





Abstract
Background: Cholinergic dysfunction is increasingly assumed to be involved in the pathophysiology of schizophrenia. Short-latency afferent inhibition (SAI) is a transcranial magnetic stimulation (TMS) paradigm that has been shown to assay central cholinergic activity from the motor cortex (M1). Recently, we established a method to index SAI from the dorsolateral prefrontal cortex (DLPFC), an area implicated in the pathophysiology of schizophrenia. We investigated SAI in M1 and DLPFC in schizophrenia. We hypothesized that modulation of N100 on TMS-evoked potentials (TEPs) from the DLPFC would be attenuated in patients with schizophrenia compared to healthy controls. Methods: SAI was examined in 12 patients, whose age was matched to controls, using TMS combined with electroencephalography (EEG). SAI was recorded with TMS applied to left M1 (M1-SAI) and DLPFC (DLPFC-SAI). For group comparison, we used the SAI data of healthy participants in our previous study. Results: In patients, N100 TEP was significantly attenuated with DLPFC-SAI, whereas P180 TEP was significantly increased with M1-SAI. Between patients and controls, there were significant differences in modulation of P180 TEP by M1-SAI (t22 = −2.748, P = .012; patients > controls) and N100 TEP by DLPFC-SAI (t22 = 5.456, P < .0001; patients < controls). Further, modulation of N100 TEP by DLPFC-SAI significantly correlated with executive function (r = −.740, P = .006, N = 12). Conclusion: Our findings suggest that DLPFC-SAI but not M1-SAI were reduced in patients with schizophrenia and this was linked to deficits in cognition. This may reflect prefrontal cholinergic deficits and represent a biomarker for cholinergic and executive dysfunction in patients with schizophrenia.
Introduction
Schizophrenia is a serious psychiatric disorder and a leading cause of disability worldwide.1 The pathophysiology of the disorder remains unclear although a variety of neurotransmitters are implicated. Among these, the dysregulation of the cholinergic system is increasingly implicated in the pathophysiology of schizophrenia and it may also contribute to cognitive impairment in patients with schizophrenia.2 Decreased expression and density of muscarinic acetylcholinergic receptors (mAChRs) has been demonstrated in the frontal, temporal and occipital cortex, caudate, putamen and thalamus of medicated and unmedicated patients with schizophrenia.3,4 Moreover, nicotine administration and smoking have been shown to improve cognitive performance among patients with schizophrenia,5–7 findings which highlight the role of cholinergic dysfunction in cognitive dysfunction which is commonly observed in this disorder. As the cholinergic system is said to facilitate a range of cognitive processes, it may be that a marker of cholinergic activity such as short-latency afferent inhibition (SAI), is able to help understand or even predict the course of the disease.
Currently, there are no reliable biomarkers that can noninvasively assess cortical cholinergic activity in neuroanatomical structures that mediate cognitive function in patients with schizophrenia. SAI is a transcranial magnetic stimulation (TMS) paradigm that indexes cortical cholinergic activity.8 In this paradigm, the motor-evoked potential (MEP) elicited by a TMS test pulse is inhibited when preceded by median nerve stimulation (MNS) at an inter-stimulus interval (ISI) of approximately 20 ms.8,9 SAI is cholinergically modulated and mediated by gamma-amino butyric acid (GABA)A receptor, implicating a direct role for central cholinergic and GABAergic activities in SAI.10–13 Previous studies showed that the muscarinic acetylcholine receptor antagonist, scopolamine decreases SAI,10 while a positive allosteric modulator of the GABAA receptor, lorazepam, reduces SAI12 among healthy participants. Previous research has demonstrated a reversal of SAI impairment in Alzheimer’s disease and mild cognitive impairment with the administration of acetylcholinesterase inhibitors such as rivastigmine11,13 and donepezil,14 which result in enhancement of cholinergic transmission, adding further support to the notion that the SAI is associated with cholinergically modulated cognitive function.15,16
More recently, TMS has been combined with electroencephalography (EEG) to allow for the direct evaluation of the inhibition of the TMS-evoked potential (TEP) instead of MEP measured in peripheral muscles. Previous TMS-EEG studies have shown that SAI administered to the motor cortex (M1) induces a significant modulation of the negative deflection of the TEP at a latency of 100 ms (N100 TEP) in the midline central area.17,18 Importantly, a significant correlation between attenuation of amplitude on N100 TEP and attenuation of MEP has been demonstrated,17,19 validating the neurophysiological link between SAI indexed with MEP and N100 TEP. Although SAI is conventionally measured from M1, ascending sensory input is relayed via the ventral nuclei of the thalamus to sensory and prefrontal regions.20 The somatosensory evoked potential (SSEP) evoked by MNS is a negative deflection at a latency of ~20 ms (N20) over somatosensory areas,21 followed shortly thereafter by a negative potential over the contralateral frontal region with a 24 ms latency (N24).22 SAI for M1 is typically delivered relative to N20. Our group recently extended this technique to the dorsolateral prefrontal cortex (DLPFC) and demonstrated that SAI could be elicited with TMS applied to DLPFC and was maximal at ISI N20 + 4 ms, at a latency similar to the N24 component. Using combined TMS-EEG, SAI administered to the DLPFC in healthy participants resulted in a significant increase in the amplitude of N100 component in the left frontal area in our prior study.19 Although there is no direct evidence of the relationship between DLPFC-SAI and cholinergic or GABAergic activity, the indirect evidence comes from the neural circuits involved in the SAI paradigm include the thalamic ventral anterior nucleus which has predominant projections to the DLPFC, the ventral lateral nucleus projects to premotor and motor areas, and the ventral posterior nucleus to somatosensory cortical regions. M1-SAI is thought to involve contributions from S1 through an indirect cortico-cortical projection,23 as well as a direct thalamocortical projection to M1. Similarly, the DLPFC-SAI is also speculated to involve the contributions from S1 and a thalamocortical projection to the DLPFC. Indeed, the ISI of M1-SAI has been shown to occur as early as N20 + 0 ms,8,24 while we have previously demonstrated that the optimal ISI of DLPFC-SAI is approximately N20 + 4 ms.19 Therefore, it is likely that SAI in M1 and DLPFC are mediated by both cortico-cortical and thalamocortical projections. Furthermore, our assertion is strongly supported by the shared electrophysiological signature observed in both regions, suggesting that they are mediated by similar mechanisms.19 Further, given that the DLPFC is enriched in AChRs and mediates cognitive function,25,26 indexing SAI from this region may improve our understanding of cholinergic dysfunction in the pathophysiology of schizophrenia.
The first objective of this study, therefore, was to index SAI from M1 and DLPFC in patients with schizophrenia compared to healthy control participants. Given that smoking has been shown to alter both cholinergic and GABAergic indices of TMS27 as well as cognitive performance,6 SAI was indexed in biochemically verified nonsmokers with schizophrenia compared to healthy controls. It was hypothesized that N100 TEP component would be significantly attenuated among patients with schizophrenia with greater impairment in the DLPFC compared to healthy controls. The second objective of this study was to examine the relationship between cognitive function and the modulation of N100 TEP indexed from both M1 and the DLPFC. We hypothesized that cognitive performance would be correlated with modulation of N100 based on previous studies that have demonstrated a relationship between M1-SAI and cognition.15,16
Methods
Participants
Twelve right-handed medicated patients with schizophrenia (8 male, mean age: 41 ± 10 y) were examined in the present study. Further, we used the SAI data of 12 healthy control participants (6 male, mean age: 39 ± 12 y).19 For patients with schizophrenia, we matched the age to healthy controls within ± 2 years difference for each pair. SAI was administered to M1 and DLPFC according to our previously published protocol.19 Participants of both groups were eligible to participate in this study if they met the following criteria: (1) between ages 18 and 59; (2) no history of neurological disorders including seizure, syncope or stroke; (3) no current alcohol or other drug abuse/dependence; (4) nonsmoker for at least 1 year prior to study which was verified by expired breath carbon monoxide (CO) determination (≤4 ppm of CO level as measured by the breath CO monitor28–30). For the healthy control participants, they also met the following criteria: (5) no history of neuropsychiatric disorders; (6) normal cognitive function; or (7) no prescription medications including anti-cholinergic medication and nicotine replacement therapy. For patients with schizophrenia, they met the criteria as follows: (8) clinically stable determined by the Positive and Negative Symptom Scale (PANSS)31 score of ≤70; (9) no specific anticholinergic drugs or benzodiazepines more than lorazepam equivalent dose of 2 mg; (10) had not been hospitalized in the past 3 months, and were on a stable dose of antipsychotic medications for at least 1 month. Chlorpromazine (CPZ) equivalents were calculated according to the technique described by Woods and colleagues.32 Demographic and clinical data shown in table 1. All participants were screened with the Structured Clinical Interview for DSM-IV Axis I Disorders prior to study participation. The experiment was conducted in accordance with the Declaration of Helsinki and was reviewed and approved by the Ethics Committee of the Centre for Addiction and Mental Health.
Demographic and Clinical Data for SCZ Participants and HC
| Mean (± 1 SD) | SCZ (n = 12) | HC (n = 12) |
|---|---|---|
| Age | 41 ± 10 | 39 ± 12 |
| Male: Female | 8: 4 | 6: 6 |
| Years of education | 15 ± 3 | 15 ± 2 |
| PANSS | ||
| Positive | 11.3 ± 3.0 | — |
| Negative | 11.7 ± 3.4 | — |
| General | 23.6 ± 2.8 | — |
| Total | 50.1 ± 6.2 | — |
| CPZ equivalent atypical (mg/d) | 330 ± 287 | — |
| Mean (± 1 SD) | SCZ (n = 12) | HC (n = 12) |
|---|---|---|
| Age | 41 ± 10 | 39 ± 12 |
| Male: Female | 8: 4 | 6: 6 |
| Years of education | 15 ± 3 | 15 ± 2 |
| PANSS | ||
| Positive | 11.3 ± 3.0 | — |
| Negative | 11.7 ± 3.4 | — |
| General | 23.6 ± 2.8 | — |
| Total | 50.1 ± 6.2 | — |
| CPZ equivalent atypical (mg/d) | 330 ± 287 | — |
Note: PANSS, positive and negative symptom scale, CPZ, chlorpromazine; SCZ, schizophrenia; HC, healthy controls.
Demographic and Clinical Data for SCZ Participants and HC
| Mean (± 1 SD) | SCZ (n = 12) | HC (n = 12) |
|---|---|---|
| Age | 41 ± 10 | 39 ± 12 |
| Male: Female | 8: 4 | 6: 6 |
| Years of education | 15 ± 3 | 15 ± 2 |
| PANSS | ||
| Positive | 11.3 ± 3.0 | — |
| Negative | 11.7 ± 3.4 | — |
| General | 23.6 ± 2.8 | — |
| Total | 50.1 ± 6.2 | — |
| CPZ equivalent atypical (mg/d) | 330 ± 287 | — |
| Mean (± 1 SD) | SCZ (n = 12) | HC (n = 12) |
|---|---|---|
| Age | 41 ± 10 | 39 ± 12 |
| Male: Female | 8: 4 | 6: 6 |
| Years of education | 15 ± 3 | 15 ± 2 |
| PANSS | ||
| Positive | 11.3 ± 3.0 | — |
| Negative | 11.7 ± 3.4 | — |
| General | 23.6 ± 2.8 | — |
| Total | 50.1 ± 6.2 | — |
| CPZ equivalent atypical (mg/d) | 330 ± 287 | — |
Note: PANSS, positive and negative symptom scale, CPZ, chlorpromazine; SCZ, schizophrenia; HC, healthy controls.
TMS Procedure and EMG Measure
Monophasic TMS pulses were administered to M1 on the left hemisphere using a 70 mm figure-of-eight coil, and 2 Magstim 200 stimulators (Magstim Company Ltd) connected via a Bistim module. Participants were seated in a chair and instructed to relax and keep their eyes open. Surface electromyography (EMG) was recorded from Ag/AgCl electrodes placed over the belly of the first dorsal interosseous muscle in the right hand. First, the optimal spot for the right first dorsal interosseous muscle to evoke the largest MEP over the M1 was confirmed, and then the individual intensity to induce 1 mV peak-to-peak MEP amplitude of the same muscle was determined. SAI was administered, while EMG and EEG were collected.
SAI Procedure
The SAI protocol was administered as previously described.8,19 The median nerve was stimulated at the right wrist using a standard bar electrode, placing a cathode proximally using a constant current stimulator (Digitimer model DS7A, Digitimer Ltd). The conditioning MNS intensity (pulse width 200 μs) was adjusted to 3 times the sensory threshold individually. SAI was delivered at the MNS-TMS ISIs relative to the SSEP at N20.18,24 Based on our previous study,19 SAI was administered at optimal ISIs of N20 + 2 ms for M1-SAI and N20 + 4 ms at the F5 electrode site33 for DLPFC-SAI. Each condition (ie, M1-SAI: TMS alone vs SAI condition [ISI N20 + 2]; DLPFC-SAI: TMS alone vs SAI condition [ISI N20 + 4]) consisted of 100 trials and was randomized. The inter-trial interval was 5 seconds and inter-block interval was about 5–10 minutes. Each block (ie, M1 stimulation or DLPFC stimulation), consisted of 200 trials in total, was also randomized across participants.
EEG Recording and Preprocessing
A Synamps 2/RT 64-channel EEG system and a Quik-Cap Electrode Placement System (Compumedics Neuroscan) were used to record TEPs. All electrodes were referenced to an electrode placed on the vertex. EEG signals were recorded at DC at 20 kHz sampling rate and with a low-pass filter of 200 Hz. EEG data were processed offline using MATLAB (MathWorks). All data were down-sampled to 1000 Hz for analyses.
SAI-TEP Data Analysis
SAI-TEP data were analyzed based on the method in our previous study.19 The continuous EEG data were epoched from −1000 ms to 2000 ms relative to the TMS pulse, and then processed EEG data were baseline-corrected with respect to the pre-stimulus interval −500 ms to −110 ms. Subsequently, the EEG data were re-segmented from 10 ms to 2000 ms post-TMS in order to avoid TMS artifacts. EEG data were visually inspected to eliminate trials and channels that were highly contaminated with artifacts such as muscle activity. After artifact removal, more than 80% of trials and 95% of channels survived. The mean (±SD) percentages of trials for the M1-SAI and DLPFC-SAI that went into the analyses were 91.4% ± 2.1% and 91.1% ± 1.8%. For the removed electrode channels, a standard interpolation method was applied.34 Then, independent component analysis (ICA) was applied to remove eye-related artifacts such as blinks and eye movements, the remaining muscle artifacts, and the TMS-related decay artifacts. In each subject and each condition, the number of ICA components that were removed from the original ICA components was not greater than 20% (ie, less than 12 components) based on previously published method19 in order to ensure that the ICA cleaning method among subjects, conditions, and groups remained consistent. Following the ICA cleaning, the Butterworth, zero-phase shift 1–55 Hz band pass filter (24 dB/Oct) for high-frequency noise and 58–62 Hz notch filter for power line noise were applied. Finally, the processed EEG data was re-referenced to the average for further analyses. SAI-TEPs were obtained after subtracting the SSEP traces from the TEP traces in the SAI condition at the last step individually, according to previous studies evaluating SAI with TMS-EEG.17,19 In the present SAI study, TEP amplitudes were measured by identifying the peaks (ie, P30, P60, and P180) and troughs (ie, N45 and N100) individually in order to obtain more accurate values for each TEP component.19
TMS-EEG SAI (ie, modulation of TEPs) was calculated as follows:
SAI = [amplitude of TEP induced by SAI condition] / [amplitude of TEP induced by single pulse test TMS]
Cognitive Battery
The following cognitive battery was administered among patients with schizophrenia: the Wechsler Test of Adult Reading (WTAR); the Letter-Number Span Test (LNST), and the Trail Making Test (TMT) Part A & B, the Hopkins Verbal Learning Test (HVLT) on the same day that SAI was measured. For the analysis of the TMT, the B/A ratio was used because this index is less affected by age and education of participants35 and assumed to be more sensitive to psychomotor factors.36
Statistical Analysis
SPSS version 19.0 was used for statistical analysis. To examine the effect of SAI on each of the TEP components, 3-way ANOVAs were performed separately for M1-SAI and DLPFC-SAI, with diagnosis (schizophrenia vs controls) as a between-subjects factor and TMS conditions (2 levels: TMS alone vs SAI condition) and TEP components (5 levels: P30, N45, P60, N100, and P180) as within-subjects factors, since both site of experiments were conducted independently. An alpha level of .05 (2-tailed) was applied for detecting significance. Post hoc independent t tests were applied for significant findings in the ANOVA with Bonferroni correction. As per previous SAI methodology,19 we clustered the specific electrodes as regions of interest for analyses of the left M1 (FC1, FC3, FC5, C1, C3, C5, CP1, CP3, CP5) and DLPFC (Fp1, AF3, AF7, F1, F3, F5, F7, FC1, FC3, FC7) regions. Moreover, to explore the relationship between the modulation of N100 by SAI paradigm and cognitive function, Pearson’s correlation analyses were performed and Bonferroni corrected for multiple tests.
Results
The Effect of SAI on MEP Between Patients With Schizophrenia and Healthy Controls
Mean intensity (±SD) to induce 1 mV peak-to-peak MEP amplitude in the healthy controls and patients with schizophrenia were 80.3% ± 11.5% and 81.8% ± 7.9%, respectively. Further, the SAI-MEP of healthy controls and patients with schizophrenia were significantly attenuated by 41.2% ± 8.0% (P < .01) and 31.7% ± 9.4% (P < .01), respectively. However, there was no significant difference in the SAI-MEP between healthy controls and patients with schizophrenia.
The Effect of SAI on TEP Among Patients With Schizophrenia vs Healthy Controls
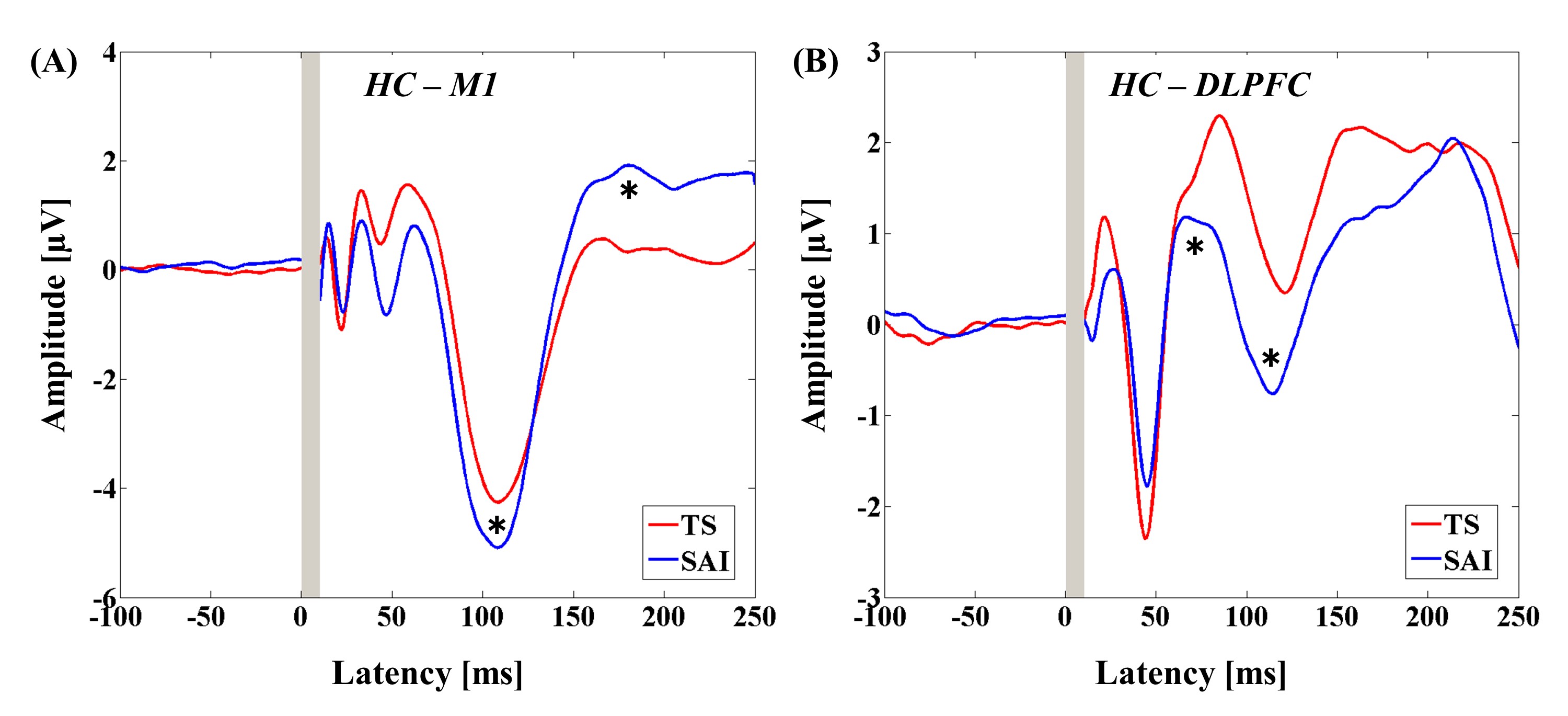
The 3-way ANOVA for M1-SAI showed a significant main effect of TEP component (F4,88 = 51.121, P < .0001) and significant interactions in TMS condition × diagnosis (F1,22 = 10.674, P = .004), TMS condition × TEP component (F4,88 = 58.353, P < .0001), and TMS condition × TEP component × diagnosis (F4,88 = 4.550, P = .002). Post hoc independent t tests indicated a significant difference in modulation of P180 TEP (t22 = −2.748, P = .012; patients > controls; see figure 1C) between the 2 groups. In contrast, the 3-way ANOVA for DLPFC-SAI demonstrated significant main effects of TMS condition (F1,22 = 14.064, P = .001) and TEP component (F4,88 = 71.609, P < .0001), and significant interactions in TEP component × diagnosis (F4,88 = 5.822, P < .0001), TMS condition × TEP component (F4,88 = 69.338, P < .0001), and TMS condition × TEP component × diagnosis (F4,88 = 9.976, P < .0001). Post hoc independent t tests showed a significant difference in modulation of N100 TEP (t22 = 5.456, P < .0001; patients < controls; figure 2C) between the 2 groups. For patients with schizophrenia, TEP traces and topographical plots for M1-SAI and DLPFC-SAI conditions are shown in figures 1 and 2.
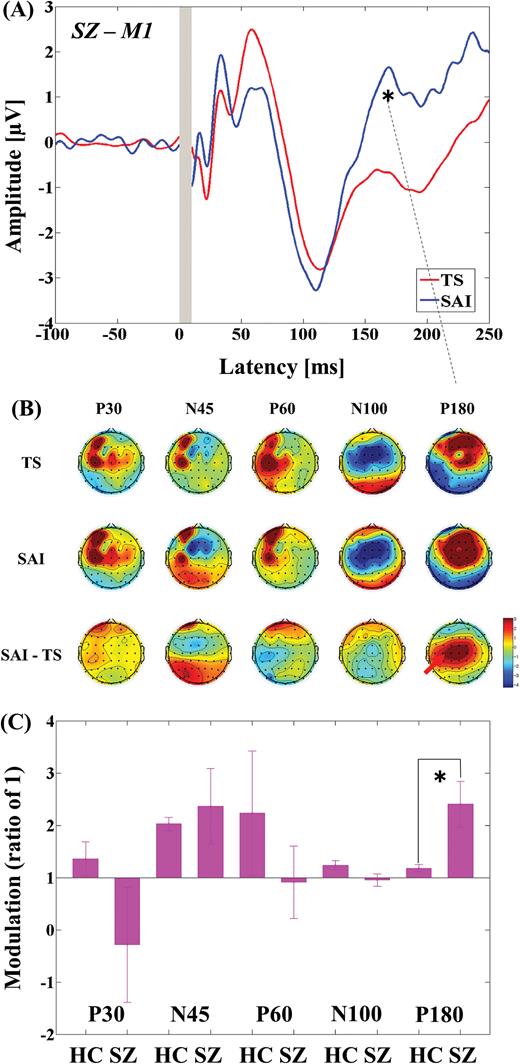
Modulation of cortical activity in the SCZ group by SAI with TMS delivered to M1 (M1-SAI). (A) Modulation of TEP traces: TEP traces following subtraction of individual somatosensory evoked potential (SSEP) components. The graphs depict TEP traces averaged across all subjects for unconditioned TMS (TS: TMS alone) and conditioned TMS (median nerve stimulation + TMS at an ISI N20 + 2 ms) at left M1. TMS was delivered at a time equivalent to 0 ms. The ANOVA and post hoc analysis indicated a significant modulation of P180 TEP component over M1 (*P < .05). (B) Topographical plots: TMS-EEG topographical plots for all TEP components in M1-SAI paradigm. The TEP topoplots are shown for (1) TS delivered alone, (2) conditioned TS at an ISI of N20 + 2 ms, and (3) the difference between TS alone and conditioned TS (SAI) at ISI N20 + 2 ms. Each vertical column depicts the TEP topoplots for P30, N45, P60, N100, and P180 component from left to right, respectively. The areas showing significant TEP changes are shown with red arrows. (C) Cross-sectional comparison of the modulation of the TEP amplitudes at M1 between the HC and SCZ group: The ANOVA and post hoc analysis demonstrated a significant difference in the modulation of N100 TEP between the HC and SCZ group (HC > SCZ; *P < .05). SCZ, schizophrenia; SAI, short-latency afferent inhibition; TMS, transcranial magnetic stimulation; M1, motor cortex; TEP, TMS-evoked potentials; ISI, inter-stimulus interval; EEG, electroencephalography; TS, test stimulus.
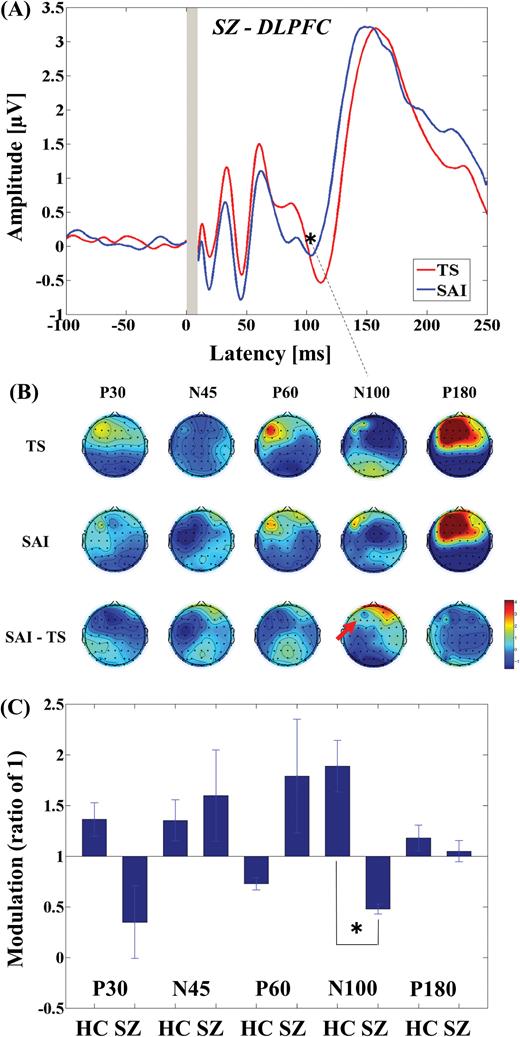
Modulation of cortical activity in the SCZ group by SAI with TMS delivered to DLPFC (DLPFC-SAI). (A) Modulation of TEP traces: TEP traces following subtraction of individual somatosensory evoked potential (SSEP) components. The graphs depict TEP traces averaged across all patients for unconditioned TMS (TS: TMS alone) and conditioned TMS (median nerve stimulation + TMS at an ISI N20 + 4 ms) at the left DLPFC. TMS was delivered at a time equivalent to 0 ms. The ANOVA and post hoc analysis indicated a significant modulation of N100 TEP component over the left DLPFC (*P < .05). (B) Topographical plots: TMS-EEG topographical plots for all TEP components in DLPFC-SAI paradigm. The TEP topoplots are shown for (1) TS delivered alone, (2) conditioned TS at an ISI of N20 + 4 ms, and (3) the difference between TS alone and conditioned TS (SAI) at ISI N20 + 4 ms. Each vertical column depicts the TEP topoplots for P30, N45, P60, N100, and P180 component from left to right, respectively. The area showing a significant TEP change is shown with red arrow. (C) Cross-sectional comparison of the modulation of the TEP amplitudes (left DLPFC) by DLPFC-SAI between the HC and SCZ group: The ANOVA and post hoc analysis demonstrated a significant difference in the modulation of N100 TEP component between the HC and SCZ group (HC > SCZ; *P < .05). DLPFC, dorsolateral prefrontal cortex; SCZ, schizophrenia; SAI, short-latency afferent inhibition; TMS, transcranial magnetic stimulation; TEP, TMS-evoked potentials; ISI, inter-stimulus interval; EEG, electroencephalography; TS, test stimulus.
In addition, a sub-analysis comparing SAI between clozapine-treated patients (4 patients; mean age: 39 ± 9.8 y, 1 female) and non-clozapine-treated patients (8 patients; mean age: 42 ± 10.5 y, 3 females) showed no significant difference in M1-SAI or DLPFC-SAI.
Correlation Analyses Between Modulation of N100 by SAI Paradigm and Cognitive Functions in Patients With Schizophrenia
Cognitive outcomes are shown in Table 3. In the M1-SAI paradigm, there was no correlation between TEP modulations and any cognitive measure. There was a significant negative correlation between modulation of N100 TEP and executive function as measured with the ratio of TMT part B to part A (r = −.740, P = .006, N = 12) in the DLPFC-SAI paradigm. This correlation remained significant after Bonferroni correction for a number of cognitive measures. Furthermore, when we controlled for age as a covariate in this correlation, it remained significant (r = −0.739, P = .009, df = 9). The scatter plots for this correlation are shown in figure 3. In addition, when we excluded the outlier who had a relatively high TMT B/A ratio (>2 SD), the significant correlation between the two is still observed (r = −.695, P = .009, N = 11; a partial correlation controlled for age: r = −.694, P = .013, df = 8). No relationships were uncovered with the other cognitive measures.
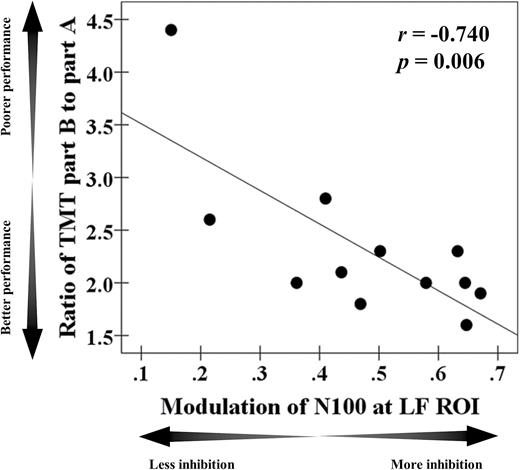
Cognitive correlation. There was a significant correlation in the SCZ group between ratio of the Trail Making Test part B to part A and modulation of N100 TEP (r = −.740, P = .006, N = 12) at the left DLPFC. Note: SCZ represents patients with schizophrenia, while HC represents healthy controls. The data of the HC group from our prior SAI study19 was used here. DLPFC, dorsolateral prefrontal cortex; SAI, short-latency afferent inhibition; TEP, TMS-evoked potentials.
Discussion
In the present study, the SAI TMS-EEG paradigm was used to index cholinergic function in cognitive (DLPFC) and motor (M1) domains in patients with schizophrenia for the first time, compared to healthy controls. Our previous study showed that SAI is primarily characterized by a robust increase of N100 amplitude in both DLPFC and M1 (supplementary figure).19 On this basis, the present findings suggest a significant reduction in SAI in patients with schizophrenia that was more dominant to DLPFC, while there was no significant modulation of N100 in M1. Further, a negative correlation was uncovered between the modulation of N100 TEP by DLPFC-SAI and executive functioning as assessed by the TMT, suggesting that reduced SAI is associated with reduced cognitive function.
Group comparison analyses between patients and controls indicated a significant difference in modulation of N100 TEP by DLPFC-SAI, specifically showing that SAI induced an attenuated modulation of N100 among patients compared to controls. Thus, the significant change of N100 in the opposite direction in patients relative to healthy controls may reflect lower cholinergic tone in the DLPFC in schizophrenia. Importantly, we did not observe a significant modulation of N100 TEP with the M1-SAI paradigm in patients group. This may be because the prefrontal cortex is more closely associated with the pathophysiology of schizophrenia compared to the M1.37–40 Recent evidence suggests that patients with schizophrenia show reductions in cholinergic receptor signaling such as in the α7 nicotinic receptor41 and cholinergic muscarinic 1 and muscarinic 4 receptors in the cortex.42–44 Furthermore, it is known that GABAAergic dysfunction is also strongly implicated in the pathophysiology of schizophrenia37–40 and indeed GABAA receptor mediated inhibition is partially associated with the mechanism of SAI.10–13 Therefore, our finding of a selective impairment of SAI in the DLPFC in patients with schizophrenia may also be related to the pathophysiology of GABAAergic function that has been demonstrated in previous studies.37–40
Given the selective attenuated modulation of N100 by the DLPFC-SAI paradigm in patients with schizophrenia, we examined if this was related to their performance on the cognitive measures. As such, a significant correlation between a measure of executive function and the modulation of N100 by DLPFC-SAI was observed, indicating N100 component attenuation by DLPFC-SAI (ie, less inhibition in SAI paradigm) was associated with reduction in performance on the executive function as measured by TMT (ie, the ratio of TMT part B to part A). In other words, in the present study, patients with schizophrenia showed impaired SAI specifically in the DLPFC that was related to poorer executive function. Thus, in patients with schizophrenia, the DLPFC-SAI may represent a possible biomarker of executive function associated with cholinergic activity in the prefrontal cortex.
Although SAI is primarily and most robustly characterized by modulation of N100 amplitude when measured using TMS-EEG, we also explored the modulation of other components. For M1 but not DLPFC-SAI, patients showed greater modulation of P180 compared to healthy controls.
The origin of P180 is not fully understood, however, it is thought to represent a late phase of the excitatory component.45 In M1 a subset of excitatory circuits are facilitated in the presence of SAI23 and SAI is followed by a period of increased excitability,24 however it is unclear whether one or both phenomena could contribute to this excitatory component in the schizophrenia group. While the etiology of P180 remains to be fully elucidated, the data indicate clearly that SAI is altered in schizophrenia. This difference probably is primarily confined to the DLPFC as neither N100, nor EMG measures of SAI were modulated in M1.
This study may provide further evidence for the dysregulation of the cholinergic system in the pathophysiology of schizophrenia. For example, decreased upregulations of high affinity nAChRs46 and decreased density of these receptors have been reported across cortical brain regions including the prefrontal cortex.46,47 Functional polymorphisms of low-affinity nAChRs that play an important role in sensory gating and physiology48 are also reported in schizophrenia. Such dysregulation of nAChRs may underlie the high prevalence of smoking rates in patients with schizophrenia that is estimated at 3-fold increase compared to the general public.49 In contrast, several lines of evidence suggest that alterations in central M1/M4 mAChRs expression are also involved in the underlying pathophysiology of schizophrenia.3,50,51 As such, dysfunction in nAChR and mAChR signaling may contribute to the pathophysiology of schizophrenia, whereas activation of nAChR and mAChR mediated system may improve the cholinergic dysfunction that is associated with cognitive impairment in schizophrenia.52
Taken together with the current study findings that patients with schizophrenia demonstrated a significant impairment in the modulation of N100 TEP by the DLPFC-SAI that was also correlated with executive function, DLPFC-SAI may be a potential biomarker for cholinergic dysfunction in schizophrenia that may represent a possible target for the treatment of cognitive deficits in this disorder.
There are some limitations in this study. First, we did not use an MRI-guided neuronavigation system to locate the DLPFC for each participant. Accuracy could have potentially been enhanced and variability reduced by using MRI neuronavigation methods rather than using the F5 electrode as an estimated DLPFC loci. However, the F5 method has been shown to be reliable in previous studies.33,53,54 Indeed, the TEPs evoked in the present study are consistent with those previously reported using either method, and further both methods produce highly comparable results in TMS-EEG studies in different groups.34,45 Furthermore, in the present study, to minimize this technical limitation, we applied an electrode clustering method for TEP data analysis to capture the representative characteristics for each ROI. Second, concomitant medications with muscarinic anticholinergic effect may have a potential confounding effect on SAI in the schizophrenia group. Further, 4 patients were taking clozapine in this study and this might have a confounding effect on the SAI. However, a sub-analysis comparing SAI between clozapine-treated patients and non-clozapine-treated patients showed no significant difference in M1 or the DLPFC. Thus future studies that apply the SAI TMS-EEG in medication naïve patients with schizophrenia could potentially clarify the more specific effects of SAI on TEPs that may be obscured by antipsychotic use. We summarized the list of concomitant antipsychotic medications in table 2. Third, since the cognitive tests were not administered in the control group, future studies should include the cognitive assessment of healthy controls. It would be more informative and useful, if we can confirm whether there is also a relationship between executive function and the DLPFC-SAI in healthy controls. Finally, 5 of 12 patients were ex-smokers while all other subjects were never smokers. While the effect of smoking history on cholinergic function is unknown, it is possible that a history of smoking may influence SAI. This represents a limitation to the study and future studies should carefully investigate the effect of smoking history on SAI measures.
List Concomitant Meds (APDs) in the SCZ Group
| Concomitant Antipsychotics |
|---|
| Clozapine (4) |
| Olanzapine (3) |
| Risperidone (2) |
| Paliperidone (1) |
| Lurasidone (1) |
| Aripiprazole (1) |
| Quetiapine (1) |
| Concomitant Antipsychotics |
|---|
| Clozapine (4) |
| Olanzapine (3) |
| Risperidone (2) |
| Paliperidone (1) |
| Lurasidone (1) |
| Aripiprazole (1) |
| Quetiapine (1) |
Note: SCZ, schizophrenia. The number in parentheses indicates the number of patients who had taken that antipsychotics during the study.
List Concomitant Meds (APDs) in the SCZ Group
| Concomitant Antipsychotics |
|---|
| Clozapine (4) |
| Olanzapine (3) |
| Risperidone (2) |
| Paliperidone (1) |
| Lurasidone (1) |
| Aripiprazole (1) |
| Quetiapine (1) |
| Concomitant Antipsychotics |
|---|
| Clozapine (4) |
| Olanzapine (3) |
| Risperidone (2) |
| Paliperidone (1) |
| Lurasidone (1) |
| Aripiprazole (1) |
| Quetiapine (1) |
Note: SCZ, schizophrenia. The number in parentheses indicates the number of patients who had taken that antipsychotics during the study.
Mean Performance Scores ± 1 SD on Cognitive Battery Among Patients With SCZ
| Mean (± 1 SD) | SCZ (n = 12) |
|---|---|
| WTAR FS IQ | 106 ± 10 |
| Letter-Number-Span | 12 ± 3 |
| HVLT | |
| Retention (%) | 82.7 ± 16.1 |
| Discrimination index | 10.5 ± 1.5 |
| TMT | |
| Trail A (s) | 30.6 ± 9.7 |
| Trail B (s) | 71.7 ± 36.5 |
| Trail B − Trail A (s) | 41.1 ± 30.1 |
| Trail B/Trail A | 2.3 ± 0.7 |
| (Trail B − Trail A)/Trail A | 1.3 ± 0.7 |
| Mean (± 1 SD) | SCZ (n = 12) |
|---|---|
| WTAR FS IQ | 106 ± 10 |
| Letter-Number-Span | 12 ± 3 |
| HVLT | |
| Retention (%) | 82.7 ± 16.1 |
| Discrimination index | 10.5 ± 1.5 |
| TMT | |
| Trail A (s) | 30.6 ± 9.7 |
| Trail B (s) | 71.7 ± 36.5 |
| Trail B − Trail A (s) | 41.1 ± 30.1 |
| Trail B/Trail A | 2.3 ± 0.7 |
| (Trail B − Trail A)/Trail A | 1.3 ± 0.7 |
Note: WTAR, Wechsler Test of Adult Reading; HVLT, Hopkins Verbal Learning Test; TMT, Trail Making Test; SCZ, Schizophrenia.
Mean Performance Scores ± 1 SD on Cognitive Battery Among Patients With SCZ
| Mean (± 1 SD) | SCZ (n = 12) |
|---|---|
| WTAR FS IQ | 106 ± 10 |
| Letter-Number-Span | 12 ± 3 |
| HVLT | |
| Retention (%) | 82.7 ± 16.1 |
| Discrimination index | 10.5 ± 1.5 |
| TMT | |
| Trail A (s) | 30.6 ± 9.7 |
| Trail B (s) | 71.7 ± 36.5 |
| Trail B − Trail A (s) | 41.1 ± 30.1 |
| Trail B/Trail A | 2.3 ± 0.7 |
| (Trail B − Trail A)/Trail A | 1.3 ± 0.7 |
| Mean (± 1 SD) | SCZ (n = 12) |
|---|---|
| WTAR FS IQ | 106 ± 10 |
| Letter-Number-Span | 12 ± 3 |
| HVLT | |
| Retention (%) | 82.7 ± 16.1 |
| Discrimination index | 10.5 ± 1.5 |
| TMT | |
| Trail A (s) | 30.6 ± 9.7 |
| Trail B (s) | 71.7 ± 36.5 |
| Trail B − Trail A (s) | 41.1 ± 30.1 |
| Trail B/Trail A | 2.3 ± 0.7 |
| (Trail B − Trail A)/Trail A | 1.3 ± 0.7 |
Note: WTAR, Wechsler Test of Adult Reading; HVLT, Hopkins Verbal Learning Test; TMT, Trail Making Test; SCZ, Schizophrenia.
In conclusion, as modulation of N100 amplitude is the most robust primary marker for SAI,18,19 the present study suggests that SAI is reduced in patients with schizophrenia in DLPFC but not M1. SAI is primarily considered a measure of cholinergic function. These data are suggestive of cholinergic dysfunction in schizophrenia, especially in light of the association with reduced executive function, and may provide a neurophysiological biomarker of schizophrenia and the associated cognitive impairment. Cognitive impairment in patients with schizophrenia has emerged as a major mediator of overall functional outcome.55,56 Cholinergic muscarinic receptors are mainly involved in the mechanism of SAI paradigm and are strongly related to learning and memory.57 The results of the present study warrant verification in medication naïve patients with schizophrenia or in prodromal phases of the illness.
Supplementary Material
Supplementary material is available at Schizophrenia Bulletin online.
Funding
This research was supported by the Temerty Centre for Therapeutic Brain Intervention, Campbell Family Research Institute through the CAMH Foundation, and Canada Foundation for Innovation.
Acknowledgments
None of the authors declare any conflict of interest. Y.N. receives postdoctoral fellowship from the Centre for Addiction and Mental Health (CAMH) Foundation. M.S.B. receives research support from the Brain and Behavior Research Foundation (formerly NARSAD) Young Investigator Grant and Schizophrenia Junior Faculty Grant from the CAMH Foundation. R.F.H.C. was supported by a Canadian Institutes of Health Research (CIHR)—Dystonia Medical Research Foundation Fellowship award. T.K.R. received research support from Brain Canada, Brain and Behavior Research Foundation, Canada Foundation for Innovation, the CIHR, Ontario Ministry of Health and Long-Term Care, Ontario Ministry of Research and Innovation, the US National Institute of Health (NIH), and the W. Garfield Weston Foundation. F.F. receives funding from NARSAD, Slaight Family Centre for Youth in Transition at the CAMH, Natural Sciences and Engineering Research Council of Canada (NSERC), the Ontario Brain Institute (OBI). T.P.G. reports that he has funding support from CIHR, NIDA/NIH, Canada Foundation for Innovation and Pfizer Global Research, and is a consultant to Novartis. Z.J.D. has received research support from the Ontario Mental Health (OMH) Foundation, the CIHR, the Brain and Behaviour Research Foundation (Formerly NARSAD), and the Temerty family and Grant family through the CAMH Foundation and the Campbell Research Institute. Z.J.D. received research and equipment in kind support for an investigator-initiated study through Brainsway Inc., and a travel allowance through Merck. Z.J.D has also received speaker funding through Sepracor Inc., and AstraZeneca, served on advisory boards for Hoffmann-La Roche Limited and Merck, and received speaker support from Eli Lilly. D.M.B. has received research support from the CIHR, NIH, Brain Canada and the Temerty Family through the CAMH Foundation and the Campbell Research Institute. He receives research support and in-kind equipment support for an investigator-initiated study from Brainsway Ltd and he is the site principal investigator for 3 sponsor-initiated studies for Brainsway Ltd. He receives in-kind equipment support from Magventure for an investigator-initiated study. He receives medication supplies for an investigator-initiated trial from Indivior. D.M.B., Y.N., M.S.B., R.F.H.C., R.C., and Z.J.D. were involved in conception and design of the study; Y.N. and M.S.B. performed experiments; Y.N. and R.Z. analyzed data; Y.N., M.S.B. R.F.H.C., R.C., Z.J.D., and D.M.B. interpreted results of experiments; Y.N. prepared figures; Y.N., M.S.B., and R.F.H.C. drafted the manuscript; M.S.B., Y.N., R.F.H.C., T.K.R, F.F., R.C., T.P.G, Z.J.D and D.M.B. edited and revised the manuscript; all authors approved final version of manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}