | ––––––––––– K ––––––––––– H ––– H–K ––––––– | −2992.3 | 210.5 | L12K G24H S28H T30K |
| 6 | ––––––––––– K –––––––––––– NK ––– K ––––––– | −2965.3 | 115.0 | L12K A25N I26K T30K |
| 7 | ––––––– Q ––– K ––––––––––––––– H–K ––––––– | −2957.7 | 191.3 | A8Q L12K S28H T30K |
| 8 | ––––––––––– K ––––––– K –––– N––––K ––––––– | −2957.2 | 122.4 | L12K S20K A25N T30K |
| 9 | –––––––– K––K ––––––––––––––– H–K ––––––– | −2954.9 | 167.7 | T9K L12K S28H T30K |
| 10 | ––––––––––– K ––––––––––– H–K–––K ––––––– | −2950.1 | 129.2 | L12K G24H I26K T30K |
| Rank | 1 ––––––– 9 –––––––– 18 ––––––– 27 –––––––– 37 | ΔG | Mutations | |
|---|---|---|---|---|
| KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | −1650.0 | 167.6 | NA | |
| 1 | ––––––––––– K ––––––––––––– K–H–K ––––––– | −3013.5 | 125.8 | L12K I26K S28H T30K |
| 2 | ––––––––––– K –––––––––––– N––H–K ––––––– | −3007.5 | 178.0 | L12K A25N S28H T30K |
| 3 | ––––––––––– K ––––––– K ––––––– H–K ––––––– | −3005.4 | 185.9 | L12K S20K S28H T30K |
| 4 | –––K ––––––– K ––––––––––––––– H–K ––––––– | −2993.3 | 246.9 | T4K L12K S28H T30K |
| 5 | ––––––––––– K ––––––––––– H ––– H–K ––––––– | −2992.3 | 210.5 | L12K G24H S28H T30K |
| 6 | ––––––––––– K –––––––––––– NK ––– K ––––––– | −2965.3 | 115.0 | L12K A25N I26K T30K |
| 7 | ––––––– Q ––– K ––––––––––––––– H–K ––––––– | −2957.7 | 191.3 | A8Q L12K S28H T30K |
| 8 | ––––––––––– K ––––––– K –––– N––––K ––––––– | −2957.2 | 122.4 | L12K S20K A25N T30K |
| 9 | –––––––– K––K ––––––––––––––– H–K ––––––– | −2954.9 | 167.7 | T9K L12K S28H T30K |
| 10 | ––––––––––– K ––––––––––– H–K–––K ––––––– | −2950.1 | 129.2 | L12K G24H I26K T30K |
values are in kcal/mol. The complete list of all 4-residue parsimonious sequences is given in Supplementary Table S4.
4.1 Constructing optimal binding analogs
As explained in the Methods section, S* will naturally have a low sequence similarity to amylin. To construct an amylin analog that is structurally conserved and binds well to amylin amyloids and extends the length of oligomers, we’ll introduce some of the amino acid variations from S* into the analog. To introduce a parsimonious selection of variations to the amylin sequence, we solve Equations (18) and (19) for a 4-residue parsimonious sequence. The assumption we make here is that the binding affinity effect of separate positions are additive. This should hold for residues that are not adjacent. We rank all the possible mutation combinations according to their global binding affinity values and organize them in Supplementary Table S4. The top 10 results are shown in Table 2. For each of the top 10 results, we calculate their ΔG values using Equation (20) to obtain a lower-bound on their amyloidogenicity potential. All of the ΔG values are positive, indicating that the analog will not spontaneously misfold. Their stability is expected to be similar to amylin because of the similarity in ΔG values. The global binding affinity score for the top 10 results are almost twice that of an amylin amyloid, suggesting that the binding between an amyloid and one of these analogs will be twice as strong as the binding between two amylin amyloids. We used TANGO (Fernandez-Escamilla etal., 2004), FoldX (Schymkowitz etal., 2005), Zyggregator (Tartaglia etal., 2008) and PASTA (Walsh etal., 2014) to explore the native stability and amyloidogenicity potential of these 10 sequences (see Supplementary Tables S3–S6). TANGO reported that all sequences were predicted to create beta-strands, were predicted to not form any alpha-helices, and expressed an extremely high propensity for aggregation. Both FoldX and Zyggregator report that the 10 results are stable in native form and possess a high aggregation propensity. Interestingly, PASTA predicts the m9 mutant to be closest in alpha-helix propensity to the native non-mutant, yet simultaneously amyloidogenic. These comparisons with the state-of-the-art tools support the high values of ΔG in the top 10 parsimonious sequences. Several other tools such as AmyloidMutants (O’Donnell etal., 2011) and BetaScan (Bryan etal., 2009) could also be used to further compare the sequence stability of the findings.
4.2 Oligomer concentrations
The values in Table 2 give good binding affinities to the amyloids. In addition to a strong binding affinity, the analog must form long fibril structures. We built the aggregate structures for all ten results and computed their free energies (Equation 1) in Figure 3. We observe that all the ten sequences in Figure 3 produce more stable oligomers than the amylin amyloid, suggesting that they would form longer aggregates since it is energetically favorable to do so. To determine the that generates the highest potential to form long aggregates, we compute molecule concentrations (Equation 27) by the law of mass action and report the volume fractions of oligomers at various lengths in Figure 4. The sequence that ranked 9th in Table 2 expressed the most stability in Figure 3 and dominated the volume fractions in Figure 4 at oligomers of size greater than 7. This suggests that this sequence creates the longest aggregates out of the 10 results. The longer it is, the more stable it gets. In contrast, we observe that the ranked 10th in Table 2 would create many short oligomers of length 4 compared to the ten sequences. It is also worth mentioning that the most stable amyloid monomer is produced by the sequence ranked 1. In Figure 4 we see that this sequence dominates the concentrations at length 1 but does not have a strong affinity to aggregate when compared to the other sequences.
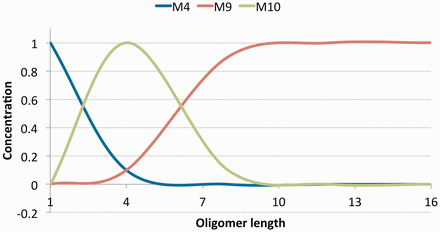
Concentration of oligomers in solution
4.3 Maintaining native form
Introducing four mutations to the sequence of amylin might destabilize it and force it to misfold. To ensure that the (T9K L12K S28H T30K) mutation preserves amylin structure and does not induce pramlintide to misfold, we ran an MD simulation of 6 ns on this amylin mutant. We report in Figure 5(a) and (b) the RMSD and RMSF graphs, respectively. The RMSD graph shows low values (<1), indicating that the mutant structure is very stable. From the RMSF graph, we see that the positions 9, 12, 28 and 30 that correspond to the mutations all have low RMSF values, indicating that the mutations do not cause structural turbulence and are not disadvantageous to amylin. This preservation of structure should preserve amylin’s functions.
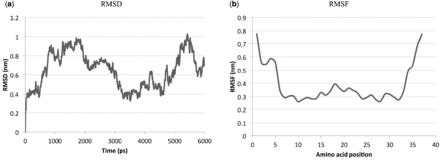
RMSD and RMSF plots for the sequence with the T9K, L12K, S28H and T30K mutations over a 6 ns simulation
5 Discussion
5.1 Binding to oligomers
It is a challenging and computationally intensive task to find a molecule A that binds well to a molecule B or subregions of B. One can perform docking simulations with millions of molecules to narrow the search of finding a good fit. In this work, we introduced the BASM method of calculating binding affinities of optimal sequences to cut down on this search process and analyze the binding preferences of B, or subregions of B. This procedure allowed us to explore engineering analogs that bind strongly to oligomers and promote their aggregation.
Applying the BASM method to engineer an amylin analog that can be administered in native, non-amyloid form with pramlintide injections resulted in discovering a mutant (T9K L12K S28H T30K) that is predicted to bind twice as strong to oligomers as amylin does to oligomers. This has been achieved by computing R-scores that bind well to oligomer surfaces and repel from pramlintide’s amyloid surface (Table 1). Using the optimal sequence constructed by these R-scores, we searched through 66 thousand combinations of parsimonious mutations to find the mutant with optimal binding and aggregation potential. This aggregation potential was both assessed by the dipolar solvent model from (Smaoui etal., 2013) and by TANGO (Fernandez-Escamilla etal., 2004). Furthermore, we validated the stability of the mutant in native form by performing an MD production of 6 ns. The results showed that the mutant was stable in native form, and hence could be a potential candidate for therapeutic injections.
The function of the mutant is intended to reduce oligomer toxicity and decrease the rate of β-cell death. The mutant is stable in native form, and forms long fibrils in amyloid form. Since its native structure is conserved and is analogous to amylin, it will carry out the normal functions of amylin and pramlintide. By design, the analog has low affinity to pramlintide and should not interfere with its function. It will act as a replacement to amylin molecules and modulate gastric emptying, inhibit glucagon, and induce satiety (Lutz et al., 1995; Rushing etal., 2000).
Once the mutant analog reaches the pancreas and makes contact with the amyloids, some of it should misfold and aggregate with the surrounding amyloids and oligomers, due to its relatively high ΔG (amyloidogenicity potential). Although long fibrils have not been observed to be toxic in the pancreas, the pharmacological effect of elongating the oligomers still needs to be further studied. If very long fibrils are observed to be unfavorable, controlling aggregation and capping the growth of oligomers at a certain length will be required and can be achieved by choosing mutants that exhibit lower oligomer concentration potentials or regulating the dosage of the injectable analogs in the body. The contribution of the BASM approach and R-scores to limit the toxicity of oligomers by favoring aggregation could be extended to explore creating potential therapeutic analog candidates for other amyloid related diseases such as Huntington and Parkinson’s.
5.2 Modeling the effect of oligomers on the insulin–glucose relationship
Upon having a meal, glucose levels increase in the bloodstream. The increase in levels signals the β-cells in the pancreas to produce insulin to instruct body organs such as the heart and muscles to absorb and metabolize glucose. This process lowers the glucose level in the blood. The amylin polypeptide is also secreted with insulin in this process at a ratio of 1:100 (Westermark etal., 2011). Together, amylin and insulin play a crucial role in maintaining glucose homeostasis.
Several mathematical models have been proposed to represent the complex process of response to glucose intake (Boutayeb and Chetouani, 2006). However, to date, no model has incorporated the production of amyloids and the effect of oligomers on β-cell death. We think that by taking into account the observed effect of oligomers on cell death the analytical solution of insulin in Equation (29) introduced by (Bolie, 1961) will be the one presented in Equation (31).
We introduce a θ and R function into Equation (31), where θ is the over burden term expressing how cells produce more insulin to compensate for their dead neighbors and R is the number of dead β-cells in the pancreas at time t. The term lowers the expected production of insulin proportional to the number of dead β-cells in R. The more β-cells die, the less insulin should be produced. However, this relationship is not linear. Upon death of β-cells, the pancreas still attempts to generate the required insulin by signalling the remaining live β-cells to secrete higher levels (Pick etal., 1998). This overburdening of cells is modelled with θ. The live cells will produce higher levels of insulin and compensate for the dead cells until a threshold d is reached, after-which the cells cannot produce any higher levels and the required level of insulin is not met. When this happens, glucose levels in Equation (30) are not returned to normal, and the pancreas is required to continue pumping insulin. More insulin produces more amylin (Equation 35) at a ratio of 1:100 (Kahn etal., 1990), and more amylin creates higher levels of amyloids (Equation 34). Again, this relationship is not linear. When amylin concentration is lower than a threshold h, minimal amyloids form, and when the threshold is exceeded, amyloid production is initiated in high concentration. Equation (33) models the percentage of amyloids in the pancreas system that form into oligomers of average length kO. This percentage is positive or 0, depending on the efficacy of any introduced oligomer inhibiting molecules. Because the death rate of β-cells due to oligomers resembles a very slow exponential-like curve (Weir and Bonner-Weir, 2004), we use the compound interest formula to model this phenomenon in Equation (32).
The key to suppress the deterioration of β-cells is to minimize the number of oligomers infecting cells, modelled in Equation (33). Producing M molecules that bind well to the oligomers achieves this result. In return, this affects Equation (32), lowering the number of dead cells and stabilizing Equation (31) to produce the right quantity of insulin. If the opposite occurs, Equation (33) produces more oligomers, increasing the dead cells in Equation (32), and in turn lowering the production of insulin in Equation (31). Glucose levels will remain high in Equation (30), prompting more insulin to be produced, triggering an increase in amylin and amyloid production in Equations (34) and (35). This in turn creates more oligomers, and the cycle repeats.
Although the behavior of the subsystems of the theoretical model presented in Equations (30)–(37) are known biologically, we do not provide a simulation at different time points because there are way too many parameters in the system of integral equations that we do not have experimental values for. The aim of the model is to provide the theoretical framework that incorporates amyloids, oligomers and toxicity of the beta cells, and to show at a high level the general effect of lowering oligomer concentration on the fitness of β-cells. It will become useful to extract data from simulating the model once many of the parameters are discovered experimentally.
5.3 Online tool
The BASM method and R-scores procedure have been packaged into a tool called Single-rEsidue Mutational based Binding Affinity (SEMBA) to probe the mutation landscape of amyloid proteins. The tool can be downloaded as a stand-alone software from http://amyloids.cs.mcgill.ca
Funding
M.R.S. was supported by a fellowship from the Canadian Institutes of Health Research System Biology Training program at McGill University and a grant from the Fonds de recherche Nature et technologies Quebec. J.W. was supported by a Discovery grant from the Natural Science and Engineering Research Council of Canada.
Conflict of Interest: none declared.
References
Author notes
Associate Editor: Anna Tramontano


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}