





Abstract
Nucleotide sequences from the c-mos proto-oncogene have previously been used to reconstruct the phylogenetic relationships between distantly related vertebrate taxa. To explore c-mos variation at shallower levels of avian divergence, we compared c-mos sequences from representative passerine taxa that span a range of evolutionary differentiation, from basal passerine lineages to closely allied genera. Phylogenetic reconstructions based on these c-mos sequences recovered topologies congruent with previous DNA-DNA hybridization–based reconstructions, with many nodes receiving high support, as indicated by bootstrap and reliability values. One exception was the relationship of Acanthisitta to the remaining passerines, where the c-mos–based searches indicated a three-way polytomy involving the Acanthisitta lineage and the suboscine and oscine passerine clades. We also compared levels of c-mos and mitochondrial differentiation across eight oscine passerine taxa and found that c-mos nucleotide substitutions accumulate at a rate similar to that of transversion substitutions in mitochondrial protein-coding genes. These comparisons suggest that nuclear-encoded loci such as c-mos provide a temporal window of phylogenetic resolution that overlaps the temporal range where mitochondrial protein-coding sequences have their greatest utility and that c-mos substitutions and mtDNA transversions can serve as complementary, informative, and independent phylogenetic markers for the study of avian relationships.
Introduction
The c-mos proto-oncogene is a highly conserved, single-copy, intronless gene that codes for a kinase involved in regulation of the cell cycle in vertebrates (Gebauer and Richter 1997 ; Sagata et al. 1988 ; Sagata 1997 ; Saint et al. 1998 ). c-mos homologs have been sequenced from a diverse array of taxa, and Graybeal (1994) identified c-mos as a potentially useful marker for investigating ancient phylogenetic relationships within vertebrates. Partial c-mos nucleotide sequences have recently been used to explore the timing of divergence among avian orders (Cooper and Penny 1997 ), to investigate phylogenetic relationships within squamate reptiles (Saint et al. 1998 ), and as part of a multilocus analysis of the branching order among reptiles, birds, and mammals (Hedges and Poling 1999 ). These studies indicate that c-mos sequences have utility at relatively deep levels of evolutionary differentiation and suggest that broader investigations of c-mos as a phylogenetic marker are warranted.
Surprisingly few studies have employed nucleotide sequences of nuclear exons as phylogenetic markers for avian systematics (e.g., Caspers et al. 1997 ; Cooper and Penny 1997 ; Groth and Barrowclough 1999 ), and these have generally focused on reconstructing relationships among highly divergent avian lineages. Here, we extend the use of c-mos to explore the more recent differentiation within the order Passeriformes, an avian group of particular interest because it is disproportionally diverse, comprising nearly three fifths of extant bird species (Sibley and Monroe 1990 ). Our sample of 15 passerine genera (table 1 ) provides comparisons across lineages spanning a hierarchical range of differentiation, from representatives of the most ancient passerine clades (e.g., oscines and suboscines) to closely allied genera (e.g., Dendroica and Basileuterus) placed in the same tribe (Sibley and Monroe 1990 ) or family (American Ornithologists' Union 1998 ) in current classifications. Our choice of particular taxa was influenced primarily by the DNA-DNA hybridization–based phylogeny of Sibley and Ahlquist (1990) , which included more than 600 passerine species, and by our own research interests on passerine systematics; figure 1 shows the Sibley and Ahlquist DNA-DNA hybridization–based reconstruction of relationships among the genera included in the present survey of c-mos differentiation.
Analysis of these avian c-mos sequences allowed us to address several interlinked issues. First, we characterized patterns of molecular evolution in the c-mos gene. Second, we compared the concordance of our c-mos trees with previous estimates of relationship based on alternative molecular techniques such as DNA-DNA hybridization. Third, we compared estimates of c-mos divergence with corresponding estimates based on mitochondrial sequences. Finally, we evaluated the utility of c-mos as a phylogenetic marker for passerine molecular systematics and compared the respective temporal windows of phylogenetic resolution offered by c-mos and mitochondrial sequences.
Materials and Methods
Laboratory Techniques
Pectoral muscle tissue was collected and stored in a DMSO/EDTA/NaCl solution (Seutin, White, and Boag 1991 ). Genomic DNA was extracted from each sample using a standard phenol/chloroform protocol (Seutin et al. 1993 ). We used primers described by Cooper and Penny (1997) to amplify a 602-nt portion of the c-mos coding sequence representing all but one of the 15 passerine taxa listed in table 1 . The c-mos fragment corresponds to positions 186–788 (including primer-binding sites) in the published chicken c-mos sequence (GenBank M19412; Schmidt et al. 1988 ). Reaction conditions included an initial 1 min at 95°C; 5 cycles of 30 s denaturation at 95°C, 45 s annealing at 48°C, and 1 min extension at 72°C; and 30 cycles of 30 s denaturation at 95°C, 45 s annealing at 54°C, and 1 min extension at 72°C. PCR products were isolated by electrophoresis in low-melting-point agarose (LMPA) gels and visualized after staining with EtBr; each reaction produced only a single band, which had the expected size. Product bands were excised from the LMPA gel and the agarose was digested with Gelase (Epicentre Technologies). Cycle sequencing reactions were conducted using the amplification primers and Dye-deoxy terminator kits (Applied Biosystems), followed by electrophoresis in a model 377 automated DNA sequencer (Applied Biosystems).
As part of an ongoing investigation of passerine relationships employing mtDNA markers, we also obtained mitochondrial sequences from 8 oscine representatives of the 15 passerine taxa listed in table 1 using protocols similar to those given above and described elsewhere (e.g., Lovette et al. 1999 ). We obtained 2,506 bp of protein-coding mtDNA sequence from each sample, which included the entire coding region of the ATP-synthase 6 (ATPase 6; 684 bp), ATP-synthase 8 (ATPase 8; 168 bp), and NADH dehydrogenase subunit II (NDII; 1,041 bp) genes and a portion of the cytochrome oxidase subunit I gene (COI; 613 bp corresponding to nucleotides 7342–7954 in the chicken mitochondrial genome sequence; GenBank X52392; Desjardins and Morais 1990 ).
c-mos Sequence and Phylogenetic Analysis
Complementary c-mos chromatograms from each individual were aligned with one another, and base calls were confirmed by eye in the program SeqEd to provide 579–582 nt of double-stranded sequence per sample. The corresponding c-mos coding regions from two outgroup species (Gallus and Struthio) and one passerine (Acanthisitta) were obtained from GenBank (Schmidt et al. 1988 ; Cooper and Penny 1997 ) (table 1 ). Except where otherwise specified, our characterizations of c-mos variation excluded the two nonpasserine outgroup taxa. Phylogenetic analyses, on the other hand, were rooted using Gallus and Struthio. Sequences were aligned by eye and imported into the program SEQUENCER (B. Kessing, personal communication) to generate nucleotide and amino acid variation statistics. We used three techniques to reconstruct the phylogenetic relationships among sequences. Maximum-likelihood (ML) searches were conducted using the quartet-puzzling method as implemented in PUZZLE, version 4.0 (Strimmer and von Haeseler 1997 ). We invoked the “exact” parameter estimation option and estimated the transition : transversion ratio, the nucleotide composition, and the gamma rate parameter from the sequence data. Maximum-parsimony (MP) and neighbor-joining (NJ) searches were conducted using Paup*, version 4.0b2 (Swofford 1999 ). Heuristic MP searches were conducted with all nucleotide substitutions weighted equally and with insertions/deletions excluded from analysis, followed by 1,000 heuristic bootstrap replications. Neighbor-joining searches were based on a distance matrix calculated using the Hasegawa-Kishino-Yano (Hasegawa, Kishino, and Yano 1985 ) method with rate variation, with the gamma parameter set to the value identified in the ML search.
Results
c-mos Nucleotide Variation
We obtained 579–582 nt of c-mos sequence corresponding to positions 957–1539 in the Gallus c-mos sequence (GenBank M19412; Schmidt et al. 1988 ) from 14 species of passerine birds, to which we added the single passerine sequence reported previously (Acanthisitta) and two nonpasserine sequences (Gallus and Struthio) (table 1 ). The presence of length variation at only a single codon position rendered the subsequent c-mos nucleotide alignment unambiguous; the resulting alignment has been archived in the EMBL alignment database (accession number DS43356). A total of 150 nucleotide sites varied across the 15 passerine sequences. Among passerines, the maximum uncorrected pairwise divergence at zerofold-degenerate codon positions was 4.9%, whereas the maximum divergence at fourfold-degenerate sites was 28.6%. In comparisons that included the Struthio and Gallus outgroup sequences, the corresponding zerofold and fourfold divergences were 5.6% and 37.6%. Consistent with the predominance of changes at silent positions, the gamma rate parameter was 0.23, indicating considerable rate variation across nucleotide sites. Proportional nucleotide composition was near parity when all codon sites were considered together, but larger biases were apparent when comparisons were made by codon position, especially at third-position sites (table 2 ). All sequences had similar base compositions and biases, and we found no evidence for differences in base composition across passerine taxa using the chi-square test implemented in Paup*, version 4.0b2 (χ2 = 7.48, df = 42, P = 1.0). Across all sites, the transition : transversion ratio estimated by ML was 3.05.
All but 2 of the 15 passerine taxa had c-mos sequences of identical length. The two wood-warblers (Dendroica and Basileuterus) shared a one-codon deletion in comparison with the other sequences in this study, including the outgroup taxa Gallus and Struthio. The deletion corresponds to positions 1263–1265 in the Gallus c-mos nucleotide sequence (GenBank M19412; Schmidt et al. 1988 ). These closely allied wood-warbler genera had almost identical c-mos sequences, and their shared deletion is a probable synapomorphy. The existence of this deletion in the two wood-warblers and not in the three other representatives of the recent nine-primaried oscine radiation indicates that this indel might prove to be a useful character for distinguishing members of the wood-warbler subfamily Parulinae from other nine-primaried oscines. However, comparisons with nonpasserine c-mos sequences indicate that the utility of this indel as a phylogenetic character may be compromised by a high degree of evolutionary lability. This same codon has also undergone deletion in two distantly related nonpasserine birds with otherwise highly dissimilar c-mos sequences, the rhea Rhea pennata (GenBank U88430; Cooper and Penny 1997 ) and the cuckoo Saurothera longinstris (unpublished data), and in one reptile, the skink Lipinia noctua (GenBank AF039465; Saint et al. 1998 ). The presence or absence of sites 1263–1265 represents the only indel observed in published avian c-mos sequences, and the independent loss of this codon in at least three avian lineages indicates that it has the potential for homoplasy, especially in comparisons of distantly related taxa.
As expected from our taxonomic sampling strategy, we noted a broad range of c-mos divergence in pairwise comparisons of passerine taxa. The most similar c-mos sequences were those of the two wood-warblers Dendroica and Basileuterus, which differed at two nucleotide sites (0.4% uncorrected divergence). Small distances also separated these warblers and three other nine-primaried oscine taxa (Icterus, Saltator, and Coereba), which differed at 5–17 nucleotide sites (0.9%–2.9%). The greatest distances among passerines were found in pairwise comparisons between oscine and suboscine genera (44–62 sites; 7.6%–10.7%) and between oscines or suboscines and Acanthisitta (44–61 sites; 7.6%–10.5%). The Gallus and Struthio outgroup taxa differed from the 15 passerines at 53–79 nucleotide sites (9.1%–14.8%).
Phylogenetic Reconstructions and Rate Variation Among Taxa
The c-mos trees reconstructed using ML, MP, and NJ techniques were nearly identical (figs. 2 and 3 ). Differences among these reconstructions involved only the placement of Acanthisitta relative to the other passerines and the branching order among several closely related nine-primaried oscine passerines. These topological differences and their implications for passerine systematics are discussed below.
We used the likelihood ratio technique of Felsenstein (1981) as implemented in PUZZLE, version 4.0, to test for differences in c-mos substitution rates among taxa. We found no evidence of significant rate variation in comparisons of the 15 passerine taxa, in which the difference between unconstrained (−Ln = 2,084.83) and clocklike (−Ln = 2,092.39) ML reconstructions was small (−2ΔLn = 15.12, df = 13, P = 0.30). Rate heterogeneity neared significance, however, in analyses that included the 15 passerines and Gallus and Struthio outgroups (unconstrained −Ln = 2,453.43; constrained −Ln = 2,465.13; −2ΔLn = 23.40, df = 15, P = 0.07) and was highly significant (unconstrained −Ln = 2,247.57; constrained −Ln = 2,296.63; −2ΔLn = 98.12, df = 13, P < 0.01) in comparisons of the 15 more distantly related avian lineages studied by Cooper and Penny (1997) . We note, however, that this apparent rate variation deep in the avian tree does not falsify the primary conclusions of Cooper and Penny (1997) , as their quartet-based technique for calibrating rates of molecular evolution is robust against lineage-specific differences in rates of nucleotide substitution. Nonetheless, although the hypothesis of rate homogeneity within passerines was not falsified, the c-mos locus does not seem to evolve in a clocklike fashion across the most divergent extant avian lineages.
The passerine c-mos trees are largely congruent with those based on DNA-DNA hybridization (fig. 1 ), and thus the c-mos phylogenetic results fit well with and contribute additional support for prevailing views regarding the evolutionary history of these taxa. Although containing several unresolved polytomies, the consensus c-mos tree generated from the results of the ML, MP, and NJ searches (which is identical to the MP consensus tree shown in fig. 3 ) differs from the DNA-DNA hybridization–based tree of Sibley and Ahlquist (1990 , fig. 1 ) only in the relative branching order in the suboscine clade comprising Conopophaga, Myrmotherula, and Formicarius. Similarly, the c-mos and mtDNA trees for oscine passerines are highly congruent (figs. 2–4 ). Despite the relatively short length (582 nt) of the c-mos fragment sequenced, many nodes in the c-mos reconstructions received strong reliability (fig. 2 ) or bootstrap (fig. 3 ) support.
Mitochondrial Variation
To compare levels of sequence variation among nuclear and mitochondrial loci, we obtained mitochondrial sequences from eight oscine passerine taxa (table 1 ). These sequences had typical avian mitochondrial characteristics, including a high transition : transversion ratio (5.8) and a biased nucleotide composition, especially at third codon positions (table 3 ). Gene-specific plots (personal observations) of pairwise mitochondrial differentiation suggested the presence of some heterogeneity in substitution rate across the four mitochondrial genes included in our survey, with the COI gene having the lowest pairwise distances and the ATPase 8 and NDII genes exhibiting the greatest, a trend that probably reflects the more modest amino acid conservation of the ATPase 8 and NDII genes.
Discussion
Phylogenetic Affinities of the Acanthisittidae
The affinities of the Acanthisittidae are a long-standing systematic enigma. Found only in New Zealand, the acanthisittid wrens share a unique suite of morphological characters (Raikow 1987 ; Millener 1988 ; Fedducia 1996) and include one of the two known flightless taxa placed within Passeriformes (Millener 1989 ; Rando, López, and Seguí 1999 ). Recent reviews have highlighted the lack of a clear relationship between the Acanthisittidae and any other group of passerine birds, although affinities with either the oscine or suboscine passerines have been proposed several times based on different morphological characters (see Sibley and Ahlquist 1990 , pp. 578–582). Previous biochemical studies of passerine relationships have also provided ambiguous results regarding the relationships of the acanthisittids to other passerines (Sibley 1970 ; Sibley, Williams, and Ahlquist 1982 ; Sibley and Ahlquist 1990 ).
The c-mos reconstructions provide further evidence that the Acanthisittidae are representatives of an ancient lineage but are ambiguous regarding the placement of the acanthisittid lineage relative to the suboscine or the oscine clades. Instead, the c-mos data suggest that the Acanthisitta lineage became evolutionarily independent at approximately the same time that the suboscine and oscine clades diverged from one another, producing the three-branch polytomy seen in our reconstructions. The uncertainty of relationship resulting from this early passeriform radiation is reflected in the topological differences between the ML, MP, and NJ reconstructions: in the ML tree (fig. 2 ), Acanthisitta is basal to a clade comprising both the oscines and the suboscines; in the MP consensus tree (fig. 3 ), Acanthisitta, the suboscines, and the oscines form a three-way polytomy; and in the NJ tree (fig. 3 ,) Acanthisitta is the sister lineage of the suboscines, with a very short internode separating it from the basal passerine bifurcation. In no reconstruction was the placement of Acanthisitta supported by high bootstrap or reliability scores. Likelihood ratio tests (Kishino and Hasegawa 1989 ) conducted in Paup* indicated no significant difference between the observed ML tree (fig 2 ; −Ln = 2,457.4), in which Acanthisitta is basal to the combined suboscine/oscine clade and trees in which Acanthisitta was constrained to be the basal lineage within either the suboscines or the oscines (for both alternatives, −Ln = 2,458.9; −ΔLn = 1.5, T = 0.6, P > 0.5). Similarly, parsimony criteria identified negligible tree length differences between these three alternative topologies: Acanthisitta (subocines, oscines) = 324 steps; oscines (Acanthisitta, suboscines) = 323 steps; suboscines (Acanthisitta, oscines) = 323 steps.
Although the c-mos reconstructions are inconclusive regarding the sister taxon of the Acanthisittidae, they provide a molecular perspective on the origin of the Acanthisittidae lineage relative to further diversification within the suboscine and oscine clades. In all reconstructions, moderately long internodes separated the root of the Acanthisitta lineage from the earliest sampled bifurcation within either the suboscine or the oscine clade, suggesting that the Acanthisittidae lineage originated before the subsequent and extensive radiations of lineages within these two groups. Although additional sequences representing several groups of enigmatic “Old World suboscines” (taxa in Sibley and Ahlquist's [1990] ‘Infraorder Eurylaimides’) are needed to test the hypothesis that the derivation of the Acanthisittidae preceded the further diversification of extant passerines, the available c-mos evidence is consistent with previous suggestions that the Acanthisittidae represent an ancient lineage relative to other extant passerines. If this hypothesis holds, it would help explain why different morphological characters have provided conflicting results regarding acanthisittid affinities.
Phylogenetic Relationships of Suboscine and Oscine Passerines
The c-mos–based topology for the other 14 passerine taxa is highly congruent both with the traditional classification of these species and with the DNA-DNA hybridization–based reconstruction of Sibley and Ahlquist (1990) . These 14 passerine taxa are divided between two well-defined clades that correspond to their traditional separation into suboscine and oscine groups.
Suboscines
In the c-mos reconstructions, the five suboscine taxa fell into two clades: the tyrannid flycatchers Elaenia and Mionectes, and the antbirds Myrmotherula and Formicarius and the gnatcatcher Conopophaga. This basal suboscine bifurcation is congruent with the phylogeny of Sibley and Ahlquist (1990) , which placed the tyrannids as the sister taxon to all other New World suboscines. The placement of Conopophaga as the sister taxon to Myrmotherula, however, conflicts with the Sibley and Ahlquist tree, in which the clade containing Myrmotherula (their parvorder Thamnophilidae) is basal to a clade (their superfamily Formicariodea) containing both Conopophaga and Formicarius. Short internodes separate these three lineages in both the c-mos (figs. 2 and 3 ) and the Sibley and Ahlquist (fig. 1 ) trees. The c-mos topology relating these three lineages was supported by a high reliability score in the ML analysis (fig. 2 ) but not by high bootstrap values in the MP or NJ reconstructions (fig. 3 ).
Oscines
One initially surprising but now widely accepted systematic revision that followed from Sibley and Ahlquist's (1990) DNA-DNA hybridization studies was the separation of the oscine passerines into two sister groups, the parvorders Corvida and Passerida. The Corvida include a number of morphologically variable taxa endemic to Australia, New Guinea, and Southeast Asia that had previously been grouped into disparate passerine families, plus the New World vireos and the panglobally distributed corvines (Sibley and Ahlquist 1990 ). In the c-mos reconstructions, Vireo and Cyanocorax together form the sister clade to the remaining oscines, thereby supporting Sibley and Ahlquist's Corvida hypothesis. The internode separating the Corvida lineage from the basal bifurcation within the Passerida, however, is short, suggesting that the Corvida lineage separated from the remaining oscines not long before further cladogenesis occurred in the Passerida.
Within the Passerida, the c-mos reconstructions identify a highly supported clade composed of Dendroica, Basileuterus, Icterus, Saltator, and Coereba. These five taxa are all representatives of the so-called “New World nine-primaried oscines” (Sibley and Ahlquist's family Fringillidae), a highly diverse radiation that includes eight tribes in their classification (Sibley and Ahlquist 1990 ). Phylogenetic relationships within the nine-primaried oscines have long been considered particularly problematic because of the recent and explosive diversification of this group (Hellmayr 1935, 1936 ; Mayr and Amadon 1951 ; Beecher 1953 ; Storer 1969 ; Sibley 1970 ; Paynter and Storer 1970 ; Lovette and Bermingham 1999 ). Both the c-mos and mtDNA trees provided high levels of support for two pairs of sister taxa, Dendroica/Basileuterus and Coereba/Saltator.
Comparative Nuclear and Mitochondrial Sequence Divergence
In comparing c-mos and mitochondrial sequences, we were primarily interested in determining relative rates of nucleotide substitution. A second goal was to use c-mos differentiation as a standard to evaluate the rate at which these mitochondrial loci approach saturation. We therefore included in our c-mos survey a set of eight oscine passerine taxa (Vireo, Cinclocerthia, Myadestes, Dendroica, Basileuterus, Saltator, Coereba, and Icterus) from which we had previously obtained long mitochondrial sequences.
The absence of significant c-mos rate variation among passerine lineages supports our underlying assumption that the c-mos gene has accumulated substitutions at an approximately constant rate in these lineages. We therefore used c-mos divergence as a standard against which to measure the relative frequencies of various classes of mitochondrial nucleotide substitution. These comparisons were valuable because they provided a calibration of mtDNA differentiation against an independent locus with a relatively low intrinsic rate of saturation. In figure 5 , we show uncorrected pairwise mitochondrial distances plotted against the corresponding gamma-corrected c-mos divergence values. A striking but expected feature of this graphic analysis is the lack of linearity in the plot of mitochondrial transitions. A plateau at approximately 10% transition distance is reached at a very low level of c-mos differentiation, suggesting that mitochondrial transitions approach complete saturation at a distance that corresponds to one or two c-mos nucleotide substitutions. On the other hand, uncorrected distances based on mitochondrial transversions appear to accumulate at an approximately constant rate for a much longer period before reaching a saturation plateau. The slope of this initial mtDNA transversion : c-mos relationship is approximately 0.8–1.0, suggesting that c-mos nucleotide substitutions accumulate at a rate similar to that of mitochondrial transversion substitutions. These differences in mtDNA transition and transversion saturation rates mirror long-standing assumptions about the molecular evolution of vertebrate mtDNA derived from comparisons of different classes of mitochondrial substitution (e.g., Kocher et al. 1995 ). As has also long been recognized, the rapid saturation of mitochondrial transitions may cause biases in genetic distances calculated by methods that do not account for these saturation effects (e.g., Yang 1997 ). These comparisons suggest that nucleotide saturation effects may cause large underestimates of sequence divergence even between avian taxa that differ by less than 10% uncorrected mitochondrial divergence, a degree of differentiation frequently observed between congeneric avian species and closely related avian genera (e.g., Helbig et al. 1995 ; Klicka and Zink 1997 ; Lovette and Bermingham 1999 ).
Phylogenetic Variation in Passerine c-mos Sequences
The results presented here demonstrate that c-mos sequences provide useful information for reconstructing relationships among the major lineages of passerine birds. This assessment is supported by several lines of evidence, including the generally high bootstrap and reliability values in our c-mos reconstructions (figs. 2 and 3 ) and the high congruence between the c-mos–based reconstructions (figs. 2–3 ) and the corresponding DNA-DNA hybridization (fig. 1 ) and mtDNA (fig. 4 ) trees. A further consideration is the relatively low level of saturation-induced homoplasy seen in our passerine c-mos trees. Given the high amino acid conservation of the c-mos gene, changes in the c-mos sequence are concentrated at synonymous codon positions, and nucleotide saturation effects are therefore likely to be more problematic for studies that address deeper levels of taxonomic divergence (Graybeal 1994 ). For example, extensive molecular evidence (e.g., Hedges and Poling 1999 ) has indicated that crocodilians and birds are sister taxa. When we compared the 375 nt of crocodile c-mos sequence available (GenBank AF039484; Saint et al. 1998 ) with the corresponding passerine c-mos fragment, we found that changes at fourfold-degenerate sites neared complete saturation, showing uncorrected pairwise divergences as high 51%. c-mos nucleotide saturation is therefore likely to introduce a confounding bias to studies of taxa more distantly related than those considered here, especially when the rate of substitution differs across lineages. This problem may be evident even in comparisons among avian orders. For example, we have conducted extensive phylogenetic analyses (unpublished data) of our novel passerine c-mos sequences in conjunction with the 22 avian c-mos sequences analyzed by Cooper and Penny (1997) . In all resulting reconstructions, very short internodes connected the 15 avian orders represented, and at the ordinal level, the topological relationships obtained were extremely sensitive to search model parameterization. These poorly resolved ordinal-level reconstructions therefore provided little information on the relative placement of the passerines relative to other avian orders, a topic of current debate (e.g., Groth and Barrowclough 1999 ; Härlid and Arnason 1999 ; Mindell et al. 1999 ; van Tuinen, Sibley, and Hedges 2000 ).
The results reported here suggest that c-mos, and, by inference, other nuclear-encoded loci, may prove particularly useful for resolving phylogenetic relationships at intermediate levels of avian divergence, as among passerine families. For comparisons within families and particularly among closely allied genera, the information content of c-mos comparisons is constrained by a lack of substitution differences; in comparisons among avian orders or vertebrate classes, homoplastic nucleotide substitutions may be common and prove problematic. Nonetheless, the phylogenetic utility of c-mos at intermediate levels of avian differentiation is auspicious, because it suggests that nuclear-encoded loci provide a temporal window of phylogenetic resolution that overlaps the upper end of the range where mitochondrial protein-coding sequences have their greatest utility (Moore and DeFilippis 1997 ). Furthermore, over a broad range of temporal overlap (fig. 5 ), c-mos substitutions and mtDNA transversion substitutions are likely to represent two informative and independent phylogenetic markers for the study of avian relationships.
Ross Crozier, Reviewing Editor
Present address: Center for Tropical Research, Department of Biology, San Francisco State University.
Keywords: c-mos, mitochondrial DNA Passeriformes phylogeny substitution rates
Address for correspondence and reprints: Irby J. Lovette, Center for Tropical Research, Department of Biology, San Francisco State University, 1600 Holloway Avenue, San Francisco, California 94132. E-mail: ilovette@sas.upenn.edu.
Table 1 Taxa Examined and Tissue Collection Numbers and GenBank Accession Numbers of c-mos and mtDNA Sequences Included in this Study
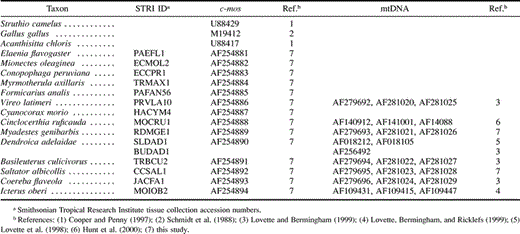
Table 1 Taxa Examined and Tissue Collection Numbers and GenBank Accession Numbers of c-mos and mtDNA Sequences Included in this Study
Table 2 Nucleotide Composition and Bias by Codon Position in Passerine c-mos Sequences
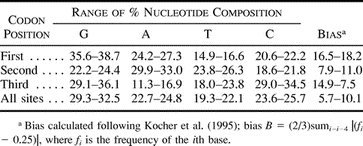
Table 2 Nucleotide Composition and Bias by Codon Position in Passerine c-mos Sequences
Table 3 Range of Nucleotide Composition and Bias in Four Oscine Passerine Mitochondrial Genes
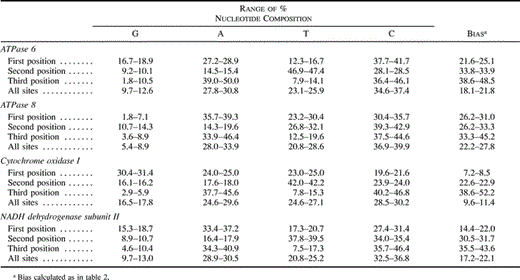
Table 3 Range of Nucleotide Composition and Bias in Four Oscine Passerine Mitochondrial Genes
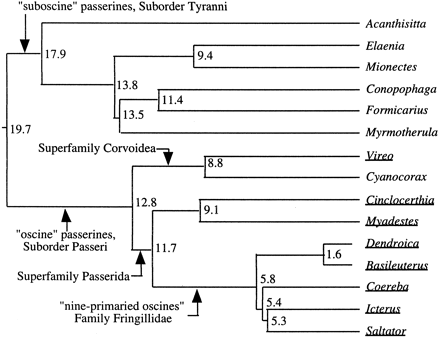
Fig. 1.—DNA-DNA hybridization–based hypothesis of relationship for the 15 passerine genera included in the present survey of c-mos variation (Sibley and Ahlquist 1990 ). Numbers adjacent to nodes indicate delta T50H values from Sibley and Ahlquist's (1990) unweighted pair grouping method with arithmetic means (UPGMA) reconstruction. Relationships of two genera (Cinclocerthia and Saltator) not included in that study are estimated from placement of closely allied genera (Toxostoma and Cardinalis, respectively). Underlined genus names indicate samples included in our mtDNA-versus-c-mos comparisons. Arrows and associated labels at internal internodes indicate some higher taxonomic categories (sensuSibley and Ahlquist 1990 ) referred to in the text
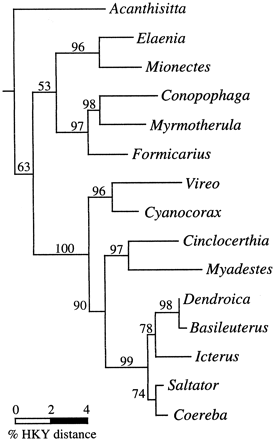
Fig. 2.—Maximum-likelihood tree for 15 passerine genera based 582 nt of exon sequence from the nuclear-encoded c-mos gene. Numbers above internodes indicate quartet puzzling reliability scores (Strimmer and von Haeseler 1996 ). The scale at bottom left indicates the gamma-corrected % Hasegawa-Kishino-Yano distance (Hasegawa, Kishino, and Yano 1985 ) along branches. The reconstruction was rooted using outgroup taxa Struthio and Gallus (not shown)
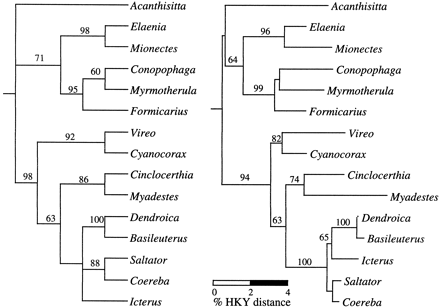
Fig. 3.—Maximum-parsimony (MP; left) and neighbor-joining (NJ; right) trees based on 582 nt of c-mos exon sequence from 15 passerine bird genera. The MP tree shown represents the strict consensus of the four equally shortest (length = 323 steps) trees found. The NJ tree was reconstructed using gamma-corrected Hasegawa-Kishino-Yano (Hasegawa, Kishino, and Yano 1985 ) distances, with branches drawn to the scale shown at bottom left. In both trees, values above internodes represent bootstrap proportions >50%. The reconstructions were rooted using outgroup taxa Struthio and Gallus (not shown)
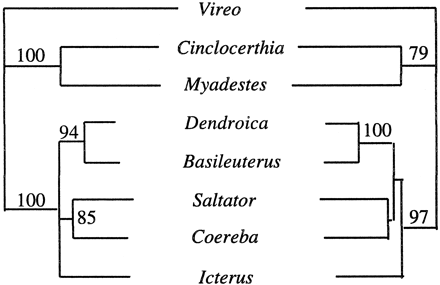
Fig. 4.—Maximum-likelihood (ML; left) and neighbor-joining (NJ; right) trees for eight oscine passerine bird genera based on 2,506 nt of protein-coding mitochondrial DNA sequence per taxon. Numbers above ML internodes indicate quartet-puzzling reliability scores (Strimmer and von Haeseler 1996 ); numbers above NJ internodes indicate bootstrap proportions
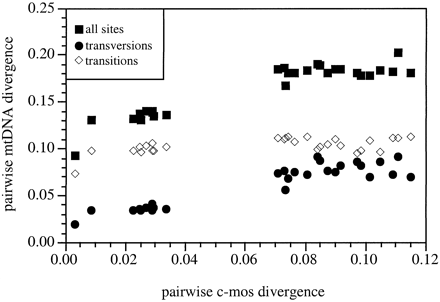
Fig. 5.—Pairwise mitochondrial distances plotted against corresponding pairwise c-mos differentiation. c-mos distances were estimated using the gamma-corrected Hasegawa-Kishino-Yano (HKY; Hasegawa, Kishino, and Yano 1985 ) metric. Uncorrected mitochondrial distances were based on 2,506 nucleotide sequences from the ATPase 6, ATPase 8, cytochrome oxidase I, and NADH dehydrogenase subunit II genes
We are grateful to the regulatory agencies of the Dominican Republic, Ecuador, Honduras, Jamaica, Montserrat, Panama, Puerto Rico, St. Lucia, and Trinidad and Tobago for granting collecting and export permits for the samples studied here. We thank L. Overton for sharing initial aliquots of the Cooper and Penny (1997) primers. J. Eberhard, B. Quinoville, M. Gonzalez, and J. Hunt provided laboratory assistance, B. Kessing shared invaluable insights on data analysis, and two anonymous reviewers provided helpful comments on the manuscript.
literature cited
American Ornithologists' Union.
Caspers, G.-J., U. Uit de Weerd, J. Wattel, and W. W. de Jong.
Cooper, A., and D. Penny.
Desjardins, P., and R. Morais.
Felsenstein, J.
Gebauer, F., and J. D. Richter.
Graybeal, A.
Groth, J. G., and G. F. Barrowclough.
Härlid, A., and U. Arnason.
Hasegawa, M., H. Kishino, and K. Yano.
Helbig, A. J., I. Seibold, J. Martens, and M. Wink.
Hellmayr, C. E.
———.
Hunt, J. S., E. Bermingham, and R. E. Ricklefs.
Kimura, M.
Kishino, H., and M. Hasegawa.
Klicka, J., and R. M. Zink.
Kocher, T. D., J. A. Conroy, K. R. McKaye, J. R. Stauffer, and S. F. Lockwood.
Lovette, I. J., and E. Bermingham.
Lovette, I. J., E. Bermingham, and R. E. Ricklefs.
Lovette, I. J., E. Bermingham, S. Rohwer, and C. Wood.
Lovette, I. J., E. Bermingham, G. Seutin, and R. E. Ricklefs.
Millener, P. R.
———.
Mindell, D. P., M. D. Sorenson, D. E. Dimcheff, M. Hasegawa, J. C. Ast, and T. Yuri.
Moore, W. S., and V. R. DeFilippis.
Paynter, R. A. Jr., and R. W. Storer.
Raikow, R. J.
Rando, J. C., M. López, and B. Segu.
Sagata, N., M. Oskarsson, T. Copeland, J. Brumbaugh, and G. F. Vande Woude.
Saint, K. M., C. C. Austin, S. C. Donnellan, and M. N. Hutchinson.
Schmidt, M., M. K. Oskarsson, J. K. Dunn, D. G. Blair, S. Hughes, F. Propst, and G. F. Vande Woude.
Seutin, G., J. Brawn, R. E. Ricklefs, and E. Bermingham.
Seutin, G., B. N. White, and P. T. Boag.
Sibley, C. G.
Sibley, C. G., and J. E. Ahlquist.
Sibley, C. G., and B. L. Monroe.
Sibley, C. G., G. R. Williams, and J. E. Ahlquist.
Strimmer, K., and A. von Haeseler.
———.
Swofford, D. L.
van Tuinen, M., C. G. Sibley, and S. B. Hedges.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}