





Abstract
Using “long-PCR,” we amplified in overlapping fragments the complete mitochondrial genome of the tapeworm Hymenolepis diminuta (Platyhelminthes: Cestoda) and determined its 13,900-nt sequence. The gene content is the same as that typically found for animal mitochondrial DNA (mtDNA) except that atp8 appears to be lacking, a condition found previously for several other animals. Despite the small size of this mtDNA, there are two large noncoding regions, one of which contains 13 repeats of a 31-nt sequence and a potential stem-loop structure of 25 bp with an 11-member loop. Large potential secondary structures were identified also for the noncoding regions of two other cestode mtDNAs. Comparison of the mitochondrial gene arrangement of H. diminuta with those previously published supports a phylogenetic position of flatworms as members of the Eutrochozoa, rather than placing them basal to either a clade of protostomes or a clade of coelomates.
Introduction
Over 100 complete metazoan mitochondrial genome sequences have been published to date (Boore 1999 ; Mitochondrial Genomics link at http://www.jgi.doe.gov). Nearly all contain genes for the same 13 proteins, 22 tRNAs, and 2 rRNAs. Among triploblastic animals, there is only one gene, atp8, that has been lost from this repertoire, three times independently: once from the mtDNAs of four nematodes (Okimoto et al. 1991, 1992 ; Keddie, Higazi, and Unnasch 1998 ), once from the mtDNAs of platyhelminths (Le et al. 2000 ), and once from the mtDNA of the bivalve mollusk Mytilus edulis (Hoffmann, Boore, and Brown 1992 ). All metazoan mtDNAs have at least one noncoding region that is thought to contain signaling elements for regulating replication and/or transcription; in some cases, potential secondary structures are associated with these signals (Shadel and Clayton 1997 ).
Although metazoan mitochondrial sequences are known to evolve rapidly, their gene arrangements often remain unchanged over long periods of evolutionary time. Comparison of the relative arrangements of these genes has several merits for reconstructing ancient evolutionary relationships, including their generally slow rate of change and the large number of potential gene orders (Boore and Brown 1998 ). With some notable exceptions, gene arrangements are relatively stable within major groups but variable between them, and arrangement comparisons have resolved several evolutionary relationships, including those among arthropods (Boore et al. 1995 ; Boore, Lavrov, and Brown 1998 ; Dowton 1999 ), annelids (Boore and Brown 2000 ), gastropods (Kurabayashi and Ueshima 2000), and echinoderms (Smith et al. 1993 ), and placed brachiopods with the eutrochozoans (Stechmann and Schlegel 1999 ).
The superphyla Deuterostomia (e.g., Chordata, Echinodermata, Hemichordata) and Protostomia (e.g., Arthropoda, Annelida, Mollusca) each appear to have evolved from an ancestor with a coelomic body cavity. Based largely on the view that the coelom evolved once, in their common ancestor, they are traditionally united in the clade Coelomata to the exclusion of the (presumably) earlier branching Platyhelminthes (Hyman 1951 ). Platyhelminths share a number of developmental features with protostomes, such as spiral cleavage, the mesoderm originating from a mesentoblast, and the mouth arising from the blastopore. In the traditional phylogenetic scheme, these shared features are interpreted as primitive for all of the Coelomata, with the Deuterostome variations being derived later for that superphylum. However, others view these shared features as indicating that the Platyhelminthes evolved within the Protostomia, requiring the interpretation either that the coeloms of deuterostomes and protostomes are of separate origins or that Platyhelminthes have lost their coelom secondarily. Most who favor this latter phylogeny place Platyhelminthes as an early offshoot within Protostomia (see Brusca and Brusca 1990 ), but some recent studies (Aguinaldo et al. 1997 ; Balavoine 1997, 1998 ; Carranza, Baguñà, and Riutort 1997 ) have concluded that Platyhelminthes (or just those other than the Acoela [Ruiz-Trillo et al. 1999] ) are, instead, allied specifically with the group of coelomate protostomes dubbed the Eutrochozoa, e.g., Mollusca and Annelida, but not Arthropoda (Ghiselin 1988 ).
Until recently, there were no complete mitochondrial sequences representing the phylum Platyhelminthes; however, these sequences are now available for the cestodes Taenia crassiceps (Le et al. 2000 ) and Echinococcus multilocularis (GenBank record AB018440) and for the trematodes Fasciola hepatica (Garey and Wolstenholme 1989 ; Le et al. 2000 ; GenBank record AF216697), Schistosoma mansoni (Blair et al. 1999 ; Le et al. 2000 ), Schistosoma japonicum, Schistosoma mekongi, and Paragonimus westermani (Le et al. 2000 ). However, these studies have emphasized specific comparative aspects without yet making a detailed genome description. We analyze and describe here the complete mitochondrial genome sequence of a platyhelminth, the tapeworm (i.e., class Cestoda) Hymenolepis diminuta, and compare aspects of this genome with those already published. Comparison of the mitochondrial gene arrangement of H. diminuta with all others available supports the view that platyhelminths are eutrochozoans.
Materials and Methods
Determination of the mtDNA Sequence
Individuals of the tapeworm H. diminuta were collected from artificially infected rat gut and preserved in hot 70% ethanol. About 10 posterior proglottids from each of five adult cestodes were longitudinally disrupted with sterilized forceps. Eggs were collected from these disrupted proglottids and pelleted by centrifugation in order to separate them from the ethanol supernatant. The eggs were then crushed in 150 μl CTAB buffer using a pestle fitting a 1.5-ml eppendorf tube, followed by addition of 450 μl CTAB buffer supplemented with 50 μg Proteinase K and incubated at 65°C for 2 h. Proteins were removed by extraction with phenol/chloroform, and the DNA was precipitated by adding 50% (v/v) 7.5 M ammonium acetate and an equal volume of 100% ethanol. After centrifugation and washing of the pellet with 70% ethanol, it was dried under a vacuum and resuspended in 50 μl ddHOH.
Initially, a short fragment (∼450 nt) of H. diminuta cob was amplified by PCR using AmpliTaq DNA polymerase (Fisher) and 40 cycles of 94°C for 15 s, 53°C for 30 s, and 72°C for 2 min, followed by an incubation at 72° for 10 min. Amplifying primers (Cytb-424F [GGW TAY GTW YTW CCW TGR GGW CAR AT] and Cytb-876R [GCR TAW GCR AAW ARR AAR TAY CAY TCW GG]) were designed based on sequences well conserved in many distantly related taxa. The amplification product was visualized on a 1% agarose gel by ethidium bromide staining and UV irradiation and then purified by three serial passages through Ultrafree spin columns (Millipore; 30,000 NMWL) and used as a template for dRhodamine dye-terminator cycle sequencing reactions (Perkin-Elmer). Sequence was resolved using an ABI 310 automated DNA sequencer.
Six oligonucleotide primers were then designed for “long-PCR” (Barnes 1994 ). The first two (Hym-Cytb-F [GCA CAA GAG TGG TAG TAA AAA TCC C] and Hym-Cytb-R [CGA ACA AGG GTG AAA CCC GTA ACA G]) matched the ends of the cob sequence as determined above. Two others (Hym-12S-F [TTT AGG ACT TGA TAG TAG GGT AGA C] and Hym-12S-R [ATC GTC CTT TAT AAC ACA CCT TCC C]) matched each end of a previously published fragment of H. diminuta rrnS (von Nickisch-Rosenegk, Lucius, and Loos-Frank 1999 ). The remaining pair (Hym-16S-F [TTA TAA ATG GCC GCA GTA TAT TGA C] and Hym-16S-R [AGG CAA TTA ATT ATG CTA CCT TYG C]) was designed from an alignment of three cestode DNA fragments (450 bp of Echinococcus multilocularis, Catenotaenia lobata, and Schistocephalus solidus; unpublished data). In each case, these primers face “out” from the fragments. These primers were used in all 12 possible combinations with rTth-XL DNA polymerase (Perkin-Elmer). Reaction conditions were optimized for Mg++ concentration and annealing temperature as necessary. Visualization, purification, and sequencing of these products were as above, except that an ABI 377 automated sequencer was used for most reactions, with additional oligonucleotides being used for “primer walking.” All sequences were determined on both strands.
Analysis
Sequences were produced and assembled using the ABI suite of programs (e.g., Sequencing Analysis and Sequence Navigator). Subsequent manipulations used MacVector, version 6.5 (Oxford Molecular Group), and GCG (Wisconsin Package, version 10.0, Genetics Computer Group, Madison, Wis.). Twelve protein-encoding genes and two ribosomal RNA genes were easily identified by comparisons with similar published sequences. Twenty-two tRNA genes were identified by eye based on their potential for forming tRNA-like secondary structures; specific identities were assigned by anticodon sequences.
Mitochondrial gene arrangements were compared by eye for gene adjacencies in all pairwise combinations for H. diminuta, E. multilocularis (GenBank record AB018440), F. hepatica (Garey and Wolstenholme 1989 ; Le et al. 2000 ; GenBank record AF216697), S. mansoni (Blair et al. 1999 ; Le et al. 2000 ), S. japonicum, S. mekongi, T. crassiceps (Le et al. 2000 ), Lumbricus terrestris (Boore and Brown 1995 ), Platynereis dumerilii (Boore and Brown 2000 ; GenBank AF178678), Helobdella robusta (partial), Galathealinum brachiosum (partial) (Boore and Brown 2000 ), Limulus polyphemus (Lavrov, Boore, and Brown 2000 ), Tetilla sp. (partial) (Watkins and Beckenbach 1999 ), and the gene arrangement previously inferred to be primitive for Vertebrata (Boore 1999 ; Boore, Daehler, and Brown 1999 ). Each of the three contending phylogenetic positions of the Platyhelminthes (see above and below) was evaluated by eye for whether these sets of shared gene boundaries were consistent or homoplastic.
Results and Discussion
Amplification of the Complete H. diminuta mtDNA
Five combinations of H. diminuta–specific primers yielded single fragments from the long-PCR reactions: Hym-Cytb-F and Hym-12S-R (9 kb); Hym-Cytb-F and Hym-16S-R (8 kb); Hym-Cytb-R and Hym-16S-F (6 kb); Hym-Cytb-R and Hym-12S-F (5 kb); and Hym-16S-F and Hym-12S-R (1 kb). Together, these five fragments and the shorter fragments of cob and rrnS jointly represent the entire mitochondrial genome in overlapping fragments. The composite sequence revealed a genome of 13,900 nt, among the smallest so far observed (GenBank accession number AF314223).
Gene Content and Organization
The 2 rRNA genes, 22 tRNA genes, and 12 of the 13 protein-encoding genes typical of metazoan mtDNAs were identified for H. diminuta by comparisons of their sequence similarity and/or potential secondary structures with those of other metazoans. Their arrangement is shown in figure 1 . One protein gene, atp8, typically found in animal mtDNAs, appears to be missing from this mtDNA (see below), as has been inferred for each of the other completely sequenced flatworm mtDNAs reported (Le et al. 2000 ; GenBank record AB018440). Gene arrangements are substantially similar among the studied flatworm mtDNAs; details are presented below.
All genes are encoded by the same strand, a condition also found for the other flatworms (Le et al. 2000 ; GenBank record AB018440), annelids, two bivalve mollusks, nematodes, brachiopods, and a sea anemone (see Boore 1999 ; Mitochondrial Genomics link at http://www.jgi.doe.gov); all other mtDNAs studied so far have these genes distributed between the two strands. The mitochondrial genes of H. diminuta are generally small. Most are comparable with the reduced sizes of those in nematodes (Okimoto et al. 1991, 1992 ; Keddie, Higazi, and Unnasch 1998 ), except for atp6 and cox3, which are each smaller in H. diminuta by about 100 nt.
Absence of atp8
After careful analysis of the H. diminuta mtDNA sequence, the only open reading frame (ORF) that is a candidate for atp8 appears to be (mostly) within the 183-nt region otherwise described as a noncoding region (below). Designated ORF52, it is equivalent to 52 codons (plus a stop codon) in length, similar to the typical size of atp8, beginning 34 nt after trnY and overlapping trnS2(uga) by 10 nt (fig. 2 ). This ORF starts with TTG, but the next codon is ATA, a more commonly used start codon for mitochondrial protein genes. It ends with TAA, although potential abbreviated stop codons (see below) are present that could truncate this ORF by one, two, or nine residues, thus requiring less or no overlap with the downstream trnS2(uga). However, there are several reasons to doubt that ORF52 is the atp8 gene. Blast searches and direct comparisons with published atp8 sequences and with the sequence of all published flatworm mtDNAs using both nucleotide and inferred amino acids reveal no similarity to this or to any other published mitochondrial gene. This lack of similarity also extends to the hydrophilicity profile, which is a well conserved feature of this protein (Watkins and Beckenbach 1999 ). Characteristic amino acid signatures of the Atp8 protein (e.g., WXW near the carboxyl terminus) are not present in ORF52. Comparisons of the entire sequence of H. diminuta with published sequences of atp8 genes, as well as with the corresponding protein sequences, reveal no similarity. The gene for atp8 is therefore assumed to be absent, and it seems unlikely that ORF52 encodes any functional peptide, considering its small size, A+T-richness, and internal repeat (see below).
Several other mtDNAs are also missing atp8, almost certainly as independent losses (see above). For each of these cases, it is unknown whether atp8 moved to the nucleus with its protein product imported to function within mitochondria or simply became dispensable.
Initiation and Termination Codons
For H. diminuta mtDNA, 10 of the 12 protein genes can be inferred to initiate with ATG without overlap of the upstream gene. The exceptions are nad4, which appears to use ATT, and cox1, which appears to use GTT. In each case, there are no other eligible start codons that are reasonable alternatives, and the inferred amino terminal residues are similar to those of their homologs in other metazoans. The inference for nad4 requires overlap with the upstream nad4L, as is common for these two genes among metazoan mtDNAs. Although GTT is not usually designated as an eligible start codon, cox1 is most extreme among mitochondrial proteins with regard to initiating with unusual start codons (Wolstenholme 1992 ).
Abbreviated stop codons (T and TA) are also common among metazoan mitochondrial protein genes, where posttranscriptional polyadenylation forms a complete UAA stop codon (Ojala, Montoya, and Attardi 1981 ). For H. diminuta mtDNA, 11 of the 12 ORFs end with a complete stop codon (nine are TAG and two are TAA). Only cox1 is inferred to end with an abbreviated stop codon, although there is an in-frame TAG stop codon that, if used, would require overlap with the downstream tRNA gene by 10 nt.
Genetic Code
The mitochondrial genomes of most studied invertebrates appear to deviate from the “universal” genetic code with regard to the identities of ATA, TGA, and AGR codons (see Wolstenholme 1992 ). Further differences have been inferred for Platyhelminthes such that AAA encodes N rather than K, ATA encodes I (as in the universal code) rather than M, and TAA encodes Y rather than termination (Bessho, Ohama, and Osawa 1992 ).
The identities of AAA and of ATA in H. diminuta mtDNA appear to be consistent with these flatworm deviations. Pairwise comparisons using the two best conserved genes, cob and cox1, identified the codons corresponding to AAA of H. diminuta for L. terrestris (AAY = 6; AAR = 2), Katharina tunicata (AAY = 4; AAR = 0), and Drosophila yakuba (AAY = 4; AAR = 0) homologs. Similar comparisons for H. diminuta ATA codons determined their matches in L. terrestris (ATY = 9; ATR = 0), K. tunicata (ATY = 6; ATR = 0), and D. yakuba (ATY = 6; ATR = 0). There are weaknesses of such inferences: the pairwise comparisons are nonindependent due to the evolutionary relationships of the taxa, and it is not clear whether mutation bias or selection for amino acid composition can be stronger influences on substitution than the tendency to synonymous substitutions. However, acknowledging these limitations, we tentatively assign AAA and ATA as N and I, respectively, for H. diminuta mtDNA.
The third case that has been suggested as a deviation for flatworm mtDNAs, that in which TAA encodes Y rather than termination, does not seem possible for H. diminuta. No TAA codon is found in-frame for any protein-encoding gene. Two genes, cox2 and nad6, are predicted to end on a TAA stop codon, and one gene, cox1, is predicted to end on an abbreviated stop codon that is completed to UAA posttranscriptionally (fig. 2 ). Each of these genes would greatly overlap their adjacent downstream gene if they were to extend to the first in-frame TAG codon.
Nucleotide and Amino Acid Composition
Hymenolepis diminuta mtDNA is very A+T-rich (71%), even for an animal mitochondrial genome. The distribution of the purines versus the pyrimidines between the strands for each TA and GC pair is highly biased, with the reported strand being very rich in T and G. The values calculated for TA-skew ([T − A]/[T + A]) and GC-skew ([G − C]/[G + C]) (Perna and Kocher 1995 ) are 0.29 and 0.33, respectively. (Skew values range from +1 to −1; the value is 0 if the strands have no skew.) The nucleotide composition of the reported strand is 25.4% A, 45.6% T, 19.3% G, and 9.6% C. Each of these values is even more extreme for the subset of nucleotides in the third positions of codons (table 1 ), which are presumably more free to change.
Transfer RNA Genes
Most of the tRNA gene sequences can be folded into conventional secondary structures with features common to mitochondrial tRNAs (fig. 3 ). Notable exceptions are tRNA(S1), tRNA(S2), and tRNA(R), which have an unpaired DHU-arm. Additional base-pairing for a longer anticodon stem is possible for each of these three tRNAs, a condition found for tRNA(S1) in other mtDNAs (e.g., Boore and Brown 1994, 2000 ).
Anticodon nucleotides are identical to those most commonly found for the corresponding tRNA genes in other mtDNAs with two exceptions. tRNA(K) for H. diminuta mtDNA, as for each of the other seven flatworms whose mtDNAs have been completely sequenced, has a CTT anticodon, rather than the more common TTT. Having C here in the wobble position would be consistent with the inferred genetic code variation such that only AAG (rather than AAR) specifies lysine. However, it is unclear how the GTT anticodon of tRNA(N) for all eight of these flatworm mtDNAs would recognize not only AAY, as for the typical code, but also AAA for flatworms. More unusually, the codon of tRNA(R) is ACG, since A in the wobble position is very uncommon. Arginine is specified by all four CGN codons, requiring this A to pair with all four nucleotides (this anticodon is normally TCG). The genes for tRNA(R) of the other two studied cestodes, E. multilocularis (GenBank AB018440) and T. crassiceps (Le et al. 2000 ), also have these two unusual features of A in the wobble position and an unpaired DHU arm (fig. 3 ). However, all of the five studied trematodes have a tRNA(R) with potential for a standard cloverleaf structure and a TCG anticodon (not shown). Whether any nucleotides of these tRNAs are posttranscriptionally modified is unknown.
Noncoding Sequences
Despite its small size, H. diminuta mtDNA contains two noncoding regions of significant size. The larger, of 444 nt, is between nad5 and trnG and consists of 12 identical repeats of a 31-nt sequence (372 nt) plus one repeat with a single-nucleotide difference. Part of this 13th repeat can form a stem-loop with the remaining 41 nt of this noncoding region. This structure has 25 canonical base pairs and a loop of 11 nt (fig. 4 ) and terminates at the beginning of trnG with an overlap of 2 nt. The single-nucleotide difference in this 13th unit is a T-to-C transition at nucleotide 16 of the repeat, creating a perfect match at this position of the stem. This is similar to reported repeat sequences of the nematode Meloidogyne javanica, which contains 11 identical repeats of a 63-nt unit, with the last copy modified by a single substitution, and which can also potentially form a stem-loop structure (Okimoto et al. 1991 ). Large potential secondary structures can be found also between nad5 and trnG (as for H. diminuta) and between trnY and trnL1 for each of the cestodes E. multilocularis and T. crassiceps (fig. 4 ). In some of these cases, there is potential for competing, mutually exclusive hairpins such as those identified in L. terrestris mtDNA (Boore and Brown 1995 ). No similar structures could be identified for the noncoding regions of any of the trematode mtDNAs. The function of this region, if any, is unknown; however, similar stem-loop structures have been shown to initiate replication in mammals (e.g., Monnerot, Solignac, and Wolstenholme 1990 ), and it is possible that the putative structure in this region serves a similar function in other metazoans.
A second region, of 183 nt, is located between trnY and trnS2(uga). Although it contains an ORF of 52 amino acids (discussed above), it seems more likely to be a noncoding region. No significantly stable potential secondary structure could be folded using these 183 nt, which are 81% A+T. A further feature of this noncoding region in H. diminuta is a 36-nt tandem repeat. Further small intergenic regions were found, with the largest being between trnW and cox1 (28 nt) and between trnL1(uag) and trnL1(uaa) (26 nt). Whether any of these noncoding regions serve any function awaits further study.
Phylogenetic Reconstruction Using Gene Arrangements
The mitochondrial gene arrangements of two cestodes, T. crassiceps and E. multilocularis (Le et al. 2000 ), are identical and differ from that of H. diminuta only by the reversal in order of trnS2 and trnL1. The trematodes S. japonicum, S. mekongi, and F. hepatica (Le et al. 2000 ) have these two tRNA genes in the same order as in T. crassiceps and E. multilocularis and otherwise differ from H. diminuta only in the positions of trnE, trnV, and trnW for the schistosomes and the position of trnE for F. hepatica. The African schistosome S. mansoni has several additional gene rearrangements (Le et al. 2000 ); these must be derived independently for this lineage based on the similarity among the cestodes and the other schistosomes, whose shared arrangements are parsimoniously inferred as primitive. Furthermore, the arrangement of the genes within the portion spanning from trnP to trnW of H. diminuta is also identical in three other cestodes whose mtDNAs have been only partially sequenced (Diphyllobothrium nihonkaiens [GenBank accession AB006205], Spirometra erinacei [GenBank accession AB006204], and S. proliferum [GenBank accession AB006203]) except for the lack of trnS1(gcu).
Hymenolepis diminuta has two pairs of adjacent genes that have been commonly found together among animal mtDNAs (see Boore 1999 ), nad4L-nad4 and trnL1(uag)-trnL2(uaa). Although the significance is doubtful, it also shares single-gene boundaries with several nematodes: trnQ-trnF, as in Caenorhabditis elegans and Ascaris suum (Okimoto et al. 1992 ), and cox1-trnT and trnV-trnA, as in M. javanica (Okimoto et al. 1991 ). The most noteworthy gene arrangement similarities, however, are to several animals within the proposed higher-level group Eutrochozoa (e.g., mollusks and annelids; Ghiselin 1988 ).
The arrangement of the genes in H. diminuta from nad1 through trnS1(gcu) is nearly identical to that found among several annelids, including one previously considered to be of the phylum Pogonophora (Galathealinum brachiosum) (Boore and Brown 1995, 2000 ) (fig. 5 ) and, except for trnS1(gcu), in an echiuran (Urechis caupo; unpublished data). Although shared single-gene boundaries are less strong as a phylogenetic character (Boore and Brown 1998 ), it is worth noting that all of these taxa also share the gene arrangement trnG-cox3.
The three most commonly proposed evolutionary positions for the Platyhelminthes are (1) basal to a clade of coelomate animals (fig. 5A ), (2) basal to protostomes only (fig. 5B ), and (3) derived eutrochozoans (fig. 5C ) (see above and below for references and significance). For several of the genes whose arrangements are identical among some platyhelminths and annelids (representative of the Eutrochozoa), a different arrangement is shared among chordates and arthropods. The positions for Platyhelminthes depicted in figure 5A or B would require either identical gene translocations or reversions to a previous arrangement in independent lineages for multiple genes. Furthermore, a small portion of a poriferan mtDNA has been reported that contains only one of the relevant genes; since this group is indisputably an outgroup to the other taxa in figure 5 , the primitive position of trnK can be inferred. All possible pairwise comparisons of the gene arrangements for the taxa considered here (see fig. 5 caption) were made. No gene arrangements are shared in a pattern to support the relationships of figure 5A or B. There are no homoplastic gene rearrangements for the tree shown in figure 5C. All of the gene arrangements identified as being informative are found in H. diminuta, T. crassiceps, E. multilocularis, and F. hepatica, and all but the single boundary nad3-trnS1 are found in S. japonicum and S. mekongi.
However, none of the informative gene boundaries are found in S. mansoni mtDNA except for trnG-cox3. This is the basis for the cautionary message of Le et al. (2000). It is important to note that if the rearranged genome of S. mansoni had been the only one sampled from platyhelminths, there would have been a lack of phylogenetic resolution but still no homoplasy in this analysis. Although the generally slow rate of gene rearrangement in animal mtDNAs (Boore 1999 ) makes it more likely that the signal of ancient relationships will be preserved, occasionally rapid rearrangements are not expected to be misleading in phylogenetic analysis. Even if there are many changes in some lineages, the great number of potential arrangements and the lack of apparent “hot spots” (Boore and Brown 1998 ) make convergence unlikely, especially for multiple rearrangements (notable exceptions to this may be in apparent movements of noncoding sequence [Mindell, Sorenson, and Dimcheff 1998] or in exchange of nearest-neighbor tRNA genes [Boore and Brown 1998 ; Boore 1999] ). Cladistic analysis as used here prevents artifactual clustering of taxa based on shared primitive characters (i.e., the “short” branches) but, rather, unites taxa only on the basis of shared, derived gene rearrangements parsimoniously inferred to have occurred only once in their common ancestor.
Arrangements of this set of genes in taxa related to those in figure 5 are consistent with this inference of evolutionary relationships. Echinoderms are thought to be the sister taxon to Chordates on the tree in figure 5C ; all echinoderm mtDNAs studied so far share the gene arrangements cox2-trnK-atp8 and trnL2(uaa)-nad1-trnI with chordates. There is slight similarity in the gene arrangements of H. diminuta with a few mollusks which would also be expected to be included in the clade of Eutrochozoa. In H. diminuta, the gene arrangement is nad1-trnN-trnP, whereas the arrangement is nad1-trnP in Dentalium eboreum, Nautilus sp. (unpublished data), Loligo bleekeri (GenBank AB009838), and Katharina tunicata (but with trnP in opposite orientation; Boore and Brown 1994 ). Finally, it may be noteworthy that three of the four tRNA genes in the cluster preceding nad3 in H. diminuta are the same, although in different arrangements, as those preceding nad3 in K. tunicata (Boore and Brown 1994 ) and Nautilus sp. (unpublished data).
The position of the Platyhelminthes in the system of Metazoa has been debated for decades (Brusca and Brusca 1990 ). Traditionally, the coelom has been viewed as a feature uniting the protostomes and deuterostomes to the exclusion of Platyhelminthes (Hyman 1951 ), although this notion is questioned by those who view the mechanisms of schizocoely (as for protostomes) and enterocoely (as for deuterostomes) as fundamentally different and therefore of separate origins. Some investigators view the simplicity of the Platyhelminthes as a secondary reduction from a coelomate ancestor (Ax 1963 ; Smith and Tyler 1985 ). Since platyhelminths share some of the basic characters of protostomes, such as spiral cleavage, the mesoderm originating (at least in part; see Boyer, Henry, and Martindale 1996 ) from the 4d mesentoblast, and the mouth arising from the blastopore, some include them in Protostomia but generally as basal to the other, presumably derived, schizocoelomate phyla (see Brusca and Brusca 1990 ).
However, an alternative interpretation is that Platyhelminthes are derived within the Eutrochozoa, a group of protostomes including annelids and mollusks, but not arthropods (Ghiselin 1988 ). This view is supported by ultrastructural analysis (Rieger 1986 ), by some sequence comparisons (Aguinaldo et al. 1997 ; Balavoine 1997 ; Carranza, Baguñà, and Riutort 1997 ; Ruiz-Trillo et al. 1999 ), and by our gene arrangement comparisons here. Further work on those flatworms comprising the Acoela may determine whether this group also occupies such a phylogenetic position or instead is basal among animal life, as has been inferred by DNA sequence comparisons (Ruiz-Trillo et al. 1999 ).
Rodney Honeycutt, Reviewing Editor
Keywords: Hymenolepis tapeworm phylogeny mitochondria evolution genome
Address for correspondence and reprints: Markus von Nickisch-Rosenegk, Molecular Parasitology, Humboldt-University at Berlin, Philippstrasse 13, 10115 Berlin, Germany. laforge@rz.hu-berlin.de.
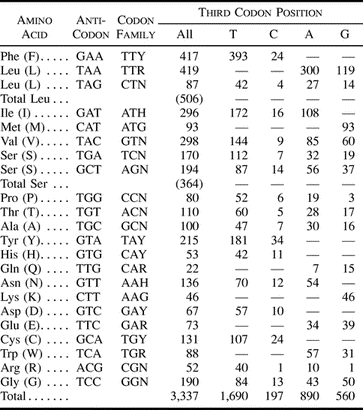
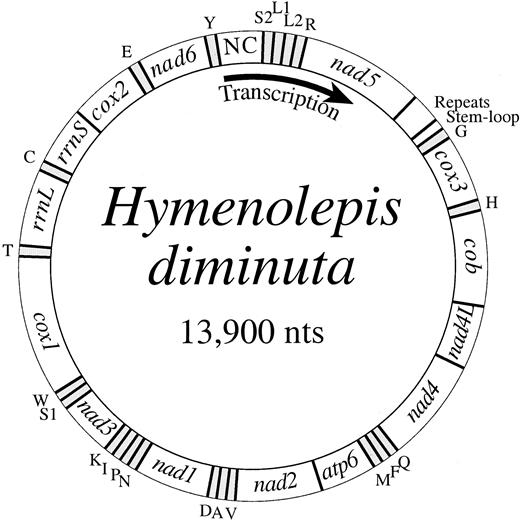
Fig. 1.—Arrangement of the mitochondrial genome of the tapeworm Hymenolepis diminuta. Gene scaling is only approximate. All genes are coded by the same DNA strand and are transcribed clockwise. The origin(s) of transcription remains undetermined. All genes have standard nomenclature except for the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the two leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGN and UCN, respectively). “NC” refers to a largest noncoding region.

Fig. 2.—A partly schematic representation of the 13,900-nt mtDNA sequence of Hymenolepis diminuta. To conserve space, most of the center portions of the larger genes have been replaced with a numeral indicating the number of omitted nucleotides. For cox1 and for nad4, the initiator methionine is placed in parentheses to indicate presumed nonconformity to the genetic code. Asterisks indicate stop codons, either complete or abbreviated (see text).
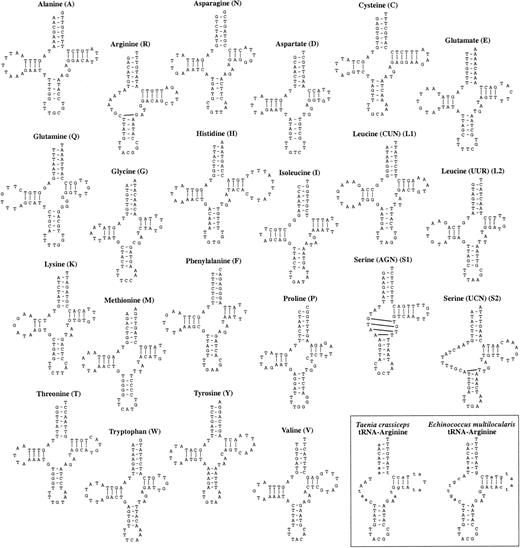
Fig. 3.—DNA sequences for the 22 tRNA genes of Hymenolepis diminuta mtDNA folded into inferred secondary structures. For the three tRNAs with unpaired DHU arms (those for R, S1, and S2), additional potential base-pairings are shown with dark lines. The H. diminuta tRNA(R) is unusual not only in having an unpaired DHU arm, but in having an A in the first anticodon position; the similar structures predicted for tRNA(R) of Taenia crassiceps and Echinococcus multilocularis mitochondrial genomes are shown in the lower right-hand box, with nucleotides that differ from H. diminuta shown in lowercase letters
Fig. 4.—Potential secondary structures found for the noncoding regions either between nad5 and trnG or between trnY and trnL1 of three cestodes. Foldings presented as triplex or quadriplex indicate alternative potential hairpins. In the case of Hymenolepis diminuta, this structure includes part of a 13th repeat element (see text), with an arrow marking the only nucleotide difference from the preceding 12 repeat elements
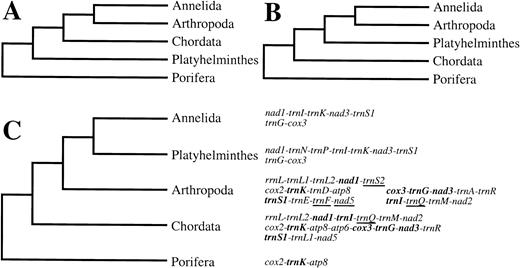
Fig. 5.—The three contending hypotheses of the phylogenetic relationships of Platyhelminthes to several other animal phyla, either as basal coelomates (A), as basal protostomes (B), or as eutrochozoans (C). Only the relationship shown in C is supported by a comparison of complete mitochondrial gene arrangements. A portion that is similar between the arrangements of annelids and Platyhelminthes is shown. A subset of these genes has an alternative arrangement shared among other phyla; these are shown in boldface along with their flanking genes. No gene arrangements are shared in a pattern to support the relationships in A or B. For these comparisons, Annelida are represented by Lumbricus terrestris (Boore and Brown 1995 ), Platynereis dumerilii (Boore and Brown 2000 ; GenBank AF178678), Helobdella robusta (partial), and Galathealinum brachiosum (partial) (Boore and Brown 2000 ), Arthropoda are represented by Limulus polyphemus (Lavrov, Boore, and Brown 2000 ) (previously inferred to be the primitive arrangement for studied arthropods; Boore et al. 1995 ; Boore, Lavrov, and Brown 1998 ; Boore 1999 ; Lavrov, Boore, and Brown 2000 ), and Chordata are represented by the gene arrangement most commonly observed (previously inferred to be primitive for Vertebrata except for the position of one tRNA that is not part of this subset of genes; Boore, Daehler, and Brown 1999 ; Boore 1999 ). Platyhelminthes are represented by H. diminuta, Echinococcus multilocularis (GenBank AB018440), Taenia crassiceps, and Fasciola hepatica (Garey and Wolstenholme 1989 ; Le et al. 2000 ), and Porifera are represented by Tetilla sp. (Watkins and Beckenbach 1999 ). The arrangements of all other nondepicted genes are unknown for the poriferan. Genes are underlined to signify opposite transcriptional orientation. Unconnected gene blocks are not adjacent
We thank André Adoutte, Kevin Helfenbein, Jonathan Henry, Dennis Lavrov, and Emili Salo for helpful comments. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) NI 559/3-1 to M.v.N.-R. and by DEB-9807100 from the NSF to J.L.B. and W.M.B. Part of this work was performed under the auspices of the U.S. Department of Energy by the Lawrence Livermore National Laboratory under contract W-7405-Eng-48.
literature cited
Aguinaldo, A. M., J. M. Turbeville, L. S. Linford, M. C. Rivera, J. R. Garey, R. A. Raff, and J. A. Lake.
Anderson, S., A. T. Bankier, B. G. Barrell et al. (14 co-authors).
Ax, P.
Balavoine, G.
———.
Barnes, W. M.
Bessho, Y., T. Ohama, and S. Osawa.
Blair, D. T., H. Le, L. Despres, and D. P. McManus.
Boore, J. L., and W. M. Brown.
———.
———.
———.
Boore, J. L., T. M. Collins, D. Stanton, L. L. Daehler, and W. M. Brown.
Boore, J. L., L. L. Daehler, and W. M. Brown.
Boore, J. L., D. Lavrov, and W. M. Brown.
Boyer, B. C., J. Q. Henry, and M. Q. Martindale.
Carranza, S., J. Baguñà, and M. Riutort.
Dowton, M.
Garey, J. R., and D. R. Wolstenholme.
Ghiselin, M. T.
Hoffmann, R. J., J. L. Boore, and W. M. Brown.
Hyman, L. H.
Keddie, E. M., T. Higazi, and T. R. Unnasch.
Kurabayashi, A., and R. Ueshima.
Lavrov, D., J. L. Boore, and W. M. Brown.
Le, T. H., D. Blair, T. Agatsuma et al. (14 co-authors).
Mindell, D., M. D. Sorenson, and D. E. Dimcheff.
Monnerot, M., M. Solignac, and D. R. Wolstenholme.
Ojala, D., J. Montoya, and G. Attardi.
Okimoto, R., H. M. Chamberlin, J. L. Macfarlane, and D. R. Wolstenholme.
Okimoto, R., J. L. Macfarlane, D. O. Clary, and D. R. Wolstenholme.
Perna, N. T., and T. D. Kocher.
Rieger, R. M.
Ruiz-Trillo, I., M. Riutort, D. T. J. Littlewood, E. A. Herniou, and J. Baguñà.
Shadel, G. S., and D. A. Clayton.
Smith, J. III, and S. Tyler.
Smith, M. J., A. Arndt, S. Gorski, and E. Fajber.
Stechmann, A., and M. Schlegel.
von Nickisch-Rosenegk, M., R. Lucius, and B. Loos-Frank.
Watkins, R. F., and A. T. Beckenbach.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}