





Abstract
Prior distributions can have a strong effect on the results of Bayesian analyses. However, no general consensus exists for how priors should be set in all circumstances. Branch-length priors are of particular interest for phylogenetics, because they affect many parameters and biologically relevant inferences have been shown to be sensitive to the chosen prior distribution. Here, we explore the use of outside information to set informed branch-length priors and compare inferences from these informed analyses to those using default settings. For both the commonly used exponential and the newly proposed compound Dirichlet prior distributions, the incorporation of relevant outside information improves inferences for data sets that have produced problematic branch- and tree-length estimates under default settings. We suggest that informed priors are worthy of further exploration for phylogenetics.
Setting priors is a necessary step in any Bayesian analysis, but the best approach to choice of priors has been a contentious issue in phylogenetics as it has in many other fields of statistical inference (Efron 2013). Approaches vary widely and different priors have been shown to influence the results of Bayesian phylogenetic analyses (e.g., Brown et al. 2010; Marshall 2010; Rannala et al. 2012; Nowak et al. 2013). Currently, prior choice in phylogenetics, including most of the default distributions used in popular software packages (MrBayes, Ronquist et al. 2011; BEAST, Drummond et al. 2012), is often justified by appeals to objectivity or robustness. Here, we consider a phylogenetic problem (branch-length inference) where the default priors can give rise to misleading conclusions and suggest an informed approach that leverages the information in previously published data to set priors.
Some researchers favor the use of reference priors, which are selected by formal rules (Kass and Wasserman 1996) to represent a lack of information about the distribution of a particular parameter and are motivated by the desire to minimally influence the posterior distribution, so that the data determine support for each hypothesis (Gelman et al. 2004). In practice, finding reference priors is often difficult. While they are noninformative for the parameter of interest, they may induce a highly informative, implicit prior on some other parameter in ways that can be difficult to predict. Whether or not reference priors can be set legitimately has been a source of controversy in statistics for centuries (Efron 2013).
Another approach to setting priors, empirical Bayes, parameterizes the prior using the focal data (Efron 2013). Empirical Bayes has the advantage of guaranteeing that parameter values near the peak of the likelihood surface will have high prior weight, but has been criticized as non-Bayesian because the priors are dependent on the focal data and lead to artificially reduced estimates of uncertainty. Consequently, many Bayesians oppose its use (e.g., Rannala et al. 2012).
If no explicit statistical framework is embraced for choice of priors, software developers often set default values for a prior that work well for data sets on which they have been tested. This approach is practical and may be effective for most analyses, but it comes with no guarantees. Critically, users may not be aware when the default is unreasonable for their data, which can lead to the publication of erroneous conclusions. As a result, some phylogenetic software packages force users to manually set priors for important parameters (e.g., BEAST; Drummond et al. 2012).
In contrast to the above methods, informed priors make use of similar data sets or expert opinion to set priors for the focal data and have been used occasionally in phylogenetics (e.g., Liang et al. 2009; Nowak et al. 2013). Informed priors are, confusingly, not necessarily informative priors—the former are set using outside information, whereas the latter means that the prior strongly influences the posterior. Informed priors have the advantage of incorporating current knowledge directly into the model, which typically leads to more precise credible intervals around parameter estimates than the reference prior approach.
The informed prior approach is not without its pitfalls. While phylogenetic databases from which relevant information could be extracted do exist (e.g., TreeBase, http://www.treebase.org; DRYAD, http://www.datadryad.org/), they are difficult to query, which makes finding relevant data sets difficult. Alternatively, for priors that should be set to reflect expert opinion, quantifying that opinion into a range of relevant parameter values can be difficult, particularly for phylogenetic analyses where wide-ranging factors such as divergence time, sampling structure, and choice of loci have a strong influence on conclusions.
B ranch-Length Priors in Bayesian Phylogenetics
To illustrate the importance of effective prior choice in phylogenetics, we consider the problem of branch-length inference. Branch-length estimates are often of direct interest in phylogenetic analyses, since they describe the amount of evolutionary change between nodes. These estimates can affect a wide variety of biological inferences, including ancestral state reconstruction, species delimitation, divergence time estimation, and rates of lineage diversification. In addition, branch-length priors can influence inferred support for different tree topologies (Yang and Rannala 2005). Therefore, researchers should be concerned by the observation that Bayesian estimates of total tree length can be an order of magnitude longer than maximum-likelihood estimates (MLEs; Brown et al. 2010; Marshall 2010) when alignments contain many closely related sequences and default branch-length priors from standard software packages (e.g., MrBayes; Ronquist et al. 2011) are used. If priors are intended to be uninformative, MLEs should be reasonable draws from the posterior.
Brown et al. (2010) investigated the issue of inflated tree lengths in a range of simulated and empirical data sets, examining whether the problem may be due to mixing problems for the Markov chain caused by 1) multiple local peaks or 2) large, nearly flat regions in the posterior. Alternatively, 3) an overly informative branch-length prior may bias the posterior toward unreasonably large branch lengths. They found support for possibilities (2) and (3). Further analysis by Rannala et al. (2012) suggested that a poorly specified branch-length prior could be the root cause of all three possibilities.
By default, MrBayes uses independent and identically distributed exponential priors with a rate () of 10 for branch lengths (Ronquist and Huelsenbeck 2003; Ronquist et al. 2011). For an unrooted tree with taxa, total tree length is the convolution of exponential densities, which has the gamma distribution with shape and rate (Fig. 1). Since the mean of the gamma distribution is /, expected tree length is (23)/. Hence, the branch-length prior sets an implicit prior on tree length that scales with the number of taxa and can be highly sensitive to changes in (see figure 7 from Brown et al. 2010 and figure 2.1 from Wang and Yang 2014). For phylogenies with recent divergences and many taxa, this sensitivity often leads to default prior tree-length distributions that effectively exclude the MLE (Fig. 1).
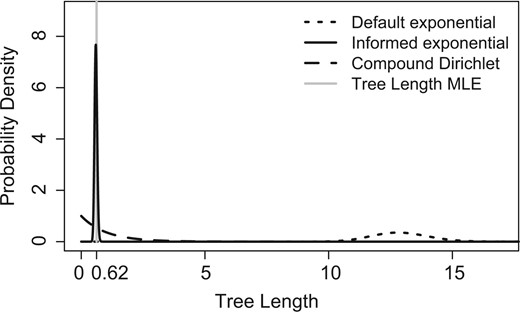
Comparison of tree-length prior distributions for default and informed exponential branch-length priors, as well as the default compound Dirichlet tree-length prior, to the MLE for Acris tree length. The informed exponential was parameterized using TreeBASE data set S1800 obtained from EmpPrior.
Several approaches have been proposed to mitigate the influence of overly informative default branch-length priors. Brown et al. (2010) recommended an empirical Bayes approach, which recovered the tree-length MLE in credible intervals, but suffers from an artificial reduction in uncertainty. Other approaches have aimed to set less informative default priors on branch- or tree-length, including the double-exponential branch-length prior (Yang and Rannala 2005) and the compound Dirichlet tree-length prior (Fig. 1; Rannala et al. 2012; Zhang et al. 2012), both of which are implemented (but not used by default) in MrBayes 3.2.2 (Ronquist et al. 2011). The double-exponential branch-length prior specifies separate exponential priors on internal and external branches, whereas the compound Dirichlet prior sets a (inverse) gamma-distributed prior on total tree length and a Dirichlet prior on the partitioning of tree length among branches that allows for different means on internal and external branches. The compound gamma Dirichlet tree-length prior (as implemented by Zhang et al. [2012] in MrBayes) has four parameters, all of which default to values of 1: tree-length shape ( and rate (, Dirichlet concentration (), and mean internal:external branch-length ratio (). The double-exponential branch-length prior marginally shrinks tree-length estimates but is still highly sensitive to prior settings, whereas the compound Dirichlet prior successfully recovers ML total tree-length estimates in 95% highest posterior density intervals (hereafter simply HPDs) for several (but not all) problematic data sets across a wide range of tree-length prior means (Zhang et al. 2012).
Here, we propose an extension to the default prior approaches mentioned above that involves setting informed priors based on outside data. We compare posteriors from informed priors to those from default priors across a range of data sets that have produced problematic branch-length estimates (Brown et al. 2010; Zhang et al. 2012). We investigate both exponential branch-length priors and recently proposed compound Dirichlet tree-length priors (Rannala et al. 2012). As we show, informed priors can greatly improve upon default settings and produce HPDs that include MLEs for both the exponential and compound Dirichlet distributions. The inclusion of MLEs in HPDs is a useful criterion for prior performance when researchers are not intending to specify strong prior beliefs, although we note (as have others, e.g., Zhang et al. 2012) that MLEs may be less reliable than Bayesian estimates in some circumstances. When the data contain little information (perhaps due to very few or very many substitutions) and when the model is poorly specified, MLEs may be extreme, have very wide confidence intervals, or be biased. In such cases, the prior can provide a mitigating influence and keep estimates within reasonable bounds. Given the wide availability of outside phylogenetic data, we recommend increased use of informed priors in Bayesian phylogenetic analyses.
Obtaining Informed Prior Estimates from Phylogenetic Databases
In order to obtain informed prior estimates, we must acquire data sets that are relevant to our focal data. We used three criteria to establish the relevance of a particular external data set. Relative to the focal data, external data sets should 1) include orthologous regions of DNA, 2) have a similar number of taxa, and 3) sample taxa with a similar degree of divergence. Properties (1) and (2) are relatively easy to test, but it may be difficult to confirm (3) without estimating parameters from the focal data. To circumvent this issue, we used taxonomic classification as a rough proxy for divergence, using external data sets only if they had similar taxonomic depth, number of species, and number of samples per species as the focal data set. Taxonomic classification is not necessarily strongly correlated with divergence, but should provide a rough approximation of tree depth and eliminate divergences that are much deeper (e.g., phylum) or much shallower (e.g., population) than a typical genus. Ideally, external data sets should not contain any data also included in the focal data set. If they do, this procedure risks the circularity inherent to empirical Bayesian approaches.
We developed a program, EmpPrior (available at http://code.google.com/p/empprior/; Andersen et al. 2015), to parameterize branch- and tree-length distributions by searching TreeBase for data sets similar to the focal data. EmpPrior comprises a Java program, EmpPrior-search, which queries TreeBase and returns matching data sets, and an R script, EmpPrior-fit, which finds ML parameter estimates for exponential branch-length and compound gamma Dirichlet tree-length distributions. We perform ML tree search on each data set returned from TreeBase, infer parameter estimates for branch- and tree-length distributions from resulting topologies, and inform focal priors based on these estimates. While the use of ML methods in a Bayesian analysis may seem unusual, it is justified in this case as a fast approximation that integrates easily into current software and often outperforms default settings (see below). To facilitate comparison of inferences across studies, we used exemplar empirical data sets previously analyzed by Brown et al. (2010) and Zhang et al. (2012) to test alternative branch- and tree-length prior distributions. The four data sets represent a diverse set of animal clades with shallow divergences and many sequences: Acris (cricket frog, 66 sequences, Gamble et al. 2008), Corbicula (freshwater clam, 93 sequences, Hedtke et al. 2008), Crinia signifera (common eastern froglet, 92 sequences, Symula et al. 2008), and Sceloporus (spiny lizard, 123 sequences, Leaché and Mulcahy 2007).
For each focal data set, we used EmpPrior to search TreeBase for data sets with regions of DNA orthologous to the focal data and a similar number of sequences (differing by no more than 20 from the number of sequences in the focal data set). Retrieved data sets with similar taxon sampling were used in downstream analyses. For each retrieved data set, we estimated ML trees using Garli v2.01 (Zwickl 2006). We used the Nelder–Mead method (Nelder and Mead 1965) as implemented in the “optim” function in R 3.0 (R Core Team 2013) and the R package “bbmle” (Bolker 2014) to perform ML estimation of parameters for exponential branch-length and compound Dirichlet tree-length distributions. In addition, we compared eight submodels of the compound Dirichlet model where , , and were either fixed at default values or estimated from the data. We set in all cases to represent a diffuse prior on tree length and because joint estimation of and was unreliable.
We used MrBayes v3.2.2 (Ronquist et al. 2011; includes compound Dirichlet tree-length prior) for Bayesian phylogenetic analyses. Each focal data set was analyzed using default and informed exponential priors, as well as default and informed compound Dirichlet priors, and a GTR+I model of sequence evolution. Each analysis was run for at least 4,400,000 generations with two independent runs and four chains per run, 25% burn-in, and samples recorded every 1000 generations. Convergence was assessed using the average standard deviation of split frequencies (ASDSFs < 0.01; Lakner et al. 2008) and trace plots in Tracer v1.5 (Rambaut and Drummond 2009). We used R package ggplot2 (Wickham 2009) to create violin plots of posterior density and R package coda (Plummer et al. 2006) to calculate HPDs. MLEs of tree length from each focal data set were estimated using 25 replicate tree searches in Garli v2.01 (Zwickl 2006).
Empirical Performance of Default and Informed Priors
The Acris data set contains 66 cyt b sequences from two species. The EmpPrior search returned 24 nexus files, 10 of which included intra-generic sampling. From the intra-generic data sets, three data sets included 10 or more sequences for multiple species and were used for further analysis. The default exponential branch-length prior did not recover the MLE of tree length in the resulting HPD (Table 1), whereas two of the three informed exponential priors did and the HPD of the third was much closer to the MLE (Fig. 2a and Table 1). Both informed and default compound Dirichlet priors recovered the MLE (Fig. 2b and Table 1), although the informed compound Dirichlet priors often resulted in HPDs with medians substantially closer to the MLE than the HPD using default values (Fig. 2b and Table 1). We focus here primarily on the effects of using compound Dirichlet priors with informed values of (Fig. 2), because these models tended to produce narrow HPDs that still included ML tree lengths from the focal data sets. Full results for compound Dirichlet priors with informed values of , or a combination of and are given in Table 1.
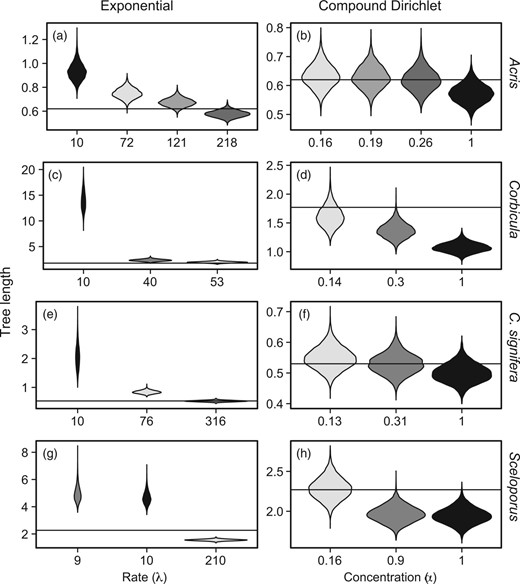
Violin plots of tree length (TL) posterior density for a–b) Acris, c–d) Corbicula, e–f) Crinia signifera, and g–h) Sceloporus. Plots resulting from default priors have a black fill whereas those from informed priors have a gray fill. Shades of gray within a row indicate informed priors derived from the same outside data. ML TL estimates are indicated with a solid horizontal line. Results from exponential priors are in the left column (a, c, e, and g) and results from compound Dirichlet priors are in the right column (b, d, f, and h).
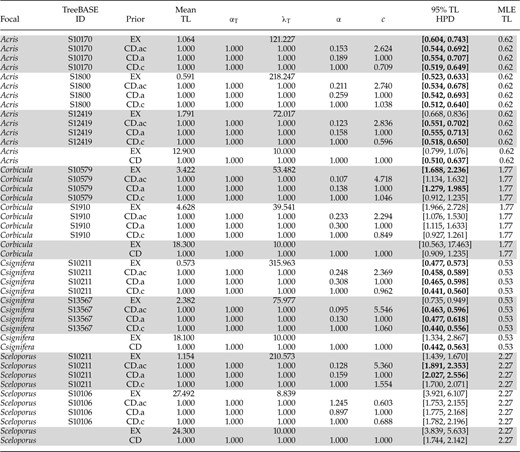
Data sets returned by EmpPrior-search, informed parameters for exponential and compound Dirichlet branch-length priors, and corresponding 95% HPDs for focal data set tree lengths
 |
|
Notes: “Focal” refers to the corresponding empirical data set and “TreeBASE ID” refers to the data set returned by EmpPrior-search. “EX” means exponential and “CD” means compound Dirichlet with suffixes a (concentration) and c (branch-length ratio) used to indicate which parameters of the compound Dirichlet are being fitted. and are the parameters for the gamma tree-length distribution, refers to concentration, is the branch-length ratio, and MLE TL is the MLE for total tree length (TL). “Mean TL” is the mean of the tree-length prior applied to the focal data set. Rows without a TreeBASE ID correspond to default MrBayes settings. Tree-length HPDs in bold contain the tree-length MLE.
Data sets returned by EmpPrior-search, informed parameters for exponential and compound Dirichlet branch-length priors, and corresponding 95% HPDs for focal data set tree lengths
|
|
Notes: “Focal” refers to the corresponding empirical data set and “TreeBASE ID” refers to the data set returned by EmpPrior-search. “EX” means exponential and “CD” means compound Dirichlet with suffixes a (concentration) and c (branch-length ratio) used to indicate which parameters of the compound Dirichlet are being fitted. and are the parameters for the gamma tree-length distribution, refers to concentration, is the branch-length ratio, and MLE TL is the MLE for total tree length (TL). “Mean TL” is the mean of the tree-length prior applied to the focal data set. Rows without a TreeBASE ID correspond to default MrBayes settings. Tree-length HPDs in bold contain the tree-length MLE.
The Corbicula data set comprised 93 COI sequences from over eight species (there is currently no consensus on the exact number of species sampled). The EmpPrior search returned 44 nexus files, 18 of them with intra-generic sampling. Two data sets included multiple sequences for at least seven species. The default exponential branch-length prior did not recover the tree-length MLE (Table 1), whereas one of the two informed exponential priors did and both informed HPDs were vastly closer to the MLE than the default (Fig. 2c and Table 1). The default compound Dirichlet prior did not recover the MLE, while one of the informed compound Dirichlet priors did, and both informed HPDs were closer to the MLE than the default (Fig. 2d and Table 1).
The C. signifera data set contained 92 concatenated 12S and 16S sequences from a single species. EmpPrior found 12 nexus files that contained both genes, none of which were entirely intraspecific. However, intraspecific sequences were extracted from two data sets to create data sets with 60 and 48 intraspecific sequences. The default exponential branch-length prior did not recover the tree-length MLE (Table 1), whereas one of the two informed exponential priors did and the other was much closer to the MLE than the default (Fig. 2e and Table 1). Both the informed and the default compound Dirichlet priors recovered the MLE, with the informed HPD medians closer to the MLE than the default (Fig. 2f and Table 1).
The Sceloporus data set contained 123 sequences for two genes, ND4 and 12S, with multiple sequences for each of 14 species. We were unable to find any relevant data sets that contained both genes, so we performed separate EmpPrior searches. EmpPrior found a single data set for each gene, which yielded over 20-fold differences in exponential rate () estimates using EmpPrior-fit ( and , respectively). The data set returned by the ND4 search contained a few Sceloporus sequences that overlapped with the focal data set, which is not ideal, but the taxonomic scale of the data sets was different and the majority of sequences were non-overlapping. We applied each of the informed priors to the concatenated alignment containing both genes. Neither informed nor default exponential branch-length priors recovered the MLE, but one informed estimate greatly reduced both the median and the width of the HPD (Fig. 2g and Table 1). One of the informed compound Dirichlet estimates yielded an HPD that included the MLE, whereas the other informed and the default HPDs fell below the MLE (Fig. 2h and Table 1).
For the exponential branch-length prior, the informed approach dramatically improved tree-length HPDs relative to MrBayes defaults in three of the analyzed data sets. Roughly half (2/3 for Acris, 1/2 for Corbicula, 1/2 for C. signifera, and 0/2 for Sceloporus) of analyses using informed priors recovered the tree-length MLE in HPDs, whereas tree-length estimates from the default exponential prior were sometimes an order of magnitude too large. The improvement in tree-length estimates using informed priors suggests that taxonomic classification was a reasonable proxy for sequence divergence for our focal data sets, but more direct measures of divergence might prove more reliable.
For the compound Dirichlet tree-length prior, the majority of informed prior HPDs included tree-length MLEs (3/3 for Acris, 1/2 for Corbicula, 2/2 for C. signifera, and 1/2 for Sceloporus). While HPDs resulting from the default compound Dirichlet tree-length prior also often included tree-length MLEs (2/4 data sets), informed HPD medians were often substantially closer to tree-length MLEs. Overall, using informed estimates of improved meaningfully upon default settings for Corbicula and Sceloporus, where the default did poorly, and improved modestly upon default settings for Acris and C. signifera.
Estimates of Dirichlet concentration () were generally less than 1, which changes the shape of the distribution from flat (at ) to U-shaped, with more prior weight on both large and small relative branch lengths. This distribution seems reasonable for the exemplar data sets, which have many short intraspecific branches and a few longer interspecific ones. Estimates of were often greater than 1, meaning that internal branches were, on average, longer than external ones. This result contradicts the expectation of Rannala et al. (2012) that the mean internal:external branch-length ratio should generally be less than 1. However, such estimates make sense for our focal data sets, which include many small, intraspecific terminal branches. This result illustrates the utility of using empirical estimates to set informed priors, since a rule of thumb that makes sense for deep divergences may not be reasonable for shallow divergences.
When inferring trees for use with informed priors, algorithmic approaches such as neighbor joining (NJ) and less thorough ML implementations such as phangorn (Schliep 2011) or FastTree 2 (Price et al. 2010) consistently yielded shorter trees than more thorough ML approaches such as Garli (Zwickl 2006). These shorter trees resulted in larger exponential rate estimates and smaller posterior mean tree lengths that often failed to include the MLE in HPDs. This downward bias may be due to difficulties inherent in estimating many short branches in retrieved data sets, since the NJ method in ape (Paradis et al. 2004) often returned negative branch lengths. However, setting a positive minimum bound on NJ branch-length estimates did not fix the issue. We recommend using a thorough ML search to inform rate estimates for the exponential branch-length prior.
The Sceloporus data set presents an interesting challenge for the application of informed priors, since a branch-length prior informed from outside data relevant to just one gene could be problematic for a data set of multiple genes with widely differing rates. EmpPrior found few data sets that contained a sufficient number of 12S and ND4 sequences, which may have led to the use of non-relevant data sets in our analysis. In particular, the external ND4 data set is suspect because it lacks intraspecific sampling, which may have contributed to its low exponential rate estimate. While both outside data sets relevant to Sceloporus produced informed compound Dirichlet priors that improved upon default settings, setting separate informed branch-length priors for separate genes or scaling priors based on gene may be appropriate in cases where genes have large differences in rate.
Relevance of Informed Priors in Bayesian Phylogenetics
Informed branch-length priors obtained with EmpPrior often dramatically improve upon default prior settings and at worst seem to cause no harm, at least for these examples. Exploring additional applications of informed priors in phylogenetics may thus prove fruitful. In particular, using previous data sets to inform priors on rate variation may improve estimates of divergence times.
While we have shown that informed branch-length priors often improve tree-length estimates for data sets with shallow divergence and many taxa, we have not tested the method on phylogenies with deeper divergences. Since the inflated tree problem seems specific to data sets with many short branches, improvements to branch-length estimates using the informed approach may be greatest for these data sets. The effectiveness of informed branch-length priors in other circumstances remains an open question.
The use of informed parameter estimates does not circumvent the need to carefully consider which prior distribution is most appropriate. Priors with informed parameterizations may be problematic if the distribution itself is overly informative or poorly specified. Prior sensitivity analyses should be helpful in identifying these circumstances and we recommend their use. If conclusions vary considerably with different, reasonable prior parameterizations, researchers may wish to interpret results with caution and/or opt for a prior distribution with less influence on the posterior (when available).
The degree of improvement provided by outside information is likely to be dependent on the prior distribution used and the parameters that are informed. Due to poor default behavior, analyses employing exponential prior distributions generally showed marked and consistent improvement with informed priors in our tests, though they remain quite sensitive to the choice of . Compound Dirichlet priors also showed frequent improvement when informing values of (Fig. 2), , or and jointly (Table 1). Informed values of alone, however, did not lead to as much improvement as informed values of . Future work should identify those parameters that are most likely to benefit from outside information.
As publicly available phylogenetic databases become larger and simpler to query, obtaining outside information to parameterize priors will become easier and these estimates may also become more accurate. EmpPrior provides a simple and effective way to query an existing database (TreeBase) for similar data sets and obtain informed branch-length prior parameter values. We have shown that informed priors can deflate excessively long Bayesian trees and are worth exploring in a wide variety of phylogenetic analyses.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.1gm13.
F unding
This work was supported partially by the National Institute of Justice [award 2011-DN-BX-K534 to M.L. Metzker and J.M.B.], as well as startup funds from the Louisiana State University College of Science and Department of Biological Sciences.
Acknowledgments
Portions of this research were conducted with high-performance computational resources provided by HPC@LSU (http://www.hpc.lsu.edu). V. Doyle, T. Heath, E.J. McTavish, M. Hellberg, B. Elderd, F. Anderson, P. Foster, C. Zhang, and C. Cox provided helpful comments that improved this manuscript.
References
Author notes
Associate Editor: Peter Foster

{kind=link}
{kind=link}