





Abstract
T4 polynucleotide kinase–phosphatase (Pnkp) exemplifies a family of enzymes with 5′-kinase and 3′-phosphatase activities that function in nucleic acid repair. The polynucleotide 3′-phosphatase reaction is executed by the Pnkp C-terminal domain, which belongs to the DxDxT acylphosphatase superfamily. The 3′-phosphatase reaction entails formation and hydrolysis of a covalent enzyme-(Asp165)-phosphate intermediate, driven by general acid–base catalyst Asp167. We report that Pnkp also has RNA 2′-phosphatase activity that requires Asp165 and Asp167. The physiological substrate for Pnkp phosphatase is an RNA 2′,3′-cyclic phosphate end (RNA > p), but the pathway of cyclic phosphate removal and its enzymic requirements are undefined. Here we find that Pnkp reactivity with RNA > p requires Asp165, but not Asp167. Whereas wild-type Pnkp transforms RNA > p to RNAOH, mutant D167N converts RNA > p to RNA 3′-phosphate, which it sequesters in the phosphatase active site. In support of the intermediacy of an RNA phosphomonoester, the reaction of mutant S211A with RNA > p results in transient accumulation of RNAp en route to RNAOH. Our results suggest that healing of 2′,3′-cyclic phosphate ends is a four-step processive reaction: RNA > p + Pnkp → RNA-(3′-phosphoaspartyl)-Pnkp → RNA3′p + Pnkp → RNAOH + phosphoaspartyl-Pnkp → Pi + Pnkp.
INTRODUCTION
T4 polynucleotide kinase–phosphatase (Pnkp) exemplifies a family of repair enzymes that heal broken termini in RNA or DNA by converting 3′-PO4/5′-OH ends into 3′-OH/5′-PO4 ends, which are then sealed by RNA or DNA ligases. During T4 infection, Pnkp thwarts an RNA-based innate immune response called “tRNA restriction,” in which the bacterium blocks viral protein synthesis by inducing site-specific breakage of host-cell tRNAs, leaving a 2′,3′-cyclic phosphate end and a 5′-OH end at the incision site. The phage evades tRNA restriction by repairing the broken tRNAs through the action of Pnkp and a T4-encoded RNA ligase (1). T4 Pnkp performs two distinct end-healing reactions in the tRNA repair pathway: (i) the transfer of the γ phosphate from ATP to the 5′-OH terminus of the 3′ tRNA fragment to generate a ligatable 5′-PO4 end; and (ii) the removal of the 2′,3′-cyclic phosphate from the 5′ tRNA fragment to yield a ligatable 3′-OH, 2′-OH end (2–7). Whereas T4 Pnkp is clearly dedicated to RNA repair in vivo (1,8,9), its biochemical activities embrace both RNA and DNA end-healing reactions in vitro, that is, T4 Pnkp is adept at phosphorylating either RNA and DNA 5′-OH ends and it hydrolyzes RNA or DNA 3′-phosphate ends. By contrast, the eukaryal homologs of Pnkp are dedicated to the healing of DNA 3′-PO4/5′-OH ends, biochemically and in vivo (10,11).
T4 Pnkp is a homotetramer of a 301-aa polypeptide, which consists of an N-terminal kinase domain (aa 1–147) of the P-loop (GxxGxGKS) phosphotransferase superfamily and a C-terminal phosphatase domain (aa 148–301) of the acylphosphatase (DxDxT) superfamily (12–15). The Pnkp tetramer is formed through pairs of phosphatase-phosphatase and kinase-kinase homodimer interfaces. Essential constituents of the separate active sites for the 5′-kinase and 3′-phosphatase activities have been identified by mutagenesis, guided initially by phylogenetic conservation of primary structure among Pnkp homologs (16,17) and later (15,18) by crystal structures of T4 Pnkp in complexes with substrates or substrate mimetics (12–15). These studies illuminated the distinctive mechanisms of the 5′-kinase and 3′-phosphomonoesterase reactions. For example, phosphoryl transfer during the 5′-kinase reaction is a relatively simple in-line attack of the polynucleotide 5′-OH on the ATP γ phosphorus (19), catalyzed by: a general base, Asp35, that abstracts a proton from the 5′-OH; and the Lys15 and Arg126 side chains and a divalent cation that stabilize the transition state of the ATP γ phosphate.
By contrast, the removal of a 3′-phosphate by the Pnkp phosphatase module is a more complex two-step “ping-pong” reaction entailing the formation and subsequent hydrolysis of a covalent enzyme-(aspartyl-Oδ-)-phosphate intermediate (Figure 1A). As with other members of the acylphosphatase superfamily (20), the first aspartate of the DxDxT signature motif (Asp165 in Pnkp) is the site of phosphoryl transfer to the enzyme. The chemistry is driven by (i) general acid–base catalysis by Asp167, which donates a proton to the ribose O3′ leaving group in the first step and accepts a proton from the water nucleophile in the second step; and (ii) transition-state stabilization by contacts to the phosphate oxygens from side chains Lys258 and Ser211 and an enzyme-bound magnesium ion (Figure 2) (15,17). The constituents of the octahedral Mg2+ coordination complex are: Asp165, Asp278, the Asp167 main-chain carbonyl, a phosphate oxygen, and two water molecules (Figure 2). The atomic contacts of these essential moieties of the T4 Pnkp phosphatase module are confidently inferred from the crystal structure of the homologous protein serine phosphatase Fcp1, captured as a covalent aspartyl–BeF3− adduct (a mimetic of the aspartyl-phosphate intermediate) with Mg2+ in the active site (Figure 2) (21). Mg2+ occupies a similar position in T4 Pnkp (15), but there are as yet no crystal structures of T4 Pnkp with a polynucleotide 3′-phosphate or phosphomimetic in the phosphatase active site.
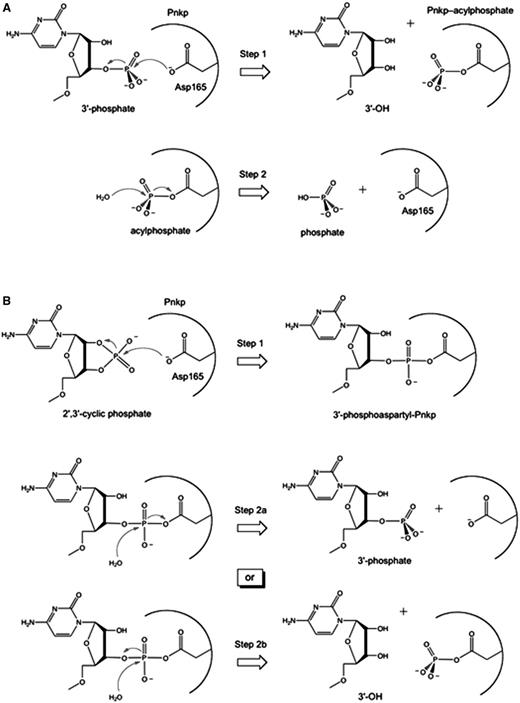
Pathways of RNA 3′ end-healing by T4 Pnkp. (A) Two-step chemical mechanism of 3′-phosphate removal through formation (Step 1) and hydrolysis (Step 2) of a covalent phosphoapartyl-enzyme intermediate. (B) Two hypothetical pathways for removal of a 2′,3′-cyclic phosphate are shown. A common first step entails attack of the aspartate nucleophile on the RNA > p end to form (Step 1) and then hydrolyze (Step 2) a covalent RNA-(phosphoaspartyl)-enzyme intermediate. In the Step 1 reaction shown, the ribose O2′ is the leaving group and the RNA 3′-phosphate is attached to the enzyme. Two variants of the Step 2 hydrolysis reaction (Steps 2a or 2b) are illustrated. In Step 2a, the enzymic aspartate is the leaving group and RNA3′p is the product. In Step 2b, the ribose O3′ is the leaving group and a phosphoaspartyl-enzyme remains.
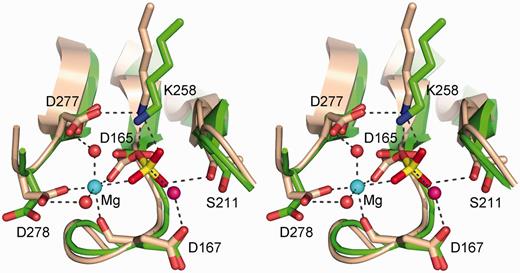
The phosphatase active site. The figure shows a stereo view of the T4 Pnkp phosphatase active site (of protomer C from pdb 2IA5; colored green) superimposed on the BeF3-modified active site of the CTD phosphatase Fcp1 (from pdb 3EF1; colored beige). The aspartyl-BeF3 adduct is depicted with the beryllium in yellow and the fluorines colored red (reflecting their mimicry of the phosphate of the aspartyl-phosphate intermediate). The Mg2+ of Fcp1 is depicted as a cyan sphere and the associated waters as red spheres. Atomic interactions in the Fcp1 active site are indicated by dashed lines. Pnkp side chains are labeled.
Whereas the mechanistic model of the 3′-phophomonoesterase reaction of Pnkp is on firm ground, it is fair to say that virtually nothing is known about the pathway by which RNA 2′,3′-cyclic phosphate ends are healed. That is because biochemical studies of the phosphatase reaction have used either 3′-phosphorylated oligodeoxyribonucleotides or deoxynucleoside 3′-monophosphates as the substrates (5–7,15–17). Recombinant T4 Pnkp has an apparent turnover number of 24 s−1 for the hydrolysis of 3′-dTMP under steady-state conditions (17). By contrast, T4 Pnkp is unable to hydrolyze ribonucleoside 3′-monophosphates in vitro (5,6). This is paradoxical given that 3′-dNMPs are effective substrates and that Pnkp is adept at hydrolyzing the 3′-phosphate of oligoribonucleotides (6). To our knowledge, there have been no studies published regarding the enzymic moieties required for RNA 3′-phosphate hydrolysis by Pnkp and no insights to the (presumably) multistep pathway by which Pnkp removes a 2′,3′-cyclic phosphate. Here we address both issues by studying the reaction of Pnkp and several phosphatase active site mutants with otherwise identical RNA oligonucleotides with 3′-phosphate versus 2′,3′-cyclic phosphate termini.
MATERIALS AND METHODS
T4 Pnkp
pET–Pnkp plasmids encoding wild-type or mutant Pnkp proteins fused to an N-terminal His10 tag (15,17) were introduced into Escherichia coli BL21(DE3). Recombinant protein production was induced by adjusting exponentially growing cultures (1000 ml) to 0.3 mM IPTG and incubating them at 17°C for 15 h with continuous shaking. The wild-type and mutant His10-Pnkp proteins were purified from soluble bacterial lysates by Ni-agarose chromatography, as described previously (16). Protein concentrations were determined by using the BioRad dye reagent with bovine serum albumin as the standard.
RNA substrates
The 20-mer RNA3′p oligonucleotide labeled with 32P at the penultimate phosphate was prepared by T4 Rnl1-mediated addition of [5′-32P]pCp to a 19-mer synthetic oligoribonucleotide, as described (22). The RNA3′p was treated with E. coli RNA 3′-terminal phosphate cyclase and ATP to generate a 2′,3′-cyclic phosphate derivative, RNA > p (22). The RNA3′p and RNA > p substrates were gel-purified before use in Pnkp phosphatase assays. A 2′-phosphate terminated derivative, RNA2′p, was produced by reacting RNA > p with purified Arabidopsis thaliana tRNA ligase (9) as follows: a reaction mixture (100 µl) containing 50 mM Tris–HCl (pH 8.0), 2 mM DTT, 10 mM EDTA, 1 µM RNA > p and 50 µM AtRNL was incubated for 30 min at 37°C. The RNA2′p product was recovered by phenol–chloroform extraction and precipitation with ethanol; the conversion of RNA > p to RNA2′p was verified by digesting the 20-mers with RNase T1 and analysing the 32P-labeled terminal fragments by urea-PAGE.
Phosphatase assay
Reaction mixtures containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 50 nM 32P-labeled RNA > p or RNAp substrate and wild-type or mutant Pnkp (specified as fmol or pmol of the enzyme monomer) were incubated at 22°C. The reactions were quenched by adding an equal volume of 100 mM EDTA. The products were digested for 30 min at 37°C with RNase T1 (1000 U; Fermentas). The samples were supplemented with 0.5 volume of 90% formamide, 0.01% bromophenol blue/xylene cyanol, 50 mM EDTA and then analysed by electrophoresis (at 50 W constant power) through a 40-cm 20% polyacrylamide gel containing 8 M urea in 45 mM Tris borate, 1.2 mM EDTA. The 32P-labeled RNAs were visualized by autoradiography of the gel and, where specified, quantified by scanning the gel with a Fuji Film BAS-2500 imager.
RESULTS
RNA 3′-phosphate and 2′,3′-cyclic phosphate removal activities of Pnkp
A goal of the present study was to assess and compare the pathways of RNA end healing by T4 Pnkp at 3′-phosphate versus 2′,3′-cyclic phosphate termini. The substrates used were otherwise identical 20-mer RNAs with 3′-phosphate (p) or 2′,3′-cyclic phosphate (>p) ends and a single radiolabel between the 3′-terminal and penultimate nucleosides (Figure 3). The RNA3′p and RNA > p substrates (0.5 pmol) were incubated for 20 min at 22°C with wild-type Pnkp. To enable resolution of the reactants and products with different 3′ ends, the mixtures were digested with RNase T1 before analysis by denaturing PAGE (Figure 3). RNase T1 incised the substrates 3′ of the most distal guanosine to yield the 32P-labeled tetranucleotides HOCUUpC3′p or HOCUUpC > p (Figure 3, lanes 0).
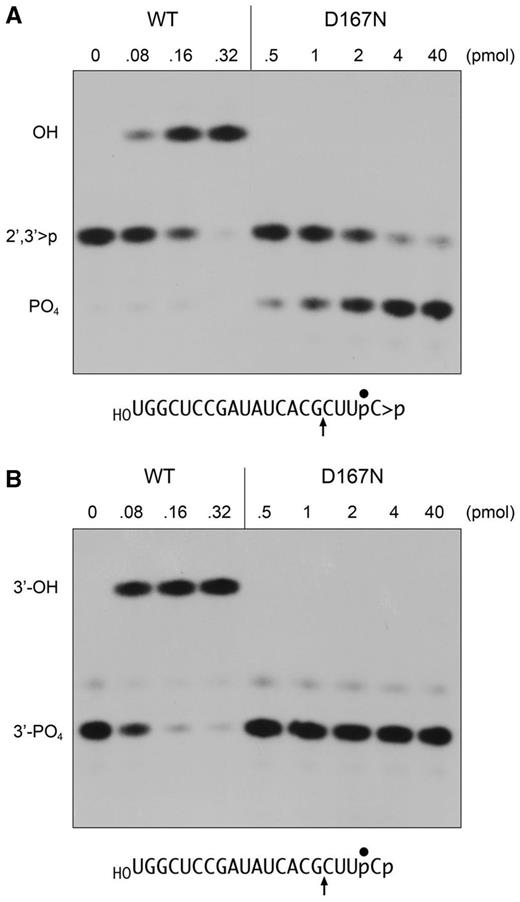
RNA 2′,3′-cyclic phosphodiesterase activity of Pnkp-D167N. (A) Reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol (50 nM) 32P-labeled RNA > p (panel A) or RNA3′p (panel B), and wild-type or D167N Pnkp as specified were incubated for 20 min at 22°C. The reactions were quenched with EDTA. The products were digested with RNase T1 and then analysed by urea-PAGE. Autoradiograms of the gels are shown. The phosphorylation states of the terminal RNase T1 fragments are indicated at left. The nucleotide sequences of the RNA > p and RNA3′p strands are shown below the autoradiograms; the position of the 32P-label is denoted by filled circle and the site of RNase T1 scission is indicated by upward arrow.
Reaction of increasing concentrations of Pnkp with the RNA3′p substrate depleted the HOCUUpC3′p T1 fragment and generated a more slowly migrating T1 fragment corresponding to the dephosphorylated product, HOCUUpCOH (Figure 3B). The same HOCUUpCOH product was generated when Pnkp reacted with RNA > p (Figure 3A). Whereas both dephosphorylation reactions proceeded to completion at saturating enzyme, the extent of RNAOH formation at limiting enzyme was higher for RNA3′p than RNA > p (Figure 3). Quantification of the enzyme titration data from three separate experiments indicated that the Pnkp specific activity was ∼3-fold higher with RNA3′p (Figure 4A) versus RNA > p (Figure 4B).
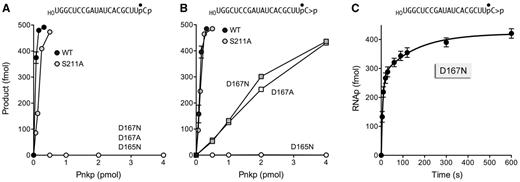
Phosphoesterase activities of wild-type and mutant Pnkp proteins. Reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol (50 nM) 32P-labeled RNA3′p (panel A) or RNA > p (panel B), and wild-type or mutant Pnkp as specified were incubated for 20 min at 22°C. The mixtures were treated with RNAse T1 and analysed by urea-PAGE. The extents of product formation are plotted as a function of input Pnkp. Each datum is the average of three independent experiments (±SEM). The kinetic profile of the D167N cyclic phosphodiesterase reaction is shown in panel C. A reaction mixture (100 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 50 nM 32P-labeled RNA > p and 500 nM Pnkp D167N were incubated at 22°C. Aliquots (10 µl, containing 0.5 pmol of RNA substrate) were withdrawn at the times specified and quenched immediately with EDTA. The RNAs were then digested with RNase T1 and analysed by urea-PAGE. The extent of RNAp formation is plotted as a function of time. Each datum is the average of three independent experiments (±SEM).
It was notable that we detected no accumulation of a phosphorylated T1 fragment HOCUUpCp (which migrates faster than HOCUUpC > p) during the reaction of sub-saturating levels of input Pnkp with the RNA > p substrate (Figure 3A). Nor did we detect a phosphorylated species when assaying the time course of the reaction of wild-type Pnkp with the RNA > p substrate (not shown). These results suggested that (i) either the pathway of 2′,3′-cyclic phosphate removal does not involve the formation of a free RNAp intermediate; or (ii) an RNAp intermediate is formed, but is rapidly hydrolyzed to RNAOH, making it difficult to detect.
The Asp165 nucleophile is required for removal of a 2′,3′-cyclic phosphate
Previous studies showed that mutation of the Asp165 nucleophile to asparagine abolished the 3′-phosphatase activity of T4 Pnkp with a 3′-dTMP mononucleotide substrate (17). Here we queried the impact of the D165N change on the removal of the terminal phosphates of the 20-mer RNA3′p and RNA > p strands. Whereas 0.16 pmol of wild-type Pnkp sufficed for near-quantitative conversion of RNA3′p to RNAOH (Figure 3B), we detected no product formation by up to 40 pmol of the D165N mutant (Figure 4A and data not shown). This result is consistent with a mechanism of 3′-phosphate hydrolysis through a covalent aspartyl-phosphate intermediate (Figure 1A). The key question is whether the same mechanism of covalent catalysis applies when Pnkp acts at a 2′,3′-cyclic phosphate, that is, does the initial step entail a nucleophilic attack by Asp165 on the cyclic phosphate to form a covalent RNA-(phosphoryl)-Asp165-Pnkp intermediate? [Note: if the ribose O2′ is the leaving group, then the initial step in cyclic phosphate removal would generate an RNA-(3′-phosphoryl)-Asp165 adduct, as depicted in Figure 1B. In the event that the ribose O3′ is the leaving group (analogous to Step 1 in Figure 1A), then an RNA-(2′-phosphoryl)-Asp165 adduct would be formed.] If a mechanism of covalent catalysis applies, then the D165N mutation should abolish the reactivity of Pnkp at a 2′,3′-cyclic phosphate end. However, one can envision an alternative mechanism whereby water is the relevant nucleophile in the initial attack on RNA > p. A hydrolysis mechanism could directly generate an RNA 3′-phosphate end in a single step that would then enter the phosphomonoesterase reaction pathway in Figure 1A. If hydrolysis is the initial event, then we might find that the D165N mutant is capable of converting RNA > p to RNAp, which ought to accumulate because Asp165 is clearly required for downstream reactions at a monophosphate end (Figure 4A). We observed that D165N was unreactive with the RNA > p substrate at up to 40 pmol of input enzyme (Figure 4B and data not shown). A simple interpretation of this result is that covalent catalysis by Asp165 is the initial step in 2′,3′-cyclic phosphate removal.
Mutating the Asp167 general acid–base allows capture of an RNAp intermediate in the pathway of 2′,3′-cyclic phosphate removal
Invocation of a covalent RNA-phosphoaspartyl-Pnkp adduct in the removal of the 2′,3′-cyclic phosphate mandates a second step in the reaction pathway in which the adduct is hydrolyzed. The proposed attack by water could, in principle, occur in either of two orientations, as illustrated in Figure 1B. In the event that the ribose oxygen is the leaving group (Step 2B), the hydrolysis reaction yields RNAOH and phosphoaspartyl-Pnkp. (The latter can be readily hydrolyzed to restore the Pnkp apoenzyme; Figure 1A). Such a pathway through Step 2B could account for our inability to detect RNAp during the 2′,3′-cyclic phosphate removal reaction. The alternative pathway (Step 2A) is that Asp165 is the leaving group in the hydrolysis of an RNA-phosphoaspartyl-Pnkp, which generates RNAp and Pnkp apoenzyme as the products. In this case, RNAp might instantly enter the phosphomonoesterase reaction pathway (Figure 1A) to be converted to RNAOH. To discriminate among these pathway options, we sought to expose the existence of an otherwise fleeting RNAp intermediate, by mutating the phosphatase active site in the hope of identifying lesions that selectively impact 3′-phosphate removal while sparing the initial Steps 1 and 2A of the hypothetical pathway of 2′,3′-cyclic phosphate removal.
Our initial attention focused on the Asp167 general acid–base catalyst, mutation of which to alanine or asparagine ablates 3′-phosphatase activity with a 3′-TMP mononucleotide substrate (17). Here we found that the D167N and D167A mutants were inert in dephosphorylating the RNA3′p substrate at up to 40 pmol of input enzyme (Figures 3B, 4A, and data not shown). By contrast, D167N and D167A were reactive with the RNA > p substrate, which they converted to RNAp as the sole product detected by PAGE (Figure 3A and data not shown). The extent of RNAp formation was proportional to the input D167N and D167A proteins (Figure 4B), with 82% of the RNA > p being converted to RNAp by 4 pmol of either mutant enzyme. The specific activity of D167N as a cyclic phosphodiesterase acting on RNA > p was ∼7% of the specific activity of wild-type Pnkp in converting RNA > p to RNAOH (Figure 4B). Thus, eliminating the proton donating/accepting ability of Asp167 selectively ablated the Pnkp 3′-phosphomonesterase, while preserving an appreciable 2′,3′-cyclic phosphodiesterase activity.
The kinetic profile of the reaction of D167N (500 nM) with RNA > p (50 nM) is shown in Figure 4C. The reaction attained an end point in 10 min with an 85% yield of RNAp product. Because increasing the D167N concentration to 1000 nM had no effect on the rate of RNAp formation or the reaction end point (not shown), we surmise that the reaction in enzyme excess is not limited by the initial rate of D167N binding to RNA > p. Nonetheless, the kinetic data for RNAp formation did not fit to a single exponential (not shown), but instead fit very well to a biphasic kinetic model (Figure 4C) in which, as calculated by non-linear regression in Prism, the rapid phase proceeded with a rate constant of 0.14 ± 0.02 s−1 (and accounted for 65% of the RNAp end-product) and the slow phase proceeded with a rate constant of 0.0065 ± 0.0017 s−1. We infer that the rapid phase reflects the rate of the chemical step(s) of the 2′,3′-cyclic phosphodiesterase reaction of D167N with RNA > p bound in the phosphatase active site. The slow phase is readily explained by the propensity of a fraction of the input RNA > p strands (which have 5′-OH termini) to bind to the phosphoacceptor site of the Pnkp kinase domain (14), where no 5′ reaction will occur in the absence of an NTP donor and where the 2′,3′ > p end is inaccessible to the phosphatase domain. The consequence is that the slow phase is limited by the rate of dissociation of the RNA > p from the kinase domain and its redistribution to the D167N phosphatase domain.
D167N sequesters the RNAp product of the cyclic phosphodiesterase reaction
We performed an order-of-addition experiment to test the susceptibility of the RNAp product generated by D167N to subsequent processing by wild-type Pnkp (per the scheme in Figure 5A). Control experiments verified that 4 pmol of D167N and 0.5 pmol of wild-type Pnkp sufficed for efficient conversion of the input RNA > p strand to RNAp and RNAOH products, respectively (Figure 5A, lanes 2 and 3). The initially surprising finding was that simply adding wild-type Pnkp to the reaction mixture after a 20-min incubation of D167N with RNA > p failed to convert any of the RNAp product to RNAOH (Figure 5A, lane 4), signifying that the RNAp end was either unreactive or shielded from reaction with wild-type Pnkp. This issue was resolved by heating the reaction mixture after the 20-min incubation with D167N and before supplementation with wild-type Pnkp. This maneuver rendered the RNAp species completely accessible to hydrolysis by wild-type Pnkp to yield RNAOH (Figure 5A, lane 5). These results suggested that the RNAp product of the D167N cyclic phosphodiesterase reaction remains bound in the phosphatase active site.
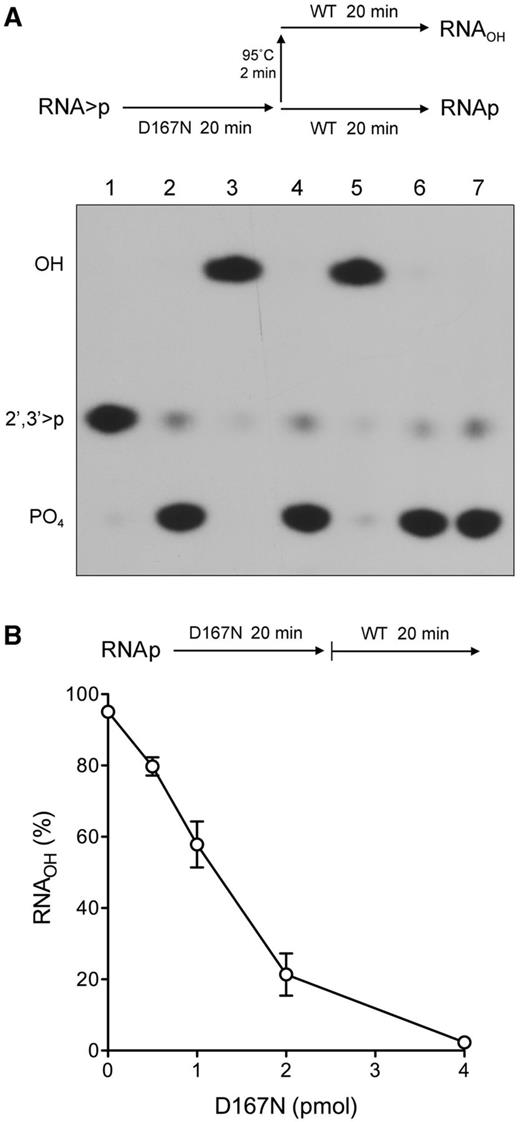
D167N sequesters the RNAp product of the cyclic phosphodiesterase reaction. (A) A schematic of the experiment is shown above the results. Six reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol (50 nM) 32P-labeled RNA > p, and either no enzyme (lane 1), 0.5 pmol (50 nM) wild-type Pnkp (lane 3), or 4 pmol (400 nM) Pnkp-D167N (lanes 2, 4, 5 and 6) were incubated for 20 min at 22°C. The reactions in lanes 1, 2, and 3 were quenched with 50 mM EDTA. The three remaining Pnkp-D167N reaction mixtures were treated as follows. Wild-type Pnkp (0.5 pmol) was either added directly (lane 4) or after heating the reaction mixture at 95°C for 2 min (lane 5), after which the wild-type Pnkp-supplemented D167N reaction mixtures were incubated for 20 min at 22°C and then quenched with EDTA. The D167N reaction mixture in lane 6 was heated at 95°C for 2 min, incubated for an additional 20 min at 22°C without wild-type Pnkp supplementation, and then quenched with EDTA. The RNAs of all six reaction mixtures were then digested with RNase T1 and analysed by urea-PAGE (lanes 1–6). An RNase T1 digest of the 32P-labeled RNA3′p 20-mer was analysed in parallel in lane 7. An autoradiogram of the gel is shown. The phosphorylation states of the terminal RNase T1 fragments are indicated at left. (B) A schematic of the experiment is shown above the results. Reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol 32P-labeled RNAp, and increasing amounts of Pnkp-D167N as specified were pre-incubated for 20 min at 22°C. The reaction mixtures were then supplemented with 0.5 pmol of wild-type Pnkp and incubated for 20 min at 22°C. The reactions were then quenched with EDTA. The RNAs were digested with RNAse T1 and analysed by urea-PAGE. The extent of RNAOH formation is plotted as a function of the amount of Pnkp-D167N included during the pre-incubation phase. Each datum is the average of three independent experiments (±SEM).
We examined this issue by asking whether pre-incubation of an RNA 3′-phosphate-terminated substrate with increasing amounts of D167N would protect the RNA3′p from subsequent hydrolysis during a 20-min reaction with 0.5 pmol of wild-type Pnkp (per the scheme in Figure 5B). We observed a progressive decline in the accessibility of the RNA3′p substrate as the level of D167N was increased, with virtually complete protection from hydrolysis being achieved at 4 pmol of input D167N. This experiment attests to the apparent stability of the D167N•RNAp complex and suggests that a pathway of 2′,3′-cyclic phosphate removal through an RNAp intermediate might be processive.
Pnkp mutant S211A transiently accumulates RNAp during 2′,3′-cyclic phosphate removal
Trapping of RNAp as a product of the D167N reaction supports the intermediacy of RNAp in cyclic phosphate removal, but the result does not rule out the possibility that RNAp formation is an “off-pathway” event prompted by the loss of the acid–base catalyst. The case for intermediacy would be fortified by identifying another change in the active site (one not affecting general acid–base catalysis) that promotes accumulation of RNAp by selectively slowing the phosphomonoesterase reaction. We considered that this might be achieved in light of the differences in the electrostatics of the presumptive pentacoordinate transition states of the 2′,3′-cyclic phosphodiesterase reaction (−2 charge) and the phosphomonoesterase reaction (−3 charge) in an associative phosphoryl transfer mechanism. If this difference pertains in the active site of the enzyme, then the 3′-phosphatase reaction might be more acutely reliant than the 2′,3′-cyclic phosphodiesterase on the Ser211 hydroxyl, which donates a hydrogen bond to one of the non-bridging phosphate oxygens of the aspartyl-phosphate adduct (Figure 2). Initial enzyme titration experiments with the S211A mutant Pnkp showed that its specific activity with the RNA > p substrate was similar to that of wild-type Pnkp (Figure 4B), whereas S211A activity with the RNA3′p substrate was 3-fold less than that of wild-type Pnkp (Figure 4A). Moreover, we readily detected RNAp when RNA > p was reacted for 20 min with limiting levels of S211A enzyme (not shown).
The kinetic profile of the reaction of excess S211A (500 nM) with RNA > p (50 nM) is presented in Figure 6A and quantified in Figure 6B. The reaction was notable for the early accumulation of RNAp, which comprised 25% and 48% of the total radiolabeled material at 5 and 10 s, respectively, at which times RNAOH comprised only 8% and 34% of the total. The abundance of RNAp declined steadily thereafter, concomitant with the accumulation of RNAOH to an extent of 93% of the total labeled RNA at 5 min (Figure 6B). These results are consistent with a precursor–product relationship between RNAp and RNAOH. By plotting the data in Figure 6B as the sum of the RNAp and RNAOH species as a function of reaction time, we derived a rate constant of 0.14 ± 0.01 s−1 for the 2′,3′-cyclic phosphodiesterase reaction of the S211A enzyme.
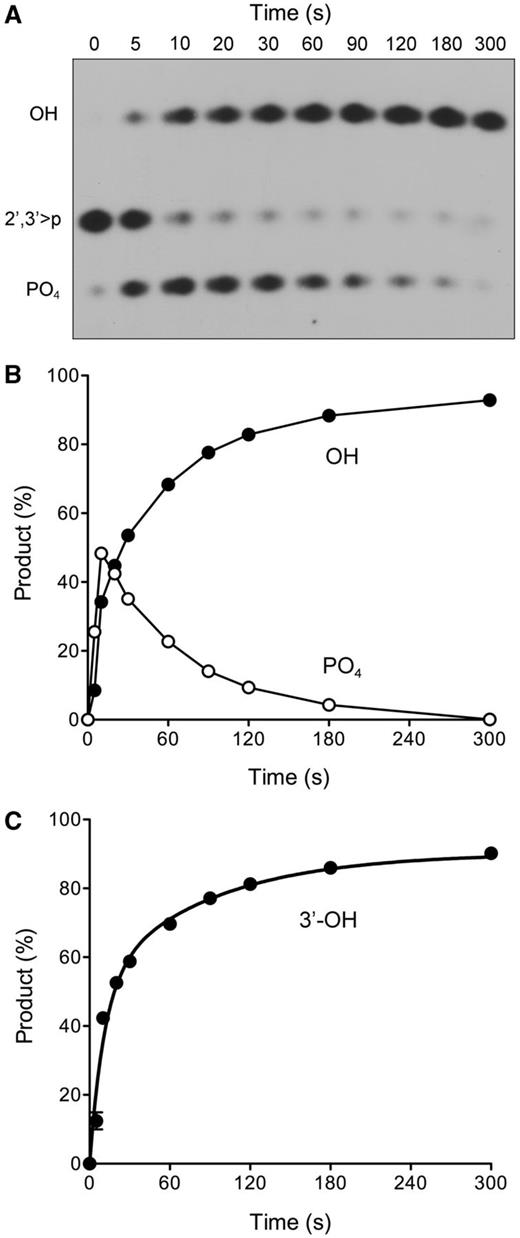
Kinetic profile of cyclic phosphate and 3′-phosphate end-healing by Pnkp-S211A. (A and B) Reaction mixtures (100 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 50 nM 32P-labeled RNA > p and 500 nM Pnkp-S211A were incubated at 22°C. Aliquots (10 µl) were withdrawn at the times specified and quenched immediately with EDTA. The RNAs were digested with RNase T1 and analysed by urea-PAGE. An autoradiograph of the gel is shown in panel A. The phosphorylation states of the terminal RNase T1 fragments are indicated at left. The quantified kinetic profile is shown in panel B, wherein the abundance of the RNAp and RNAOH species (as the percentage of the total labeled RNA) is plotted as a function of time. Each datum in the graph is the average of three independent experiments (±SEM). (C) A 3′-phosphatase reaction mixture (100 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 50 nM 32P-labeled RNA3′p, and 500 nM Pnkp-S211A was incubated at 22°C. Aliquots (10 µl) were withdrawn at the times specified and quenched immediately with EDTA. The RNAs were digested with RNase T1 and analysed by urea-PAGE. The extent of RNAOH formation (expressed as the percent of total labeled RNA) is plotted as a function of time. Each datum in the graph is the average of three independent experiments (±SEM).
The reaction of excess S211A with the RNA3′p substrate displayed biphasic kinetics with an end point of 91% RNAOH formation (Figure 6C). Non-linear regression curve fitting of the data in Prism highlighted a fast phase with an apparent rate constant of 0.092 ± 0.019 s−1 that accounted for 57% of the reaction product. (The rate of the slow phase was 0.012 ± 0.0026 s−1). Taken together, the kinetic data for the two substrates indicate that the rate of the S211A 2′,3′-cyclic phosphodiesterase reaction is ∼1.5-fold faster than that of the 3′-phosphatase step, thereby accounting for the accumulation of the RNAp intermediate during the reaction of S211A with RNA > p.
RNA 2′-phosphatase activity of Pnkp
The PAGE system used for analysis of the T1 fragments does not discriminate whether the RNAp product formed by the D167N cyclic phosphodiesterase reaction has a 3′-phosphate or 2′-phosphate terminus. Given that wild-type Pnkp is competent to hydrolyze an RNA 3′-phosphate, and that the RNAp formed in the cyclic phosphodiesterase reaction is also hydrolyzed by wild-type Pnkp, it is tempting to assume that the CPDase reaction product is itself a 3′-phosphomonoester. However, as far as we know, there is no report in the literature concerning the capacity of T4 Pnkp to remove an RNA 2′-phosphate. Here we prepared a 20-mer RNA2′p substrate by treating the 20-mer RNA > p with plant tRNA ligase (AtRNL) under conditions in which only the RNA 2′,3′-cyclic phosphodiesterase domain is active. The plant CPDase hydrolyzes the RNA > p end exclusively to a 3′-OH, 2′-phosphate (23–25).
The 32P-labeled RNA2′p strand was reacted with wild-type or mutant T4 Pnkp and the reaction mixtures were digested with RNase T1 before analysis by PAGE (Figure 7A). Wild-type Pnkp catalyzed near-quantitative conversion of the 2′-phosphate end to a more slowly migrating 2′-OH product. The D165N and D167N mutants were inactive. Thus, Pnkp can hydrolyze an RNA 2′-phosphomonoester in a reaction that requires both the aspartate nucleophile and the aspartate general acid–base. Enzyme titration showed that Pnkp had similar specific activity with the RNA2′p and RNA > p substrates (Figure 7B).
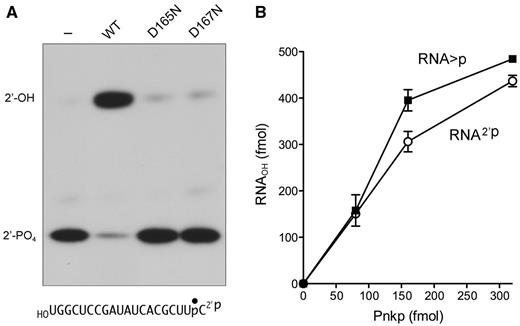
RNA 2′-phosphatase activity of Pnkp. (A) Reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol 32P-labeled RNA2′p, and 1 pmol of wild-type, D165N or D167N Pnkp as specified were incubated for 20 min at 22°C. A control reaction mixture from which Pnkp was omitted in shown in lane dash. The mixtures were treated with RNase T1 and analysed by urea-PAGE. (B) Reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol of 32P-labeled RNA > p or RNA2′p substrate, and Pnkp as specified were incubated for 20 min at 22°C. The mixtures digested with RNase T1 and analysed by urea-PAGE. The extent of product formation is plotted as a function of input Pnkp. Each datum is the average of three independent experiments (±SEM).
The RNAp product of the cyclic phosphodiesterase reaction is a 3′-phosphate
To determine the nature of the RNAp end generated when D167N hydrolyzes RNA > p, we exploited the distinctive end-specificities of two RNA ligases: plant AtRNL and E. coli RtcB. AtRNL is a three-domain enzyme that joins 2′,3′-cyclic phosphate and 5′-OH RNA ends through the following healing and sealing steps: (i) a 5′-kinase transfers the γ phosphate from ATP to the 5′-OH RNA end to yield a 5′-phosphate; (ii) a CPDase converts the 2′,3′-cyclic phosphate to a 3′-OH, 2′-PO4; and (iii) an ATP-dependent ligase joins the healed 3′-OH, 2′-PO4 and 5′-PO4 ends to yield a 3′,5′-phosphodiester, 2′-PO4 splice junction (26). The 2′-PO4 is strictly required for the sealing reaction of plant tRNA ligase (23–26). RtcB joins 2′,3′-cyclic phosphate and 5′-OH RNA ends by a different set of chemical steps entailing (i) Mn2+-dependent hydrolysis of the RNA > p end to a 3′-PO4, 2-OH; and (ii) GTP/Mn2+-dependent joining of the 3′-PO4 and 5′-OH ends to generate a conventional 3′,5′ phosphodiester splice junction (22,27,28). RtcB strictly requires a 3′-PO4 end for ligation; it does not seal a 2′-PO4 end (22).
In the experiment in Figure 8, the 20-mer RNA > p substrate was reacted with D167N to generate RNAp and the reaction was heated to dissociate D167N from the RNA product. Direct analysis of the radiolabeled RNAs by PAGE (without RNase T1 digestion) revealed no appreciable change in the mobility of the linear RNA as a consequence of D167N opening the cyclic phosphodiester (compare lanes 1 and 2). The reactivity of the heated D167N products with AtRNL and RtcB was tested by supplementing the reaction mixtures with the respective ligases and the requisite NTPs and metal cofactors. The outcome of successful ligation is intramolecular end joining to yield a circular RNA product that migrates faster than the linear RNA substrate. Whereas AtRNL effected virtually complete circularization of the RNA > p strand (see the leftmost lane in Figure 8), it failed to circularize the RNAp species formed by D167N (Figure 8, lane 3). Note that the slight increase in the electrophoretic mobility of the linear RNAp after the reaction with AtRNL reflected its efficient 5′-phosphorylation by the AtRNL kinase domain to yield a linear pRNAp product. By contrast, RtcB readily circularized the RNAp species formed by D167N (Figure 8, lane 4). These results indicate that the CPDase reaction of D167N generates a 3′-PO4 product.
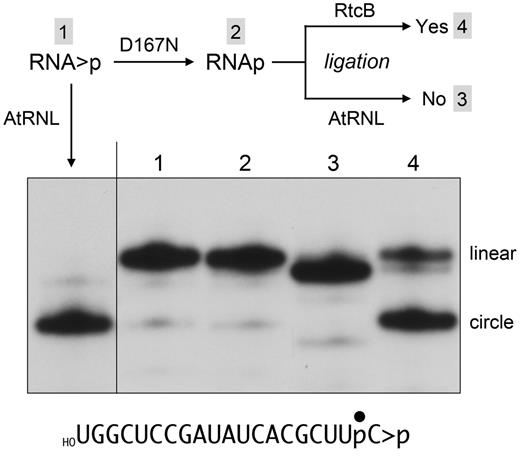
The product of the D167N cyclic phosphodiesterase reaction is a 3′-phosphate. A schematic of the experiment is shown above the results. Four reaction mixtures (10 µl) containing 100 mM Tris-acetate (pH 6.0), 10 mM MgCl2, 2 mM DTT, 0.5 pmol (50 nM) of 32P-labeled RNA > p (shown at the bottom), and either no enzyme (lane 1) or 4 pmol (400 nM) Pnkp-D167N (lanes 2–4) were incubated for 20 min at 22°C. The no enzyme control (lane 1) and one of the D167N Pnkp reaction mixtures (lane 2) were quenched immediately with 50 mM EDTA/90% formamide. The remaining two D167N reaction mixtures were heated at 95°C for 2 min and then supplemented with either the 2′-PO4-requiring plant tRNA ligase AtRNL (lane 3) or the 3′-PO4-requiring ligase E. coli RtcB (lane 4). For the AtRNL reaction, the mixture was adjusted to 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 2 mM DTT, 0.1 mM ATP, 0.1 mM GTP, and 5 µM AtRNL in a final volume of 20 µl. For the RtcB reaction, the mixture was adjusted to 50 mM Tris-HCl (pH 8.0), 2 mM MnCl2, 0.1 mM GTP, and 10 µM RtcB in a final volume of 20 µl. The ligation reaction mixtures were incubated for 30 min at 37°C and then quenched with EDTA/formamide. The samples were analysed by urea-PAGE. A “positive control” reaction of AtRNL with the 32P-labeled RNA > p substrate was included in the left-most lane. An autoradiograph of the gel is shown. The positions of the linear 20-mer and the ligated circular RNA are indicated on the right.
DISCUSSION
The present study advances our understanding of the mechanism by which the T4 Pnkp phosphatase enzyme heals RNA ends. We show that Pnkp is adept at hydrolyzing RNA 3′-phosphomonoesters and RNA 2′-phosphomonoesters to yield the respective hydroxyl termini in a reaction pathway that relies on both the Asp165 nucleophile and the Asp167 general acid–base. The ability of either the ribose O3′ or O2′ to be the leaving group during the attack of Asp165 on the terminal phosphate suggests some flexibility in the active site, whereby (i) the terminal ribose can bind in alternative conformations that place either the O2′ or O3′ close to the Asp167 general acid; or (ii) the terminal ribose binds in a fixed orientation and the Asp167 general acid has some range of motion to access either a 2′ bridging or a 3′ bridging phosphate oxygen.
It is well-established that T4 Pnkp is an RNA repair enzyme in vivo, dedicated to the healing and sealing of broken tRNAs with 2′,3′-cyclic phosphate and 5′-OH ends (1,2,9). Our studies of the Pnkp-D167N and Pnkp-S211A enzymes reveal that removal of a 2′,3′-cyclic phosphate occurs through sequential cyclic phosphodiesterase and phosphomonoesterase reactions. Pnkp-D167N activity arrests after conversion of RNA > p to RNA3′p, which remains sequestered in the phosphatase active site. The D167N CPDase reaction outcome (a 3′-PO4) suggests that the terminal 2′,3′-cyclic phosphate binds in a specific orientation that places the O2′ leaving group apical to the Asp165 nucleophile. Apparently, the O2′ leaving group can be expelled when Asp167 is not available as a proton donor, albeit slowly. The S211A mutation, affecting the coordination of the scissile phosphate (Figure 2), results in transient accumulation of high levels of an RNAp intermediate that is subsequently hydrolyzed to RNAOH. It is our assumption that cyclic phosphate removal by wild-type Pnkp also proceeds through an RNAp intermediate, but we have been unable to confirm this because the rate of 3′ end healing by wild-type Pnkp under conditions of enzyme excess is too fast to measure by manual methods.
It is worthwhile to compare and contrast the properties of T4 Pnkp phosphatase (a Mg2+-dependent acylphosphatase) with those of three other types of RNA repair enzymes that act on 2′,3′-cyclic phosphate ends (Table 1). Clostridium thermocellum (Cth) Pnkp exemplifies a family of RNA end-healing and sealing enzymes found in diverse bacteria from many phyla. CthPnkp is composed of three catalytic modules: an N-terminal polynucleotide 5′-kinase; a central 2′,3′-phosphatase; and a C-terminal ligase (29). The CthPnkp phosphatase domain, which belongs to the binuclear metallophosphoesterase superfamily, catalyzes the release of Pi from 2′-PO4, 3′-PO4 or 2′,3′-cyclic phosphate ribonucleotides and requires either Mn2+ or Ni2+ as the metal cofactor (29–33). As with T4 Pnkp, CthPnkp converts a 2′,3′-cyclic phosphate to 3′-OH, 2′-OH end-product through sequential diesterase and monoesterase reactions. However, unlike T4 Pnkp, which generates an exclusive 3′-PO4 product of its CPDase reaction (as shown for “diesterase-only” mutant D167N), a diesterase-only mutant of CthPnkp opens a 2′,3′-cyclic phosphodiester to form an exclusive 2′-PO4 product (32,33). The chemical mechanism of the CthPnkp phosphoesterase is through attack of a metal-bound water on the scissile phosphate, that is, the reaction does not involve a covalent phosphoenzyme intermediate.
Comparison of RNA repair enzymes that heal 2′,3′-cyclic phosphate ends
| Enzyme | Family | Metal | End-product | CPDase product | 3′-Pase | 2′-Pase |
|---|---|---|---|---|---|---|
| T4 Pnkp | Acylphosphatase | Mg2+ | 3′-OH, 2′-OH | 3′-PO4, 2′-OH | Yes | Yes |
| CthPnkp | Binuclear metallophosphoesterase | Mn2+ Ni2+ | 3′-OH, 2′-OH | 3′-OH, 2′-PO4 | Yes | Yes |
| Yeast and plant tRNA ligase | 2H phosphoesterase | None | 3′-OH, 2′-PO4 | 3′-OH, 2′-PO4 | No | No |
| RtcB | RtcB | Mn2+ | 3′-PO4, 2′-OH | 3′-PO4, 2′-OH | No | ? |
| Enzyme | Family | Metal | End-product | CPDase product | 3′-Pase | 2′-Pase |
|---|---|---|---|---|---|---|
| T4 Pnkp | Acylphosphatase | Mg2+ | 3′-OH, 2′-OH | 3′-PO4, 2′-OH | Yes | Yes |
| CthPnkp | Binuclear metallophosphoesterase | Mn2+ Ni2+ | 3′-OH, 2′-OH | 3′-OH, 2′-PO4 | Yes | Yes |
| Yeast and plant tRNA ligase | 2H phosphoesterase | None | 3′-OH, 2′-PO4 | 3′-OH, 2′-PO4 | No | No |
| RtcB | RtcB | Mn2+ | 3′-PO4, 2′-OH | 3′-PO4, 2′-OH | No | ? |
Comparison of RNA repair enzymes that heal 2′,3′-cyclic phosphate ends
| Enzyme | Family | Metal | End-product | CPDase product | 3′-Pase | 2′-Pase |
|---|---|---|---|---|---|---|
| T4 Pnkp | Acylphosphatase | Mg2+ | 3′-OH, 2′-OH | 3′-PO4, 2′-OH | Yes | Yes |
| CthPnkp | Binuclear metallophosphoesterase | Mn2+ Ni2+ | 3′-OH, 2′-OH | 3′-OH, 2′-PO4 | Yes | Yes |
| Yeast and plant tRNA ligase | 2H phosphoesterase | None | 3′-OH, 2′-PO4 | 3′-OH, 2′-PO4 | No | No |
| RtcB | RtcB | Mn2+ | 3′-PO4, 2′-OH | 3′-PO4, 2′-OH | No | ? |
| Enzyme | Family | Metal | End-product | CPDase product | 3′-Pase | 2′-Pase |
|---|---|---|---|---|---|---|
| T4 Pnkp | Acylphosphatase | Mg2+ | 3′-OH, 2′-OH | 3′-PO4, 2′-OH | Yes | Yes |
| CthPnkp | Binuclear metallophosphoesterase | Mn2+ Ni2+ | 3′-OH, 2′-OH | 3′-OH, 2′-PO4 | Yes | Yes |
| Yeast and plant tRNA ligase | 2H phosphoesterase | None | 3′-OH, 2′-PO4 | 3′-OH, 2′-PO4 | No | No |
| RtcB | RtcB | Mn2+ | 3′-PO4, 2′-OH | 3′-PO4, 2′-OH | No | ? |
Yeast and plant tRNA ligases are composed of an N-terminal ligase domain, a central polynucleotide kinase domain and a C-terminal 2′,3′-cyclic phosphodiesterase domain (9,25,26). The CPDase domain of yeast/plant tRNA ligase belongs to the 2H phosphoesterase superfamily (26,34). The tRNA ligase CPDase reaction generates an exclusive 3′-OH, 2′-PO4 product. Unlike T4 Pnkp or CthPnkp, the tRNA ligase CPDase cannot hydrolyze 2′-PO4 or 3′-PO4 termini (24). And unlike T4 Pnkp, the plant tRNA ligase CPDase reaction is independent of a metal cofactor (B. Remus and S. Shuman; unpublished data) and does not involve a covalent intermediate.
The recently elucidated pathway of 2′,3′-cyclic phosphate/5′-OH end joining by E. coli RtcB involves an initial manganese-dependent 2′,3′-cyclic phosphodiesterase reaction that converts RNA > p to RNA3′p and a subsequent manganese-dependent 3′-PO4/5′-OH ligation reaction that requires GTP and proceeds through covalently activated RtcB-(histidinyl-N)–GMP and RNA(3′)ppG intermediates (22,27,28). Available evidence suggests that the RtcB CPDase and ligase reactions are executed at a single active site. Whereas T4 Pnkp and RtcB both generate 3′-PO4, 2′-OH ends through their CPDase activities, RtcB, unlike T4 Pnkp, does not further hydrolyze the 3′-PO4 end.
It is apparent that nature has evolved many different chemical and structural solutions to the problem of 3′ end healing in RNA repair pathways (Table 1). In the cases of T4 Pnkp, CthPnkp and yeast/plant tRNA ligase, the phosphoesterase components are members of widely distributed enzyme superfamilies that have been harnessed to RNA repair, by the acquisition of substrate specificity determinants and by fusion of the phosphatase domain to one or more other catalytic components of the repair pathway. By contrast, RtcB exemplifies a novel enzyme family and a stand-alone catalytic unit. Too little is known at present about the RtcB family to speculate whether the many bacterial, archaeal and eukaryal RtcB homologs are devoted to RNA repair/splicing or whether their unique chemistry might be applied to other biological purposes. However, we are willing to speculate that the four flavors of cyclic phosphate end-healing enzymes in Table 1 are not the end of the story and that new enzymes and strategies for repairing broken ends await discovery.
FUNDING
Funding for open access charge: NIH [GM63611].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments