




Abstract
Pyruvate decarboxylase from Zymomonas mobilis (PDC) and benzoylformate decarboxylase from Pseudomonas putida (BFD) are thiamine diphosphate-dependent enzymes that decarboxylate 2-keto acids. Although they share a common homotetrameric structure they have relatively low sequence similarity and different substrate spectra. PDC prefers short aliphatic substrates whereas BFD favours aromatic 2-keto acids. These preferences are also reflected in their carboligation reactions. PDC catalyses the conversion of benzaldehyde and acetaldehyde to (R)-phenylacetylcarbinol and predominantly (S)-acetoin, whereas (R)-benzoin and mainly (S)-2-hydroxypropiophenone are the products of BFD catalysis. Comparison of the X-ray structures of both enzymes identified two residues in each that were likely to be involved in determining substrate specificity. Site-directed mutagenesis was used to interchange these residues in both BFD and PDC. The substrate range and kinetic parameters for the decarboxylation reaction were studied for each variant. The most successful variants, PDCI472A and BFDA460I, catalysed the decarboxylation of benzoylformate and pyruvate, respectively, although both variants now preferred the long-chain aliphatic substrates, 2-ketopentanoic and 2-ketohexanoic acid. With respect to the carboligase activity, PDCI472A proved to be a real chimera between PDC and BFD whereas BFDA460I/F464I provided the most interesting result with an almost complete reversal of the stereochemistry of its 2-hydroxypropiophenone product.
Received March 25, 2005; revised and accepted May 9, 2005 Edited by Bauke Dijkstra
Introduction
Pyruvate decarboxylase (E.C. 4.1.1.1) from Zymomonas mobilis (PDC) and benzoylformate decarboxylase (E.C. 4.1.1.7) from Pseudomonas putida (BFD) are structurally similar homotetrameric enzymes whose main reaction is the non-oxidative decarboxylation of 2-keto acids. With the exception of a few amino acid residues at the active site, which are involved in the binding of the cofactor thiamine diphosphate (ThDP) and Mg2+, their sequence similarity is low (Hasson, 1998; McLeish, 2004). In line with their distinct metabolic functions, PDC prefers pyruvate as a substrate and, to a lesser extent, other aliphatic 2-keto acids (Pohl, 1998), whereas BFD prefers benzoylformate and derivatives thereof (Reynolds, 1988; Weiss, 1988). Both enzymes are also able to catalyse the carboligation reaction of two aldehyde molecules resulting in chiral 2-hydroxy ketones (Figure 1), which are useful building blocks for organic chemistry. It is still unclear whether the carboligase activity is of physiological importance or just a relic of an ancestral protein. The carboligase activity of pyruvate decarboxylase from Saccharomyces cerevisiae (ScPDC) has been used for decades in the synthesis of (R)-phenylacetylcarbinol [(R)-PAC], the chiral step in the commercial production of ephedrine (Hildebrandt, 1934). During this biotransformation, which is still a matter of investigation (Rosche et al., 2002, 2003, 2005), ScPDC has two catalytic functions: the decarboxylation of pyruvate and the ligation of the resulting ThDP-bound acetaldehyde to benzaldehyde (Figure 1). Unlike the yeast enzyme, Z.mobilis PDC does not require the decarboxylation of pyruvate for carboligase activity. Instead, it is able to use acetaldehyde, which proved to be advantageous for the development of an improved biocatalytic process (Goetz et al., 2001). Apart from the mixed carboligation product, (R)-PAC, PDC also catalyses the formation of acetoin by carboligation of two acetaldehyde molecules (Figure 2).
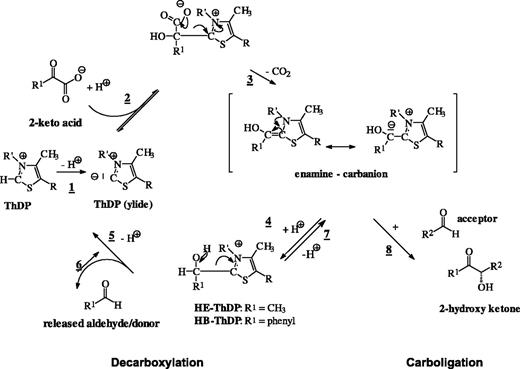
Reaction mechanism of BFD and PDC. The reaction cycle is started by deprotonation of ThDP bound to the enzyme (1). The ylide is able to bind a 2-keto acid (pyruvate, R1 = methyl; benzoylformate, R1 = phenyl) via nucleophilic addition to the carbonyl group (2). The resulting adduct decarboxylates to an enamine carbanion (3). Protonation of this species yields hydroxyethyl-(HE)-ThDP or hydroxybenzyl-(HB)-ThDP (4), which subsequently eliminates the corresponding aldehyde (PDC, acetaldehyde; BFD, benzaldehyde) upon protonation and regenerated the ThDP ylide (5). Decarboxylation and carboligation are assumed to have a common intermediate, the enamine carbanion. In addition to the decarboxylation route (2 and 3) this species can be generated by direct addition of a (donor) aldehyde to the ThDP ylide (route 6 and 7). Addition of a further (acceptor) aldehyde leads to the formation of 2-hydroxy ketones.
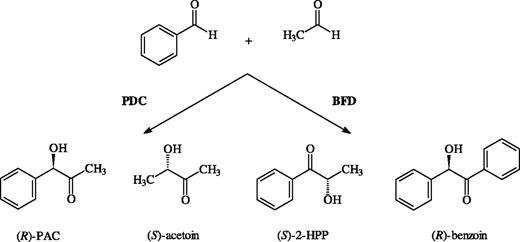
Carboligation products of acetaldehyde and benzaldehyde via PDC and BFD catalysis, respectively.
The carboligase reaction catalysed by BFD was first described by Ward and co-workers (Wilcocks et al., 1992). As a result of an inverse binding of substrates in BFD relative to PDC, the carboligase product is (S)-2-hydroxypropiophenone (2-HPP), which is isomeric and enantiomeric with (R)-PAC (Figure 2). We have reported earlier that BFD, like PDC, does not require the decarboxylation of benzoylformate to catalyse the ligation reaction, as it is able to catalyse the conversion of two molecules of benzaldehyde to (R)-benzoin (Figure 2) (Iding et al., 2000). Further, a mechanistic model was developed to account for the interesting and hitherto unknown dependence of the enantioselectivity of (S)-2-HPP formation on the benzaldehyde concentration (Iding et al., 2000). The carboligase activity of BFD, using acetaldehyde, benzaldehyde and derivatives thereof, has also been studied intensively. With the exception of ortho-substituted derivatives, BFD accepts a variety of benzaldehydes as both donors (first substrate) and acceptors (second substrate) (Figure 1) (Demir et al., 1999; Dünnwald et al., 2000a,b; Dünkelmann et al., 2002). Unfortunately, biotransformations in aqueous media are hampered by the low solubility of the aromatic benzaldehydes. To overcome this problem, stabilization of BFD towards DMSO, a water-miscible organic solvent used to enhance the solubility of aromatic aldehydes, was achieved by directed evolution (Lingen et al., 2002). The same method was applied to evolve BFD variants which accept ortho-substituted benzaldehyde derivatives as donors (Lingen et al., 2003). Interestingly, these mutations are not directly involved in the active site.
During the last decade a number of mutations in the active site of PDC (Bruhn et al., 1995; Candy et al., 1996; Pohl et al., 1998; Chang et al., 1999, 2000; Huang et al., 2001) and BFD (Siegert, 2000; Dünkelmann et al., 2002; Polovnikova et al., 2003) have been examined for their effects on both the decarboxylation reaction and the carboligation reaction.
One of the intriguing features of PDC and BFD is that, with the exception of residues involved in binding ThDP, there is little strict conservation of other active site residues, at least in terms of sequence identity (Hasson et al., 1998; McLeish et al., 2004). This is surprising as the structural differences between the two natural substrates are not great. However, there appears to be some positional conservation of residues which suggests that the chemistry of the cofactor and reaction intermediates, in addition to active site architecture, have played a dominant role in the evolution of the two enzymes (Hasson et al., 1998). To explore the evolutionary relationships between the two enzymes, we are attempting to interconvert the activities of the two enzymes, using a combination of site-directed and directed evolution techniques. In this study, we focused on those amino acid residues influencing the substrate range of both enzymes [parts of this work have been published in advance as a proceedings contribution (Siegert et al., 2004)]. Based on sequence alignments and the X-ray structures (Dobritzsch et al., 1998; Hasson et al., 1998; Polovnikova et al., 2003) several mutants were created by site-directed mutagenesis in an attempt to interconvert the activities of the two enzymes. Kinetic data was then used to characterize the enzymes' properties, substrate ranges and enantioselectivity.
Materials and methods
All buffer salts were of p.a. quality, purchased from Fluka, Roth and Merck. 2-Keto acids were purchased from Sigma-Aldrich or were a generous gift from G.Krix (Krix et al., 1997).
Construction, expression and purification of His-tagged PDC variants
Wild-type PDC (WT PDC) and PDCI472A were available from a previous study (Pohl et al., 1998). Other PDC point mutations were introduced by PCR using the overlap extension technique. All fragments were amplified using the following outer primers:
PDC (bp 879) 5′-GGTGGACGGATATCCCTGATCC-3′ (sense);
PDC (bp 1717) 5′-AGTAAGCTTCTAGAGGAGCTTGTTAAC-3′ (antisense).
The PCR-generated fragments were initially cloned into pUC18 and sequenced. Fragments (635 bp) carrying the desired mutation were excised using EcoRV and StuI and cloned into the similarly digested expression vector pPDC-His6 (Pohl et al., 1998).
The forward primers used in the mutagenesis were as follows:
PDCI476F: 5′-ACACCATCGAAGTTATGtTCCATGATGG-3′;
PDCI472A/I476F: 5′-ACACCgcCGAAGTTATGtTCCATGATGG-3′.
In both cases the mutated codons are underlined, with the lower-case letters indicating a base change relative to WT.
All PDC variants were expressed and purified to homogeneity as described previously (Pohl et al., 1998). The only modification was that Ni-chelate chromatography was carried out in 50 mM potassium phosphate, pH 6.5, containing 2 mM MgSO4 and 0.1 mM ThDP and not in Mes–KOH buffer as used in the earlier study.
Construction, expression and purification of the BFD variants
The BFD variants BFDA460I, BFDF464I and BFDA460I/F464I were prepared using Pfu DNA polymerase and the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The DNA template for the single mutants was pBFDtrc (Tsou et al., 1990), while pBFDF464I served as the template for the construction of the double mutant. The following forward primers were used for the mutagenesis:
BFDA460I: 5′-ATGAACAACGGCACgTACGGTatcTTGCGATGG-3′;
BFDF464I: 5′-GCGTTGCGATGGaTTGCaGGCGTTCTCGAA-3′.
The mutated codons are underlined and the lower-case letters indicate a base change relative to WT. In addition to creating the appropriate mutations the change in sequence for BFDA460I resulted in the loss of a BanI restriction site, while the F464I mutation resulted in the loss of an NaeI restriction site. Following mutagenesis, the template DNA was removed by treatment with DpnI and the remaining PCR products transformed into Escherichia coli strain XL1-Blue (Stratagene). Single colonies were picked and their DNA isolated and screened for the desired mutation by restriction analysis using the appropriate combination of restriction enzymes. For all mutants, the fidelity of the PCR amplification and the presence of the mutation were confirmed by sequencing.
After IPTG-induced expression in E.coli SG13009, the cells were harvested and disrupted with glass beads. The expression levels of all variants were similar to that of WT BFD, ∼20% soluble cell protein. The cell-free extracts of all BFD variants were purified using a Q-Sepharose FF column equilibrated with 50 mM potassium phosphate buffer, pH 6.0, containing ThDP (0.1 mM), MgSO4 (2 mM) and KCl (150 mM). After elution of non-bound proteins with the equilibration buffer, a linear gradient of 200–400 mM NaCl was used to elute bound proteins. BFD eluted from the column at 280–300 mM NaCl. After this step, the level of protein purity was sufficiently high to perform carboligation experiments. For detailed kinetic characterization, all the variants were purified to homogeneity by hydrophobic interaction chromatography (HIC). Initially the NaCl was removed and the protein concentrated by ultrafiltration. Ammonium sulfate was added to a final concentration of 0.25 M before the protein was loaded on to a phenyl Sepharose high-substitution column (Pharmacia), which had been equilibrated with 50 mM potassium phosphate buffer containing 0.25 M ammonium sulfate. Elution of BFD variants was accomplished using a linear gradient of 0.25–0 M ammonium sulfate. The BFD variants eluted towards the end of the gradient (0.05–0 M) and the active fractions were collected, pooled, desalted by ultrafiltration and lyophilized.
Protein concentration
All protein concentrations were determined using the Bradford method (Bradford, 1976) with BSA as a standard.
Assay of decarboxylase activity
PDC variants
The decarboxylation of pyruvate was measured at 30°C using a coupled enzymatic assay as described elsewhere (Pohl et al., 1994). The assay mixture contained 16.9 mM sodium pyruvate, 0.18 mM NADH, 10 U/ml yeast ADH in 50 mM Mes–KOH or 50 mM potassium phosphate, pH 6.5, 2 mM MgSO4, 0.1 mM ThDP. The substrate spectrum was examined in the same assay mixture using 20 mM of the various 2-keto acids and 5 U/ml yeast alcohol dehydrogenase (ADH).
BFD variants
A similar continuous coupled spectrophotometric assay for BFD was performed at 30°C in 50 mM potassium phosphate buffer, pH 6.0, 0.1 mM ThDP, 2.5 mM MgSO4, 10 mM benzoylformate, 0.35 mM NADH and 5 U/ml horse liver alcohol dehydrogenase (HL ADH). For determination of the substrate specificity the same photometric assay was performed with 20 mM 2-keto acid replacing the benzoyl-formate.
One unit is defined as the amount of enzyme that catalyses the decarboxylation of 1 µmol of 2-keto acid per minute under standard conditions.
Determination of kinetic data
To analyse the kinetic data, initial enzymatic activities were measured in the range 0.03–50 mM 2-keto acid, when possible, using a Thermomax microplate reader (Molecular Devices). Data were collected in duplicate. The deviation of the respective duplicate values was in the region of 10%. Values of Km and Vmax were calculated by fitting of the hyperbolic curves to the Michaelis–Menten equation using the program Origin 3.5 (Microcal). Determination of substrate inhibition of BFD was calculated according to the Dixon equation (Bisswanger, 2002). kcat values were calculated based on the tetramer with four active centres.
Enzymatic synthesis, isolation and characterization of acetoin
Acetoin was synthesized in a batchwise process using either PDC or BFD variants as the catalysts. For reactions catalysed by PDC variants, the reaction mixture consisted of pyruvate (35 mM) in 50 mM Mes–KOH buffer, pH 6.5, containing MgSO4 (2 mM), ThDP (0.1 mM) and 10–100 µg/ml PDC variant. For reactions catalysed by BFD variants, the reaction mixture comprised acetaldehyde (500 mM) in 50 mM potassium phosphate buffer, pH 7.0, containing MgSO4 (2 mM), ThDP (0.1 mM) and 100 µg/ml BFD variant.
In both cases the reactions were incubated for 20 h at 30°C before being halted by heat inactivation of the enzyme for 2 min at 94°C. After filtration and chloroform extraction of the samples, the enantiomeric excess (ee) was determined by gas chromatography using a chiral column as described previously (Pohl et al., 1998). At a column temperature of 90°C the retention times of (R)-acetoin and (S)-acetoin were 5.5 and 6.0 min, respectively, and that of acetaldehyde was 3.0 min. Racemic acetoin was used as a standard. The identity of the enantiomers was confirmed using polarimetry according to Crout and Morrey (1983).
Enzymatic synthesis and isolation of benzoin
As with acetoin, benzoin was synthesized in a batchwise process using either PDC or BFD variants as catalysts. For reactions catalysed by PDC variants, benzaldehyde (30–70 mM) was incubated at 30°C with up to 100 µg/ml PDC variant in 50 mM Mes–KOH buffer, pH 6.5, containing MgSO4 (2 mM) and ThDP (0.1 mM), for periods between 16 h and 4 days. For reactions catalysed by BFD, benzaldehyde (50 mM) was incubated at 30°C with 100 µg/ml BFD variant in 50 mM potassium phosphate buffer, pH 7.0, containing MgSO4 (2 mM) and ThDP (0.1 mM), for 16 h.
The reaction was stopped by heat inactivation of the enzyme (2 min, 94°C) and the reaction mixture was extracted with diethyl ether, the precipitated protein was removed by filtration and the ether phase was collected and dried over MgSO4. After reduction of the organic phase in vacuo, benzoin was crystallized from diethyl ether–n-hexane (1:1, v/v; −20°C). Any residual benzaldehyde was removed by washing the crystals with cold n-hexane. Finally, the purified benzoin dried in vacuo.
Enzymatic synthesis, isolation and characterization of PAC and 2-HPP
For reactions catalysed by PDC variants, pyruvate (35 mM) or acetaldehyde (35 mM) and benzaldehyde (30–70 mM) in 50 mM Mes–KOH buffer, pH 6.5, MgSO4 (2 mM), ThDP (0.1 mM) were incubated with 10–100 µg/ml PDC variant for 20 h at 30°C. The reaction starting from pyruvate was used to distinguish between PDC-catalysed carboligation products and non-catalysed product formation (e.g. interconversion of PAC to 2-HPP). The excess of acetaldehyde produced during decarboxylation was removed enzymatically using a coupled enzymatic system with formate dehydrogenase as described elsewhere (Bruhn et al., 1995).
For reactions catalysed by BFD, 100 µg/ml BFD variant was incubated with acetaldehyde (500 mM) and benzaldehyde (0.1–50 mM) in 50 mM potassium phosphate buffer, pH 7.0, containing MgSO4 (2 mM) and ThDP (0.1 mM), for 20 h at 30°C.
After 20 h, the reaction was stopped by heat inactivation of the enzyme (2 min, 94°C). The reaction mixture was extracted with diethyl ether, any precipitated protein was removed by filtration and the ether phase was collected and dried over MgSO4. After reduction of the organic phase in vacuo, benzaldehyde was removed from the product mixture by silica gel chromatography (silica gel 60, 230–400 mesh, Merck) using diethyl ether–hexane (3:1, v/v) as eluent. Subsequently, the levels of PAC and 2-HPP were determined by RP-HPLC as described elsewhere (Pohl et al., 1998). If either PAC or 2-HPP was found to be present in the reaction mixture, chiral analysis was performed by chiral GC according to Pohl et al. (1998) at a column temperature of 130°C. Under these conditions, the retention times of (R)-2-HPP, (S)-2-HPP, (S)-PAC and (R)-PAC were 16, 18, 18 and 20 min, respectively. If simultaneous analysis of both products was required, the column temperature was set to 110°C, at which point the retention times increased to 37, 47, 49 and 59 min, respectively.
Results and discussion
Identification of active site residues involved in substrate binding
An alignment of the sequences of BFD (PDB: 1BFD) and PDC (PDB: 1ZPD) shows that the proteins shared about 18% sequence identity and 22% sequence similarity (Figure 3). Two highly conserved residues, Ile472 and Ile476, previously identified as playing a role in the binding of substrates to PDC (Pohl et al., 1998), had BFD counterparts in Ala460 and Phe464. Superimposition of the X-ray structures of the active sites regions of BFD (Polovnikova et al., 2003) and PDC (Dobritzsch et al., 1998) confirmed the suggestion that Ala460/Phe460 were indeed analogues of Ile472/Ile476 in PDC (Figure 4). In addition to Ile472 and Ile476, several PDC residues have already been investigated by site-directed mutagenesis. These include Trp392, which restricts the substrate binding site for aromatic substrates (Bruhn et al., 1995), and Glu473, which is essential for the catalytic activity and the structural integrity of the enzyme (Pohl et al., 1998).

Sequence alignment of BFD and PDC. The major residues interacting with ThDP are highlighted in bold. The active site histidine residues are highlighted in bold italic. Note that the histidine residues arise from distinctly different regions of sequence and are not identified by standard sequence alignments. The substrate recognition residues described in the text are printed in bold and are marked by asterisks. The ThDP-binding motif is underlined.
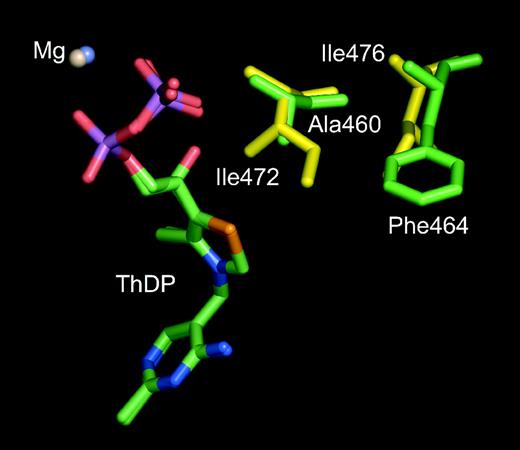
Putative substrate binding residues of PDC (gold) and BFD (green). The figure was obtained with the program QUANTA (http://www.accelrys.com) using coordinates from PDB 1ZPD (Dobritzsch et al., 1998) and 1BFD (Hasson et al., 1998) for PDC and BFD, respectively. The close superimposition of the ThDP cofactors demonstrate the similarities of the active site.
Motivated by the observation that the PDCI472A variant exhibited some BFD-like activity (Pohl et al., 1998), we extended our studies on further mutants of both enzymes. In order to test whether analogous residues have similar effects in the substrate specificity and enantioselectivity of both enzymes, we replaced the PDC residues Ile472 and Ile476 with alanine and phenylalanine, respectively. Similarly, the BFD residues, Ala460 and Phe464, were both replaced with isoleucine. In addition to the individual mutations, variants containing both mutations were also prepared.
Expression and purification
Mutagenesis and gene expression were carried out using standard procedures as described in Materials and methods. The PDC variants were all purified to homogeneity using metal chelate chromatography followed by gel filtration. The BFD variants were also purified in a two-step process using anion-exchange chromatography followed by hydrophobic interaction chromatography. None of the mutations appeared to affect the chromatographic properties of the enzymes, suggesting that, at the very least, the gross structure was retained.
Stability of cofactor binding
To investigate the stability of the cofactor binding, the PDC variants were incubated in potassium phosphate buffer, pH 6.5, in the absence of Mg2+ and ThDP. The residual activity was followed over 20 h. Unlike WT PDC and PDCI472A, which lost only 10% of their activity in this time period, the PDC variants containing the I476F mutation were found to lose activity rapidly. Inactivation occurred within 1 min for PDCI472A/I476F and within 100 min for PDCI476F. All variants proved to be stable in the presence of 2 mM MgSO4 and 0.1 mM ThDP. Clearly, it is imperative for biocatalytic studies to ensure that both cofactors are present in the reaction buffer.
The BFD variants were tested similarly. In contrast to the PDC variants, the mutations in Ala460 and Phe464 appeared to have no effect on the cofactor binding of the BFD variants. Nonetheless, both cofactors were maintained in reaction buffers.
Decarboxylase activity
In the first instance, the substrate spectrum of WT and mutant PDC and BFD was examined by comparing their specific activities with a variety of aliphatic and aromatic 2-keto acids under standard assay conditions. The results are given in Table I.
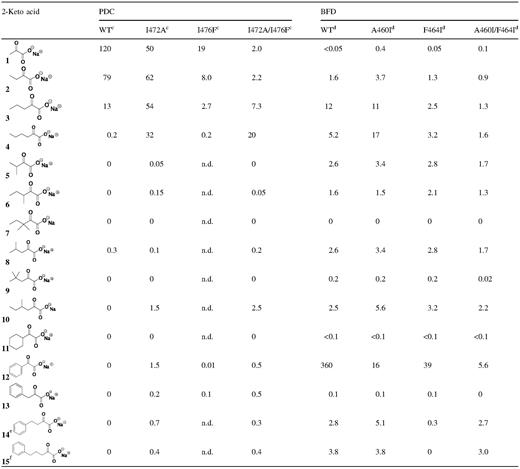
Reactions were carried out at 30°C at a substrate concentration of 20 mM as described in Materials and methods. Specific activities are reported in U/mg.
n.d.: not determined.
Data obtained using the standard PDC assay.
Data obtained using the standard BFD assay.
The solubility of this substrate permits only 5 mM in the assay.
The solubility of this substrate permits only 10 mM in the assay.
Reactions were carried out at 30°C at a substrate concentration of 20 mM as described in Materials and methods. Specific activities are reported in U/mg.
n.d.: not determined.
Data obtained using the standard PDC assay.
Data obtained using the standard BFD assay.
The solubility of this substrate permits only 5 mM in the assay.
The solubility of this substrate permits only 10 mM in the assay.
PDC variants
In line with earlier observations (Pohl et al., 1998), we found that the activity of the enzyme decreases as the length of the aliphatic chain increased from three to six carbons. Benzoylformate 12 is not a substrate for PDC, nor are compounds 5–7, the 2-keto acids branched at C3. However, a small level of activity towards compound 8, a 2-keto acid branched at C4, is observed (Table I). The I472A mutation results in a 2-fold decrease in pyruvate decarboxylase activity but greatly enhanced activity towards 2-ketopentanoic acid 3 and 2-ketohexanoic acid 4, the linear C5 and C6 substrates, respectively. In fact, both 3 and the C4 substrate, 2-ketobutanoic acid 2, are now preferred over pyruvate 1. Further, albeit to a much lesser extent, PDCI472A also shows an increased ability to decarboxylate branched-chain aliphatic substrates. More importantly, this variant shows activity with several aromatic substrates (12–15) and is able to decarboxylate benzoylformate 12 at about 3% of the rate at which pyruvate 1 is decarboxylated.
In some respects, replacement of Ile476 with phenylalanine has the most deleterious impact on the decarboxylase activity of PDC. The rate of decarboxylation decreases for all substrates and the selectivity for longer chain length is no longer observed. However, PDCI476F does show some marginal activity towards 12 and phenyl pyruvate 13. While the PDCI472A/I476F double mutant shows the greatest overall decrease in PDC activity, it also exhibits a marked preference for the linear C6-keto acid 4. Whereas WT PDC exhibits only very low activity towards 4, by contrast, in the standard assay, the PDCI472A/I476F double mutant is able to decarboxylate substrate 4 40 times faster than the WT enzyme and 10 times more rapidly than it can decarboxylate 1. Further, the C4-branched substrate 10 is decarboxylated by the double mutant at a rate similar to pyruvate and benzoylformate is decarboxylated at a rate about 25% that of pyruvate. Both the PDC variants containing the I472A mutation are able to decarboxylate aromatic 2-keto acids 12–15 with the double mutant, PDCI472A/I476F, being less rigid in its structural requirements. Finally, none of the PDC variants are able to decarboxylate the saturated analogue of benzoylformic acid, cyclohexaneglyoxylic acid 11.
To ascertain whether the alterations in specificity were due to changes in substrate binding affinity or catalytic activity, the Michaelis–Menten parameters were obtained for each of the PDC variants with some of their preferred substrates. The data in Table II indicate that changes in the value of kcat/Km, often termed the specificity constant (Fersht, 1998), have contributions from changes in both Km (substrate affinity) and kcat (catalytic efficiency). The PDC variants, PDCI472A and PDCI472A/I476F, both show increases in substrate binding affinity and specificity as the chain length of the keto acid increases from three to six carbons. For the I472A variant, the increased specificity for the longer chain substrates is due almost entirely to substrate binding affinity as values of kcat remain approximately the same or decrease marginally as the chain becomes longer, whereas the value of Km decreases 40-fold. Conversely, WT PDC shows a 10-fold decrease in binding affinity for the C6 substrate 4. Both the WT and the I476F variant show steady decreases in value of kcat as the chain length is increased, culminating in a similar 10-fold decrease for the C6 substrate 4. Surprisingly, for the double mutant, the reverse is the case with the value of kcat increasing 10-fold over the corresponding increase in chain length. Taken together, these results suggest that Ile472 plays a significant role in substrate recognition but not catalysis. Replacement of Ile472 by alanine permits substrates larger than pyruvate to enter the active site with only a limited effect on the catalytic rate. Ile476, on the other hand, although clearly influencing substrate binding, plays a more significant role in positioning the substrate for catalysis. Further, the active site of PDC favours straight-chain keto acids as 4-methyl-2-ketohexanoic acid 10 has a greater Km value with WT PDC than does 4, its straight-chain analogue (Table III). A similar increase is observed for both variants containing the I472A mutation. Even when 10 is bound, it is not well positioned for catalysis as, for all variants, its kcat value is 10-fold lower than that of 4 (Table III).
| Compound | Parameter | WT | I472A | I476F | I472A/I476F |
|---|---|---|---|---|---|
| Pyruvate (1) | Km (mM) | 1.1 | 7.8 | 2.6 | 50 |
| kcat (s−1) | 486 | 200 | 77 | 8.0 | |
| kcat/Km (mM−1 s−1)c | 442 (100) | 26 (6) | 30 (7) | 0.2 (2) | |
| 2-Ketobutanoic acid (2) | Km (mM) | 4.7 | 6.7 | n.d. | 50 |
| kcat (s−1) | 320 | 250 | 32d | 9.0 | |
| kcat/Km (mM−1 s−1)c | 68 (100) | 37 (54) | n.d. | 0.2 (0.3) | |
| 2-Ketopentanoic acid (3) | Km (mM) | 7.6 | 2.5 | n.d. | 11 |
| kcat (s−1) | 53 | 220 | 11d | 30 | |
| kcat/Km (mM−1 s−1)c | 7.0 (100) | 87 (1260) | n.d. | 2.7 (39) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 12.7 | 0.2 | n.d. | 0.5 |
| kcat (s−1) | 4.0 | 130 | 0.8d | 82 | |
| kcat/Km (mM−1 s−1)c | 0.3 (100) | 650 (216 666) | n.d. | 164 (54 666) | |
| Benzoylformate (12) | Km (mM) | 0 | 1.8 | n.d. | 4.4 |
| kcat (s−1) | 0 | 6.9 | n.d. | 1.2 | |
| kcat/Km (mM−1 s−1) | 0 | 3.8 | n.d. | 0.3 |
| Compound | Parameter | WT | I472A | I476F | I472A/I476F |
|---|---|---|---|---|---|
| Pyruvate (1) | Km (mM) | 1.1 | 7.8 | 2.6 | 50 |
| kcat (s−1) | 486 | 200 | 77 | 8.0 | |
| kcat/Km (mM−1 s−1)c | 442 (100) | 26 (6) | 30 (7) | 0.2 (2) | |
| 2-Ketobutanoic acid (2) | Km (mM) | 4.7 | 6.7 | n.d. | 50 |
| kcat (s−1) | 320 | 250 | 32d | 9.0 | |
| kcat/Km (mM−1 s−1)c | 68 (100) | 37 (54) | n.d. | 0.2 (0.3) | |
| 2-Ketopentanoic acid (3) | Km (mM) | 7.6 | 2.5 | n.d. | 11 |
| kcat (s−1) | 53 | 220 | 11d | 30 | |
| kcat/Km (mM−1 s−1)c | 7.0 (100) | 87 (1260) | n.d. | 2.7 (39) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 12.7 | 0.2 | n.d. | 0.5 |
| kcat (s−1) | 4.0 | 130 | 0.8d | 82 | |
| kcat/Km (mM−1 s−1)c | 0.3 (100) | 650 (216 666) | n.d. | 164 (54 666) | |
| Benzoylformate (12) | Km (mM) | 0 | 1.8 | n.d. | 4.4 |
| kcat (s−1) | 0 | 6.9 | n.d. | 1.2 | |
| kcat/Km (mM−1 s−1) | 0 | 3.8 | n.d. | 0.3 |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
In parentheses are the relative values expressed as a percentage of the WT PDC value.
Value obtained using substrate concentration of 30 mM.
| Compound | Parameter | WT | I472A | I476F | I472A/I476F |
|---|---|---|---|---|---|
| Pyruvate (1) | Km (mM) | 1.1 | 7.8 | 2.6 | 50 |
| kcat (s−1) | 486 | 200 | 77 | 8.0 | |
| kcat/Km (mM−1 s−1)c | 442 (100) | 26 (6) | 30 (7) | 0.2 (2) | |
| 2-Ketobutanoic acid (2) | Km (mM) | 4.7 | 6.7 | n.d. | 50 |
| kcat (s−1) | 320 | 250 | 32d | 9.0 | |
| kcat/Km (mM−1 s−1)c | 68 (100) | 37 (54) | n.d. | 0.2 (0.3) | |
| 2-Ketopentanoic acid (3) | Km (mM) | 7.6 | 2.5 | n.d. | 11 |
| kcat (s−1) | 53 | 220 | 11d | 30 | |
| kcat/Km (mM−1 s−1)c | 7.0 (100) | 87 (1260) | n.d. | 2.7 (39) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 12.7 | 0.2 | n.d. | 0.5 |
| kcat (s−1) | 4.0 | 130 | 0.8d | 82 | |
| kcat/Km (mM−1 s−1)c | 0.3 (100) | 650 (216 666) | n.d. | 164 (54 666) | |
| Benzoylformate (12) | Km (mM) | 0 | 1.8 | n.d. | 4.4 |
| kcat (s−1) | 0 | 6.9 | n.d. | 1.2 | |
| kcat/Km (mM−1 s−1) | 0 | 3.8 | n.d. | 0.3 |
| Compound | Parameter | WT | I472A | I476F | I472A/I476F |
|---|---|---|---|---|---|
| Pyruvate (1) | Km (mM) | 1.1 | 7.8 | 2.6 | 50 |
| kcat (s−1) | 486 | 200 | 77 | 8.0 | |
| kcat/Km (mM−1 s−1)c | 442 (100) | 26 (6) | 30 (7) | 0.2 (2) | |
| 2-Ketobutanoic acid (2) | Km (mM) | 4.7 | 6.7 | n.d. | 50 |
| kcat (s−1) | 320 | 250 | 32d | 9.0 | |
| kcat/Km (mM−1 s−1)c | 68 (100) | 37 (54) | n.d. | 0.2 (0.3) | |
| 2-Ketopentanoic acid (3) | Km (mM) | 7.6 | 2.5 | n.d. | 11 |
| kcat (s−1) | 53 | 220 | 11d | 30 | |
| kcat/Km (mM−1 s−1)c | 7.0 (100) | 87 (1260) | n.d. | 2.7 (39) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 12.7 | 0.2 | n.d. | 0.5 |
| kcat (s−1) | 4.0 | 130 | 0.8d | 82 | |
| kcat/Km (mM−1 s−1)c | 0.3 (100) | 650 (216 666) | n.d. | 164 (54 666) | |
| Benzoylformate (12) | Km (mM) | 0 | 1.8 | n.d. | 4.4 |
| kcat (s−1) | 0 | 6.9 | n.d. | 1.2 | |
| kcat/Km (mM−1 s−1) | 0 | 3.8 | n.d. | 0.3 |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
In parentheses are the relative values expressed as a percentage of the WT PDC value.
Value obtained using substrate concentration of 30 mM.
| Compound | Parameter | PDC WT | PDCI472A | PDCI472A/I476F | BFD WT | BFDA460I |
|---|---|---|---|---|---|---|
| 2-Keto-4-methylpentanoic acid (8) | Km (mM) | n.d. | n.d. | n.d. | 9.6 | 3.4c |
| kcat (s−1) | n.d. | n.d. | n.d. | 11 | 38 | |
| kcat/Km (mM−1 s−1)d | n.d. | n.d. | n.d. | 1.1 (100) | 11 (975) | |
| 2-Keto-4-methylhexanoic acid (10) | Km (mM) | >40 | 1.7 | 3.7 | ≫20 | 1.1e |
| kcat (s−1) | 0.4f | 7.7 | 8.1 | 8.6f | 9.8 | |
| kcat/Km (mM−1 s−1)d | <0.01 | 4.5 | 2.2 | n.d. | 8.9 |
| Compound | Parameter | PDC WT | PDCI472A | PDCI472A/I476F | BFD WT | BFDA460I |
|---|---|---|---|---|---|---|
| 2-Keto-4-methylpentanoic acid (8) | Km (mM) | n.d. | n.d. | n.d. | 9.6 | 3.4c |
| kcat (s−1) | n.d. | n.d. | n.d. | 11 | 38 | |
| kcat/Km (mM−1 s−1)d | n.d. | n.d. | n.d. | 1.1 (100) | 11 (975) | |
| 2-Keto-4-methylhexanoic acid (10) | Km (mM) | >40 | 1.7 | 3.7 | ≫20 | 1.1e |
| kcat (s−1) | 0.4f | 7.7 | 8.1 | 8.6f | 9.8 | |
| kcat/Km (mM−1 s−1)d | <0.01 | 4.5 | 2.2 | n.d. | 8.9 |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
Displays substrate inhibition with Ki = 60 mM.
In parentheses are the relative values expressed as a percentage of the WT value.
Displays substrate inhibition with Ki = 48 mM.
Value obtained at a substrate concentration of 30 mM.
| Compound | Parameter | PDC WT | PDCI472A | PDCI472A/I476F | BFD WT | BFDA460I |
|---|---|---|---|---|---|---|
| 2-Keto-4-methylpentanoic acid (8) | Km (mM) | n.d. | n.d. | n.d. | 9.6 | 3.4c |
| kcat (s−1) | n.d. | n.d. | n.d. | 11 | 38 | |
| kcat/Km (mM−1 s−1)d | n.d. | n.d. | n.d. | 1.1 (100) | 11 (975) | |
| 2-Keto-4-methylhexanoic acid (10) | Km (mM) | >40 | 1.7 | 3.7 | ≫20 | 1.1e |
| kcat (s−1) | 0.4f | 7.7 | 8.1 | 8.6f | 9.8 | |
| kcat/Km (mM−1 s−1)d | <0.01 | 4.5 | 2.2 | n.d. | 8.9 |
| Compound | Parameter | PDC WT | PDCI472A | PDCI472A/I476F | BFD WT | BFDA460I |
|---|---|---|---|---|---|---|
| 2-Keto-4-methylpentanoic acid (8) | Km (mM) | n.d. | n.d. | n.d. | 9.6 | 3.4c |
| kcat (s−1) | n.d. | n.d. | n.d. | 11 | 38 | |
| kcat/Km (mM−1 s−1)d | n.d. | n.d. | n.d. | 1.1 (100) | 11 (975) | |
| 2-Keto-4-methylhexanoic acid (10) | Km (mM) | >40 | 1.7 | 3.7 | ≫20 | 1.1e |
| kcat (s−1) | 0.4f | 7.7 | 8.1 | 8.6f | 9.8 | |
| kcat/Km (mM−1 s−1)d | <0.01 | 4.5 | 2.2 | n.d. | 8.9 |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
Displays substrate inhibition with Ki = 60 mM.
In parentheses are the relative values expressed as a percentage of the WT value.
Displays substrate inhibition with Ki = 48 mM.
Value obtained at a substrate concentration of 30 mM.
BFD variants
It is immediately apparent from the data in Table I that, in its standard assay, BFD is much more selective for its natural substrate, benzoylformate, than is PDC for pyruvate. For example, the specific activity of BFD with its best alternative substrate 3 is only 1.4% of that with benzoyl-formate 12. Replacement of Ala460 with isoleucine results in a 22-fold decrease in activity with 12, but activity with most other substrates is relatively unaffected. The mutation brings about some change in specificity as there is one example where BFDA460I has greater activity with alternative substrates. For instance, the rate of decarboxylation of 4 increases more than 3-fold and is now slightly greater than that of 12, whereas that with 3 is unaffected. In fact, BFDA460I is able to decarboxylate the C6-keto acid 4 at a rate (17 U/mg) similar to that shown by PDCI472A (32 U/mg) (Table I). In addition, activity with the branched-chain 2-keto acids, 5, 8 and 10, is also higher with this variant than with WT PDC. Overall the activity decreases as the length of the carbon chain decreased but it is still measurable even with 1 whose decarboxylation rate was increased 8-fold over that with the WT enzyme. With a few exceptions, the F464I mutation also results in a significant decrease in decarboxylase activity. Slight increases in activity are observed for branched-chain substrates (6, 8 and 10), but the rate of decarboxylation of 12 decreases 9-fold and the enzyme is virtually inactive with other aromatic substrates. The double mutant, BFDA460I/F646I, exhibits a 60-fold decrease in activity with 12 and little activity with any other substrate including, disappointingly, pyruvate. Finally, unlike PDC, which is unable to decarboxylate cyclohexaneglyoxylic acid, WT BFD and all of its variants show consistent but very low levels of activity with this substrate.
The Michaelis–Menten data for the BFD variants are shown in Table IV. The benzoylformate Km values for both single mutants, and also the double mutant, do not differ greatly from that of WT BFD, indicating that the ability of the aromatic substrate to bind to the active site is still relatively high. This is in marked contrast to PDC, where the double mutant showed a 50-fold increase in Km value for its native substrate (Table II). It is also surprising as it may have been expected that enlargement of the residue in position 460 may have rendered the active site less accessible for aromatic substrates. However, with all mutants, the catalytic activity with benzoylformate is greatly impaired. This suggests that, although the substrates are able to bind, they are not positioned precisely for catalysis. Not surprisingly, for WT BFD the Km values for the aliphatic substrates increases as the chain length decreases (essentially the reverse of the trend for PDC). However, more unexpectedly, the A460I variant has lower values of Km for each of the various substrates, so that its Km values for 3 and 4 were similar to those of the WT enzyme with benzoylformate. The kcat values with the WT enzyme dropped markedly with aliphatic substrates but, unlike PDC, the drop-off did not seem to correlate directly with Km values or chain length; rather, it seemed that benzoylformate was the optimal substrate and, although others were tolerated, their kcat values were uniformly 1–2 orders of magnitude lower. It is intriguing that, for BFDA460I, the kcat values for 3, 4 and 12 are broadly similar and only 2–3 times that for compound 2. Its counterpart, PDCI472A, has a similar trend but, instead, favouring the smaller substrate, i.e. the values of kcat for 1, 2 and 3, similar and only about double that for compound 4. In addition, when compared with the WT enzymes, each of these variants showed an increased specificity (as measured by kcat/Km) for 3 and 4, and also two branched chain substrates, 8 and 10 (Table III).
| Compound | Parameter | WT | A460I | F464I | A460I/F464I |
|---|---|---|---|---|---|
| Benzoylformate (12) | Km (mM) | 0.8 | 0.2 | 2.3 | 1.1 |
| kcat (s−1) | 1349 | 62 | 123 | 21 | |
| Kcat/Km (mM−1 s−1)c | 1686 (100) | 310 (18) | 53 (3) | 19 (1) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 4.1d | 1.0e | n.d. | n.d. |
| kcat (s−1) | 21.0 | 71.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | 5.1 (100) | 71.8 (1408) | n.d. | n.d. | |
| 2-Ketopentanoic acid (3) | Km (mM) | 6.0f | 1.2f | n.d. | n.d. |
| kcat (s−1) | 44.3 | 40.6 | n.d. | n.d. | |
| kcat /Km (mM−1 s−1)c | 7.4 (100) | 33.8 (457) | n.d. | n.d. | |
| 2-Ketobutanoic acid (2) | Km (mM) | >35 | 18.3 | n.d. | n.d. |
| kcat (s−1) | 17.3g | 18.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | n.d. | 1.0 | n.d. | n.d. | |
| Pyruvate (1) | Km (mM) | ≫50 | ≫50 | n.d. | n.d. |
| kcat (s−1) | 0.1g | 5.6g | n.d. | n.d. | |
| kcat/Km (mM−1 s−1) | n.d. | n.d. | n.d. | n.d. |
| Compound | Parameter | WT | A460I | F464I | A460I/F464I |
|---|---|---|---|---|---|
| Benzoylformate (12) | Km (mM) | 0.8 | 0.2 | 2.3 | 1.1 |
| kcat (s−1) | 1349 | 62 | 123 | 21 | |
| Kcat/Km (mM−1 s−1)c | 1686 (100) | 310 (18) | 53 (3) | 19 (1) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 4.1d | 1.0e | n.d. | n.d. |
| kcat (s−1) | 21.0 | 71.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | 5.1 (100) | 71.8 (1408) | n.d. | n.d. | |
| 2-Ketopentanoic acid (3) | Km (mM) | 6.0f | 1.2f | n.d. | n.d. |
| kcat (s−1) | 44.3 | 40.6 | n.d. | n.d. | |
| kcat /Km (mM−1 s−1)c | 7.4 (100) | 33.8 (457) | n.d. | n.d. | |
| 2-Ketobutanoic acid (2) | Km (mM) | >35 | 18.3 | n.d. | n.d. |
| kcat (s−1) | 17.3g | 18.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | n.d. | 1.0 | n.d. | n.d. | |
| Pyruvate (1) | Km (mM) | ≫50 | ≫50 | n.d. | n.d. |
| kcat (s−1) | 0.1g | 5.6g | n.d. | n.d. | |
| kcat/Km (mM−1 s−1) | n.d. | n.d. | n.d. | n.d. |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
In parentheses are the relative values expressed as a percentage of the WT BFD value.
Displays substrate inhibition with Ki = 25 mM.
Displays substrate inhibition with Ki = 10 mM.
Displays substrate inhibition with Ki = 20 mM.
Value obtained at a substrate concentration of 100 mM.
| Compound | Parameter | WT | A460I | F464I | A460I/F464I |
|---|---|---|---|---|---|
| Benzoylformate (12) | Km (mM) | 0.8 | 0.2 | 2.3 | 1.1 |
| kcat (s−1) | 1349 | 62 | 123 | 21 | |
| Kcat/Km (mM−1 s−1)c | 1686 (100) | 310 (18) | 53 (3) | 19 (1) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 4.1d | 1.0e | n.d. | n.d. |
| kcat (s−1) | 21.0 | 71.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | 5.1 (100) | 71.8 (1408) | n.d. | n.d. | |
| 2-Ketopentanoic acid (3) | Km (mM) | 6.0f | 1.2f | n.d. | n.d. |
| kcat (s−1) | 44.3 | 40.6 | n.d. | n.d. | |
| kcat /Km (mM−1 s−1)c | 7.4 (100) | 33.8 (457) | n.d. | n.d. | |
| 2-Ketobutanoic acid (2) | Km (mM) | >35 | 18.3 | n.d. | n.d. |
| kcat (s−1) | 17.3g | 18.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | n.d. | 1.0 | n.d. | n.d. | |
| Pyruvate (1) | Km (mM) | ≫50 | ≫50 | n.d. | n.d. |
| kcat (s−1) | 0.1g | 5.6g | n.d. | n.d. | |
| kcat/Km (mM−1 s−1) | n.d. | n.d. | n.d. | n.d. |
| Compound | Parameter | WT | A460I | F464I | A460I/F464I |
|---|---|---|---|---|---|
| Benzoylformate (12) | Km (mM) | 0.8 | 0.2 | 2.3 | 1.1 |
| kcat (s−1) | 1349 | 62 | 123 | 21 | |
| Kcat/Km (mM−1 s−1)c | 1686 (100) | 310 (18) | 53 (3) | 19 (1) | |
| 2-Ketohexanoic acid (4) | Km (mM) | 4.1d | 1.0e | n.d. | n.d. |
| kcat (s−1) | 21.0 | 71.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | 5.1 (100) | 71.8 (1408) | n.d. | n.d. | |
| 2-Ketopentanoic acid (3) | Km (mM) | 6.0f | 1.2f | n.d. | n.d. |
| kcat (s−1) | 44.3 | 40.6 | n.d. | n.d. | |
| kcat /Km (mM−1 s−1)c | 7.4 (100) | 33.8 (457) | n.d. | n.d. | |
| 2-Ketobutanoic acid (2) | Km (mM) | >35 | 18.3 | n.d. | n.d. |
| kcat (s−1) | 17.3g | 18.8 | n.d. | n.d. | |
| kcat/Km (mM−1 s−1)c | n.d. | 1.0 | n.d. | n.d. | |
| Pyruvate (1) | Km (mM) | ≫50 | ≫50 | n.d. | n.d. |
| kcat (s−1) | 0.1g | 5.6g | n.d. | n.d. | |
| kcat/Km (mM−1 s−1) | n.d. | n.d. | n.d. | n.d. |
Reactions were carried out at 30°C as described in Materials and methods. Errors are estimated to be ±10% from duplicate measurements of 24 data points in the range 0.03–30 mM of the respective 2-keto acid.
n.d.: not determined.
In parentheses are the relative values expressed as a percentage of the WT BFD value.
Displays substrate inhibition with Ki = 25 mM.
Displays substrate inhibition with Ki = 10 mM.
Displays substrate inhibition with Ki = 20 mM.
Value obtained at a substrate concentration of 100 mM.
The kinetic measurements have indicated that, for WT BFD and BFDA460I, the Km values for pyruvate are ≫50 mM. This indicates that the WT enzyme has very little affinity for this small aliphatic substrate and the affinity is not greatly affected by the mutation (Table IV) There appears to be a small increase in value of kcat for the A460I variant, which gave rise to the 8-fold increase in specific activity (Table I), but the high values of Km precluded any possibility of determining accurate kcat values. Similarly, the low activity of BFDF464I and the A460I/F464I double mutant made it difficult to obtain sufficiently reliable data to determine values of Km and kcat for these variants.
Finally, plotting the initial velocity against substrate concentration, i.e. v vs [S], gave rise to hyperbolic curves. However, for the C5 and C6 2-keto acids, both WT BFD and the BFDA460I variant showed lower than expected velocities at high substrate concentrations, indicating substrate inhibition (Table IV), which usually occurs when a second molecule of substrate can bind to the enzyme–substrate (E–S) complex, to form an inactive S–E–S ternary complex (Bisswanger, 2002). Given that BFD carries out carboligation reactions and is able to accommodate benzoin within its active site (Iding et al., 2000), it is not entirely surprising that a second molecule of an alternative substrate can also accommodated. That said, PDC also carries out carboligation reactions and shows no sign of substrate inhibition, although it is subject to inactivation by its product, acetaldehyde (Bruhn et al., 1995). The inhibition constants (Ki), listed in Tables III and IV, are all at least 3-fold higher than the corresponding value of Km and it is notable that the branched-chain substrates only inhibit BFDA460I.
Carboligase activity
PDC and BFD are examples of enzymes exhibiting catalytic promiscuity as they can catalyse a second reaction, the carboligation reaction, at the same active site as decarboxylation (Figure 1) (Bornscheuer and Kazlauskas, 2004). Using acetaldehyde and benzaldehyde as substrates, PDC and optimized mutants thereof catalyse the formation of enantiopure (R)-PAC and predominantly (S)-acetoin (Figure 2) (Pohl, 1997; Pohl et al., 1998). Formation of both acyloins requires the initial binding of acetaldehyde (acting as the donor substrate) to the active site. Both benzaldehyde and acetaldehyde can act as the second (acceptor) substrate yielding PAC and acetoin, respectively (Figure 2). (S)-2-Hydroxypropiophenone [(S)-2-HPP] and (R)-benzoin, typical products for the BFD-catalysed carboligation reaction, arise when benzaldehyde is the donor substrate and acetaldehyde or benzaldehyde is the acceptor, respectively. It is not unreasonable to expect that, as with the donor substrate, the binding of the acceptor will also be influenced by the amino acid residues lining the active site. As has been described previously (Pohl et al., 1998), (S)-2-HPP and benzoin are not accessible by WT PDC catalysis. However, replacement of Ile472 with alanine has significant impact on the carboligase reaction, influencing both the substrate range and the enantioselectivity. The PDCI472A variant is able to catalyse the formation of the ‘BFD-like’ products, 2-HPP and benzoin, in addition to PAC and acetoin. On that basis, this variant could be viewed as chimera of PDC and BFD. Given the changes in substrate specificity of the decarboxylase activity detailed in Tables II–IV, it was of interest to examine the effects of these active site mutations on the carboligation reactions catalysed by PDC and BFD.
PDC variants
The reaction products accessible from acetaldehyde (obtained via decarboxylation of pyruvate) and benzaldehyde were compared for all PDC variants. Pyruvate was chosen as a substrate as WT PDC has proved to be sensitive to high acetaldehyde concentrations (Bruhn et al., 1995). The choice of pyruvate has the additional advantage in that the continuous increase of acetaldehyde resulting from pyruvate decarboxylation allows one to distinguish enzymatic and possible non-enzymatic side reactions (see below). As shown in Table V, in contrast to PDCI472A, the I476F mutation does not influence the enantioselectivity of the PAC synthesis. Further, like the WT enzyme, the PDCI476F variant does not catalyse the formation of 2-HPP and benzoin, suggesting that it does not accept benzaldehyde as a donor aldehyde. However, both the I472A and the I476F mutations improve the enantioselectivity of (S)-acetoin formation whereas the double mutant, PDCI472A/I476F, is even more effective, showing the highest enantioselectivity for (S)-acetoin (61%) in this series (Table V). Unfortunately, although the enantioselectivity is improved, the yield with both of these variants is 40-fold lower than that of the WT PDC.
| PDC variant | Acetoin (ee) | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | 4.0 (20 S) | 2.4 (>98 R) | 0 | 0 |
| I472A | 4.0 (50 S) | 1.6 (70 R) | 0.4 (70 S) | 0.1 (>98 R) |
| I476F | <0.1 (44 S) | 1.3 (>98 R) | 0 | 0 |
| I472A/I476F | <0.1 (61 S) | 2.0 (70 R) | 0.3 (70 S) | ≪0.1 (n.d.) |
| PDC variant | Acetoin (ee) | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | 4.0 (20 S) | 2.4 (>98 R) | 0 | 0 |
| I472A | 4.0 (50 S) | 1.6 (70 R) | 0.4 (70 S) | 0.1 (>98 R) |
| I476F | <0.1 (44 S) | 1.3 (>98 R) | 0 | 0 |
| I472A/I476F | <0.1 (61 S) | 2.0 (70 R) | 0.3 (70 S) | ≪0.1 (n.d.) |
Yields are reported in mM with enantiomeric excess (%) in parentheses; n.d., not determined.
Reaction conditions: 35 mM pyruvate, 70 mM benzaldehyde and 50 µg/ml enzyme in 50 mM Mes–KOH buffer, pH 6.5, 2 mM MgSO4, 0.1 mM ThDP were incubated for 20 h at 30°C. The reaction was stopped by heat inactivation of the enzyme (2 min, 94°C) and aromatic products were determined by HPLC. Detection of acetoin was performed by gas chromatography.
| PDC variant | Acetoin (ee) | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | 4.0 (20 S) | 2.4 (>98 R) | 0 | 0 |
| I472A | 4.0 (50 S) | 1.6 (70 R) | 0.4 (70 S) | 0.1 (>98 R) |
| I476F | <0.1 (44 S) | 1.3 (>98 R) | 0 | 0 |
| I472A/I476F | <0.1 (61 S) | 2.0 (70 R) | 0.3 (70 S) | ≪0.1 (n.d.) |
| PDC variant | Acetoin (ee) | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | 4.0 (20 S) | 2.4 (>98 R) | 0 | 0 |
| I472A | 4.0 (50 S) | 1.6 (70 R) | 0.4 (70 S) | 0.1 (>98 R) |
| I476F | <0.1 (44 S) | 1.3 (>98 R) | 0 | 0 |
| I472A/I476F | <0.1 (61 S) | 2.0 (70 R) | 0.3 (70 S) | ≪0.1 (n.d.) |
Yields are reported in mM with enantiomeric excess (%) in parentheses; n.d., not determined.
Reaction conditions: 35 mM pyruvate, 70 mM benzaldehyde and 50 µg/ml enzyme in 50 mM Mes–KOH buffer, pH 6.5, 2 mM MgSO4, 0.1 mM ThDP were incubated for 20 h at 30°C. The reaction was stopped by heat inactivation of the enzyme (2 min, 94°C) and aromatic products were determined by HPLC. Detection of acetoin was performed by gas chromatography.
With optimized variants (concerning carboligation) of PDC, we have already demonstrated that the carboligation products, acetoin and PAC, are direct products of the enzyme-catalysed carboligation (Pohl, 1997). At pH 6.5, there is no evidence that the reaction is reversible, i.e. the enzyme is unable to catalyse the cleavage of these acyloins under conditions where their synthesis occurs, nor is there evidence that acetoin or PAC is able to inhibit the carboligase reaction (data not shown). Furthermore, although (R)-PAC was sensitive towards slightly alkaline pH, resulting in racemization and isomerization to 2-HPP, no enzyme-catalysed racemization or isomerization was observed during batch reactions. However, there are ThDP-dependent enzymes, such as benzaldehyde lyase (BAL), whose main activity is to catalyse the cleavage of 2-hydroxy ketones (Demir et al., 1999; Pohl et al., 2002, 2004). This motivated us to investigate whether the full spectrum of products obtained using PDCI472A are a direct result of the enzymatically catalysed carboligation reaction or a consequence of subsequent enzymatic or non-enzymatic reactions.
Since the decarboxylation of pyruvate is a proton-consuming process (Figure 1) and the liberated CO2 further increases the pH of the batch reaction, we examined the possibility that the reaction is influenced by the pH. pH control was performed by a pH-stat. The products of a pH-controlled batch reaction (pH 6.5) were monitored over a 2-day period. The enantiomeric excess (ee) of the reaction products was unchanged from those of a similar non-pH-controlled reaction. Not surprisingly, it was also observed that PAC is formed quickly at the beginning of the reaction when pyruvate is still available. However, 2-HPP formation increased with the concentration of free acetaldehyde in solution. If the acetaldehyde is continuously removed using formate dehydrogenase, no acetaldehyde accumulates and no 2-HPP was observed (data not shown). No hint of enzymatic or non-enzymatic interconversion of PAC and 2-HPP was seen. Taken together, these results suggest that the isomeric products (R)-PAC and (S)-2-HPP are directly formed from pyruvate (acetaldehyde) and benzaldehyde in a reaction catalysed by PDCI472A. It was shown earlier (Tables I and II) that PDCI472A can utilize benzoylformate as a substrate. It is now apparent that, analogously, this variant can use benzaldehyde as a donor substrate in the carboligation reaction.
Since PDCI472A also decarboxylates longer chain aliphatic, in addition to aromatic, 2-keto acids (Tables I and II), it was possible that the corresponding aldehydes are also substrates for the carboligase reaction. To explore this possibility, batch experiments were carried out in which 2, 3 and 4 were reacted with equimolar amounts of benzaldehyde. The results in Figure 5 show that, under the given conditions, the aliphatic aldehydes are selective acyl donors for PDCI472A. In line with the observation that the affinity for PDCI472A increases with the chain length of the 2-keto acid (Table II), we observed an increasing activity in carboligation reactions using 2, 3 and 4 as an acyl donor and benzaldehyde as acyl acceptor (data not shown). All products were unambiguously identified by GC–MS. Given their relatively low levels of decarboxylase activity, no attempt was made to examine the carboligase activity of the PDCI476F and PDCI472A/I476F variants.
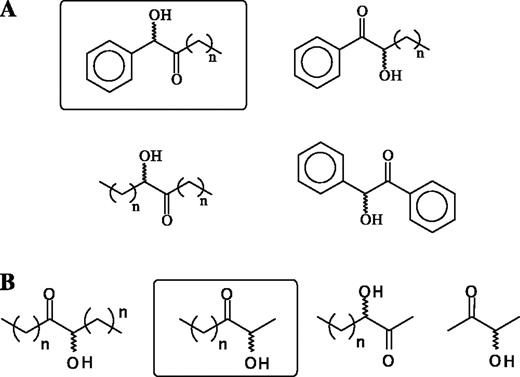
Potential products from (A) the PDCI472A-catalysed carboligation of C4, C5 and C6 aliphatic 2-ketoacids with benzaldehydea,b and (B) the carboligation of C4, C5 and C6 aliphatic 2-keto acids or the corresponding aldehyde, with acetaldehyde catalysed by BFDA460Ia,c. The major product observed for each reaction is highlighted. an = 3–5. bReaction conditions for A: 40 mM of 2, 3 and 4 (as their sodium salts) were reacted with 40 mM benzaldehyde in 50 mM Mes–KOH buffer, pH 6.5, 2 mM MgSO4, 0.1 mM ThDP with 5 U/ml PDCI472A for 16 h to 4 days at 30°C. cReaction conditions for B: 50 mM of 2, 3 or 4 or their corresponding aldehyde were reacted with 500 mM acetaldehyde in 50 mM potassium phosphate buffer, pH 7.0, 2 mM MgSO4, 0.1 mM ThDP, with 10 U/ml BFDA460I at 30°C for 16 h. Subsequently, the products formed were extracted with chloroform and analysed by GC–MS.
BFD variants
As shown in Table VI, all possible products except PAC can be obtained from acetaldehyde and benzaldehyde in a reaction catalysed by BFD. When both aldehydes are present benzaldehyde is favoured as a donor substrate and 2-HPP and benzoin are the major products. PAC, the prototypical PDC product, which requires the formation of hydroxyethyl-ThDP (HE-ThDP) by addition of acetaldehyde to ThDP (Figure 1), was not observed under the conditions tested, whereas the formation of small amounts of acetoin was observed in the absence of benzaldehyde. This demonstrates that the BFD variants are all able to stabilize HE-ThDP and suggests that reaction with benzaldehyde is simply much faster than reaction with acetaldehyde. The replacement of Ala460 and Phe464 by isoleucine had no effect on the range of products obtained via the carboligation reaction, nor did it affect the yield and enantiospecificity of the benzoin synthesis. However, the yield of 2-HPP decreased about 3-fold and the enantioselectivity of acetoin and 2-HPP formation was also altered appreciably (Table VI). WT BFD and both variants containing single mutations catalyse the synthesis of predominantly (S)-2-HPP. The replacement of Ala460 has the greatest impact on enantioselectivity as evidenced by BFDA460I giving rise to (S)-2-HPP with only 10% enantiomeric excess.
| BFD variant | Acetoin (ee)c | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | n.d. (14 R) | 0 | 2.8 (90 S) | 0.4 (>99 R) |
| A460I | n.d. (53 R) | 0 | 0.7 (90 S) | 0.4 (>99 R) |
| F464I | n.d. (71 R) | 0 | 0.9 (90 S) | 0.4 (>99 R) |
| A460I/F464I | n.d. (82 R) | 0 | 1.1 (50 R) | 0.4 (>99 R) |
| BFD variant | Acetoin (ee)c | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | n.d. (14 R) | 0 | 2.8 (90 S) | 0.4 (>99 R) |
| A460I | n.d. (53 R) | 0 | 0.7 (90 S) | 0.4 (>99 R) |
| F464I | n.d. (71 R) | 0 | 0.9 (90 S) | 0.4 (>99 R) |
| A460I/F464I | n.d. (82 R) | 0 | 1.1 (50 R) | 0.4 (>99 R) |
Yields are reported in mM with enantiomeric excess (%) in parentheses; n.d., not determined.
Reactions were carried out in 500 mM acetaldehyde, 50 mM benzaldehyde, 100 µg/ml enzyme in 50 mM potassium phosphate, pH 7.0, 2 mM MgSO4, 0.1 mM ThDP. The mixture was incubated for 20 h at 30°C before being stopped by heat inactivation of the enzyme (2 min, 94°C). Aromatic products were determined by HPLC. Detection of acetoin was performed by gas chromatography.
Reaction was carried out in 500 mM acetaldehyde only, 1 U/ml enzyme, 16 h, 30°C.
| BFD variant | Acetoin (ee)c | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | n.d. (14 R) | 0 | 2.8 (90 S) | 0.4 (>99 R) |
| A460I | n.d. (53 R) | 0 | 0.7 (90 S) | 0.4 (>99 R) |
| F464I | n.d. (71 R) | 0 | 0.9 (90 S) | 0.4 (>99 R) |
| A460I/F464I | n.d. (82 R) | 0 | 1.1 (50 R) | 0.4 (>99 R) |
| BFD variant | Acetoin (ee)c | PAC (ee) | 2-HPP (ee) | Benzoin (ee) |
|---|---|---|---|---|
| WT | n.d. (14 R) | 0 | 2.8 (90 S) | 0.4 (>99 R) |
| A460I | n.d. (53 R) | 0 | 0.7 (90 S) | 0.4 (>99 R) |
| F464I | n.d. (71 R) | 0 | 0.9 (90 S) | 0.4 (>99 R) |
| A460I/F464I | n.d. (82 R) | 0 | 1.1 (50 R) | 0.4 (>99 R) |
Yields are reported in mM with enantiomeric excess (%) in parentheses; n.d., not determined.
Reactions were carried out in 500 mM acetaldehyde, 50 mM benzaldehyde, 100 µg/ml enzyme in 50 mM potassium phosphate, pH 7.0, 2 mM MgSO4, 0.1 mM ThDP. The mixture was incubated for 20 h at 30°C before being stopped by heat inactivation of the enzyme (2 min, 94°C). Aromatic products were determined by HPLC. Detection of acetoin was performed by gas chromatography.
Reaction was carried out in 500 mM acetaldehyde only, 1 U/ml enzyme, 16 h, 30°C.
By contrast, the WT enzyme gives >90% (S)-2-HPP. Intriguingly, the product obtained with the double mutant has inverted chirality, i.e. the product is (R)-2-HPP. Not only is there a change in stereospecificity, but also the product is obtained in reasonable yield and enantiomeric excess (ee 50%). This variant may therefore provide a strong lead for development of a new chiral catalyst with potential synthetic utility. Surprisingly, given the result just described, the corresponding mutation has no effect on the overall stereochemistry of acetoin formation, although the stereospecificity of the reaction increased from 14% R with WT enzyme to 82% R with BFDA460I/F464I.
It has been shown previously that the enantioselectivity of the (S)-2-HPP synthesis catalysed by BFD is dependent on the benzaldehyde concentration and that the enantioselectivity is significantly reduced when the benzaldehyde concentration is high (Iding et al., 2000). If the carboligation reaction is started using benzoylformate and acetaldehyde as substrates, the benzaldehyde concentration in the batch reaction will increase continuously. This is because decarboxylation is 15-fold faster than carboligation and, potentially, this could result in a progressive decline of the enantioselectivity. To avoid this problem, studies of the carboligase activity of BFD variants were performed using varying amounts of benzaldehyde (rather than benzoylformate) and/or acetaldehyde. This allowed us to determine whether the enantioselectivity of the carboligation reaction catalysed by the BFD variants showed a similar dependence on the benzaldehyde concentration to that catalysed by WT BFD. The results in Figure 6 demonstrate that this effect is even more pronounced with the BFD variants, with the greatest effect occurring as the benzaldehyde concentration was increased from 0.1 to 10 mM. No further change was observed even at 50 mM benzaldehyde. For WT BFD, the ee of (S)-2-HPP decreases by ∼8%, presumably owing to an increase in the rate of (R)-2-HPP formation (Iding et al., 2000). In the BFD variants, the change in ee also corresponds to an increase in formation of (R)-2-HPP and occurs in the order BFDA460I (Δee = 24%) < BFDF464I=BFDA460I/F464I (Δee = 33%). The last, of course, now forms predominantly (R)-HPP (ee = 50% R). It is also of interest that the stereoselectivity of the benzoin synthesis did not exhibit any such dependence on benzaldehyde concentration and (S)-benzoin was never detected as a reaction product, nor has this effect ever been observed with PDC-catalysed carboligations.
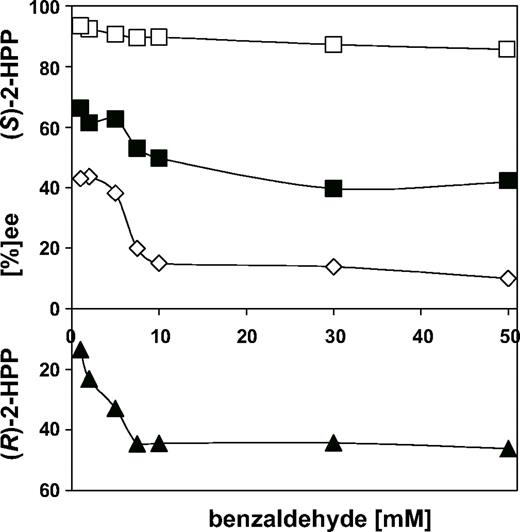
Influence of the benzaldehyde concentration on the enantioselectivity of the 2-HPP synthesis catalysed by BFD variants. Open squares, WT BFD; closed squares, BFDA460I; open diamonds, BFDF464I, closed triangles, BFDA460I/F464I.
These effects can be explained by the model which had been developed earlier for BFD (Iding et al., 2000) and which was recently supported by crystallographic studies (Polovnikova et al., 2003; McLeish et al., 2004). Simplified, the ThDP-bound benzaldehyde (HB-ThDP, Figure 1) can adopt several orientations in the active centre. The lowest energy conformation is one that has favourable aromatic edge-to-face contacts with Phe464 and low steric hindrance. According to the model, addition of acetaldehyde to the low-energy conformation gives rise to the (S)-2-HPP, whereas the R-enantiomer arises as a consequence of acetaldehyde adding to the high-energy conformation. The A460I mutation and, in particular, the F464I mutation removes some of the favourable interactions that stabilize the low-energy conformation, reducing the energy barrier between the two conformations. In the first instance this results in an increase in production of (R)-2-HPP for the BFD mutants. It has also been seen and confirmed in this study that free benzaldehyde in solution can play a role in stabilizing the high-energy conformation. If the energy barrier between the two HB-ThDP conformations is reduced in the BFD variants, it is not unreasonable to expect that any stabilization by benzaldehyde will be more significant for the mutants than for the WT enzyme. Consequently, the larger increases in (R)-2-HPP shown in Figure 6 are not surprising. However, other BFD variants such as H70A, F397A and L473Q (Lingen et al., 2002) do not show this concentration-dependent effect.
Finally, since the BFDA460I showed a significant decarboxylase activity with aliphatic 2-keto acids other than pyruvate (Tables I and IV), the corresponding aldehydes were tested as acyl donors using acetaldehyde as acyl acceptor in carboligation studies. Of four possible products, only the mixed 2-HPP analogue product is formed under these conditions (Figure 5).
Conclusions
With the I472A mutation we were able to introduce some benzoylformate decarboxylase activity into PDC and, although the kcat value for PDCI472A with benzoylformate, 6.9 s−1, is well below that for pyruvate, 200 s−1, the rate is still respectable. The Km value of PDCI472A for benzoylformate is 1.8 mM, which is not dissimilar to the Km values of benzoylformate with WT BFD and pyruvate with WT PDC. Further, we demonstrated that Ile472 plays a significant role in substrate specificity in PDC, but has a limited role in catalysis. Conversely, although Ile476 does have a role in substrate recognition and binding, its more critical role is in positioning the substrate for catalysis.
This approach was also successful in introducing pyruvate decarboxylase activity into BFD. The variant BFDA460I able to decarboxylate pyruvate with the respectable kcat of 5.6 s−1, which is only 10-fold lower than its kcat for benzoylformate. However, pyruvate is bound with only a very low affinity and the kcat value is almost 250-fold lower than that of the WT enzyme with benzoylformate. This suggests that there are more residues which are responsible for specificity towards small aliphatic 2-keto acids. As implied by the structure-based alignment, we have shown that Ala460 in BFD has also a significant impact an the substrate selectivity, as was demonstrated by the changes in Km for aliphatic 2-keto acids.
Overall, by exchanging structurally equivalent active site residues in PDC and BFD we created two effective medium-chain 2-keto acid decarboxylases, PDCI472A and BFDA460I. It is interesting to note a very recent report of the characterization of a branched-chain 2-keto acid decarboxylase (KdcA) from Lactococcus lactis (Smit et al., 2005). This enzyme, KdcA, was able to decarboxylate 4-methyl-2-ketopentanoic acid (8, Table I), the straight-chain keto acids 2, 3 and 4, and also phenyl pyruvate (13). Sequence alignments indicate that the catalytic residues, in addition to the substrate recognition residues of KdCA, are identical with those of PDC, with the exception of Ile472, which has been replaced by a valine (Smit et al., 2005). Although there has been an earlier study on Ile472 variants, PDCI472V was not prepared (Pohl et al., 1998). Clearly this substitution warrants further investigation.
In addition to its decarboxylase activity, PDCI472A is also able to catalyse stereospecifically the ligation of C3–C6 aldehydes with benzaldehyde with moderate yields. Clearly this requires further investigation, but this activity may prove to be valuable in providing building blocks for chiral synthesis. Similarly, the variant BFDA460I/F464I, which shows a reversal of enantioselectivity (Figure 6), may also prove to be of synthetic utility. Our data unambiguously confirm that the decarboxylation and carboligation reactions take place at the same active centre and are influenced by the same active site residues. On that basis, we demonstrate that evaluation of the substrate range for the decarboxylation reaction will yield important hints for the substrate range of the acyl donor in carboligation reactions.
Although we have already demonstrated the power of directed evolution to adopt special enzymatic properties to synthetic requirements (Lingen et al., 2002, 2003), we still regard site-directed mutagenesis based on structural and experimental data as a powerful tool to elucidate evolutionary and mechanistic detail and to design useful catalysts for bioorganic chemistry.
This project was supported, in part, by a Frontiers in Integrative Biological Research grant from the National Science Foundation and by the Deutsche Forschungsgemeinschaft in the frame of SFB 380.
References
Demir,A.S., Dünnwald,T., Iding,H., Pohl,M. and Müller,M. (
Dünkelmann,P., Kolter-Jung,D., Nitsche,A., Demir,A.S., Siegert,P., Lingen,B., Baumann,M., Pohl,M. and Müller,M. (
Fersht,A.R. (ed.) (
Hasson,M.S., Muscate,A., McLeish,M.J., Polovnikova,L.S., Gerlt,J.A., Kenyon,G.L., Petsko,G.A. and Ringe,D. (
Iding,H., Dünnwald,T., Greiner,L., Liese,A., Müller,M., Siegert,P., Grötzinger,J., Demir,A.S. and Pohl,M. (
Krix,G., Bommarius,A.S., Drauz,K., Kottenhahn,M., Schwarm,M. and Kula,M.-R. (
Lingen,B., Kolter-Jung,D., Dünkelmann,P., Feldmann,R., Grötzinger,J., Pohl,M. and Müller,M. (
McLeish,M.J., Kenyon,G.L., Polovnikova,E.S., Bera,A.K., Anderson,N.L. and Hasson,M.S. (
Polovnikova,E.S., McLeish,M.J., Sergienko,E.A., Burgner,J.T., Anderson,N.L., Bera,A.K., Jordan,F., Kenyon,G.L. and Hasson,M.S. (
Siegert,P., Pohl,M., Kneen,M.M., Pogozheva,I.D., Kenyon,G.L. and McLeish,M.J. (
Smit,B., van Hylckama Vlieg,E., Engels,W., Meijer,L., Wouters,J. and Smit,G. (
Tsou,A.Y., Ransom,S.C., Gerlt,J.A., Buechter,D.D., Babbitt,P.C. and Kenyon,G.L. (
Author notes
1Institute of Molecular Enzyme Technology, Heinrich-Heine University of Düsseldorf, Research Centre Jülich, D-52426 Jülich, Germany and 3College of Pharmacy, University of Michigan, Ann Arbor, MI 48109-1065, USA
2Present address: Henkel KgaA, Research Technology, Enzyme Technology, Henkelstrasse 67, D-40191 Düsseldorf, Germany
4Present address: Royal Institute of Technology, Alba Nova, Wood-Biotechnology, Roslagstullsbacken 21, SE-106 91 Stockholm, Sweden
5Present address: Hoffmann-La Roche AG, Emil-Barell-Strasse 1, D-79639 Grenzach-Wyhlen, Germany

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}