



Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive decline in memory and cognitive function. Pathological hallmark of AD includes aberrant aggregation of amyloid beta (Aβ) peptide, which is produced upon sequential cleavage of amyloid precursor protein (APP) by β- and γ -secretases. On the contrary, α-secretase cleaves APP within the Aβ sequence and thereby prevents Aβ generation. Here, we investigated the role of ubiquitin ligase Ube3a (involved in synaptic function and plasticity) in the pathogenesis of AD using APPswe/PS1δE9 transgenic mouse model and first noticed that soluble pool of Ube3a was age-dependently decreased in AD mouse in comparison with wild type controls. To further explore the role of Ube3a in AD patho-mechanism, we generated brain Ube3a-deficient AD mice that exhibited accelerated cognitive and motor deficits compared with AD mice. Interestingly, these Ube3a-deficient AD mice were excessively obese from their age of 12 months and having shorter lifespan. Biochemical analysis revealed that the Ube3a-deficient AD mice had significantly reduced level of Aβ generation and amyloid plaque formation in their brain compared with age-matched AD mice and this effect could be due to the increased activity of α-secretase, ADAM10 (a disintegrin and metalloproteinase-10) that shift the proteolysis of APP towards non-amyloidogenic pathway. These findings suggest that aberrant function of Ube3a could influence the progression of AD and restoring normal level of Ube3a might be beneficial for AD.
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorders clinically characterized by progressive loss of normal memory and cognition. It is also a prevalent form of dementia in elderly people. Pathological feature of AD includes extracellular deposition of amyloid beta (Aβ) plaques and cytoplasmic aggregation of hyper-phosphorylated tau as well as Aβ peptide (1,2). Amyloid precursor protein (APP, a transmembrane protein) is sequentially cleaved by β-secretase (beta-site APP cleaving enzyme 1, BACE1) and γ-secretase to produce Aβ1-40 and highly neurotoxic Aβ1-42 (3). On the other hand, proteolysis of APP by α-secretase, a disintegrin and metalloproteases (ADAM) 9, 10, 17 prevents Aβ production (non-amyloidogenic pathway), because the cleavage site is located within the Aβ sequence(4). Wide range of mutations in APP or γ-secretase subunits, presenilin 1 (PS1) and PS2 are known to cause early onset familial AD (FAD) and thereby strongly linking the role of Aβ in AD pathogenesis (5,6). Since, FAD constitutes only about 5–10% of total AD, it is believed that various other cellular pathways could be involved in the regulation of Aβ metabolism. Those pathways could not only be linked with altered amyloidogenic or non-amyoloiedogenic processing of APP but also might prevent clearance of extracellular or intracellular Aβ (2). Extensive studies using cellular models, transgenic mice models and post-mortem AD brain samples have demonstrated that how neurotoxic small Aβ oligomers could trigger neuronal dysfunction and degeneration that are connected in the pathogenesis of AD. Some of the neuronal defect includes hyper-phosphorylation of microtubule associated protein tau and formation of neurofibrillary tangles, oxidative stress, inflammation, synaptic dysfunction and inhibition of hippocampal long-term potentiation etc. (2,7–10).
The trafficking of APP is tightly regulated as it travels through secretory and endo/lysosomal compartments of cells, where proteolytic processing by α-/β- and γ-secretases take place. Several lines of evidence suggest that ubiquitination of APP regulates its trafficking and the generation of Aβ (11–13). Similarly, autophagic degradation pathway is also implicated in the metabolism of Aβ and autophagosomes are shown to generate and comprise Aβ (14–16). These findings indicate that ubiquitination of APP might be playing a critical role in the metabolism of Aβ. In this context, identification of ubiquitin ligase linked with the ubiquitination of APP is very crucial. So far, several ubiquitin ligases have been shown to be involved in the ubiquitination of APP and metabolism of Aβ including CHIP (C-terminus of HSP70 interacting protein), parkin, FBL2 (F-box and leucine rich repeat protein 2) and HRD1 (17–19). Overexpression of those ligases in the cell culture system promotes the ubiquitination and degradation of APP with a concomitant decrease in Aβ generation. The level of some of these ligases like FBL2 and HRD1 were also significantly decreased in AD brain implicating their possible involvement in the development of AD (20,21).
Recently, ubiquitin ligase Ube3a has been shown to promote the clearance of various disease-linked misfolded protein and also implicated in the progression of mouse model of Huntington’s disease (HD) (22–24). Ube3a also plays an important role in synaptic function and plasticity (25,26). Moreover, the expression of paternally inherited UBE3A gene is imprinted in the brain and the loss of function of maternally inherited UBE3A causes Angelman syndrome (AS), which is characterized by intellectual and developmental disabilities (27–29). Here, we aimed to investigate the role of Ube3a in the metabolism of Aβ and progression of AD using its APPswe/PSEN1δE9 mouse model. We first found that the level of Ube3a was age-dependently decreased in these AD mice compared with age-matched controls. Next, we generated Ube3a-maternal deficient AD mice by taking the advantage of imprinted Ube3a expression in the brain. These Ube3a-maternal deficient AD mice exhibited aggravated AD-related behavioral phenotypes, although the level of Aβ plaque is significantly reduced in their brain compared with AD mice. Finally, we demonstrated that deficiency of Ube3a leads to increased expression of ADAM10 resulting in non-amyloidogenic processing of APP and decreased formation of Aβ.
Results
Age-dependent decline in Ube3a level in AD mice brain
In order to investigate the possible link of Ube3a in AD pathogenesis, we first tested age-dependent alteration in the expression of Ube3a in the brain of AD mice in comparison with age-matched wild type controls. Cortical samples from wild type and AD mice of different age groups were collected, soluble and insoluble fractions were made and then soluble fractions were subjected to immunoblot analysis using antibody against Ube3a. As shown in the Figure 1A and B, Ube3a level was almost at the similar level between wild type and AD mice at 4 months of age. However, its level was significantly diminished at 8- and 12-months old AD mice when compared with controls and the decrease was progressive. Even in wild type mice, Ube3a level was considerably decreased at 12 months of age with respect to 4 months. Interestingly, the mRNA level of Ube3a was unaltered between wild type and AD mice brain at 4, 8 and 12 months of age (Fig. 1C). This result indicates that the stability of Ube3a might be affected in older AD mice brain as well as in normal aging mice. To further confirm the stability of Ube3a in AD mice brain, we analyzed Ube3a level in the insoluble fraction using dot blot assay. Insoluble fractions prepared from AD mice cortical samples showed significantly increased accumulation of Ube3a compared with wild type controls particularly at 12 months of age (Fig. 1D and E) and this result was well correlated with the accumulation of Aβ plaque (Fig. 1D and F). Insoluble fractions obtained from cortical lysates of 12-months old wild type and AD mice were further analyzed by immunoblot analysis and results were very similar to dot blot assay (Fig. 2A and B). Next, we performed immunohistochemical staining of Ube3a to study the expression and localization of Ube3a in different brain regions of wild type and AD mice. The expression and localization pattern of Ube3a in cortical and hippocampal neurons were very similar at 4-months old wild type and AD mice brain. Ube3a was localized in both cytoplasmic and nuclear compartment with predominant nuclear staining. However, at 12 months age, Ube3a staining was considerably reduced in many cortical and hippocampal neurons of AD mice brain in comparison with age-matched controls (Fig. 2C). The reason for reduced Ube3a immunoreactivity in older AD mice brain is not clear at present. Ube3a could form self-associated homo multimers leading to blockade of antibody-binding site.
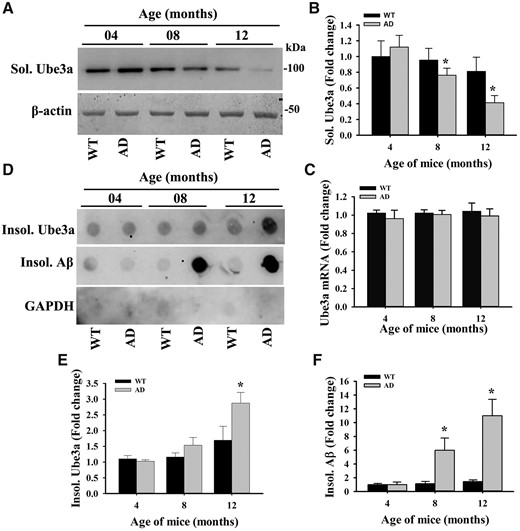
AD mice showed age-dependent decrease in Ube3a level in their brain. Cortical brain lysates obtained from wild type and AD mice of different age groups were centrifuged at 15 000g for 15 min and soluble and insoluble fractions were separated. Soluble fractions were subjected to immunoblot analysis using Ube3a and β-actin antibodies (A, B), while insoluble fractions were processed for dot blot analysis using Ube3a and 6E10 (that detect Aβ) antibodies (D–F). Band intensities of soluble Ube3a (B) were quantified using NIH Image analysis software, normalized against β-actin and expressed as fold change. Spot intensities of insoluble Ube3a (E) and insoluble Aβ (F) were also quantified and expressed as fold change. (C) Quantitative real time RT-PCR analysis of Ube3a mRNA in the cortical sample. Values are mean ± SD with five mice in each age group. *P < 0.01 compared with wild type control (one-way ANOVA followed by Holm-Sidak post-hoc test).
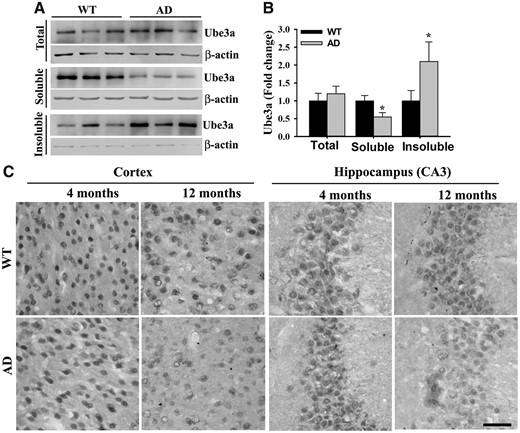
(A) Increased accumulation of Ube3a in the insoluble fraction. Cortical brain lysates obtained from wild type and AD mice at 12 months of age were subjected to the preparation of soluble and insoluble fractions as described in the ‘Materials and Methods’ section. Total lysates, soluble and insoluble fractions (after solublization with SDS) were then subjected to immunoblot analysis using Ube3a and β-actin antibodies. (B) Band intensities of Ube3a present in different fractions were quantified using NIH Image analysis software, normalized against β-actin and expressed as fold change. Each lane represents sample from different mice. Values are mean ± SD; n = 3. (C) Representative immunohistochemical staining of Ube3a. Brain sections obtained from wild type and AD mice of different age group were kept on same slide and processed for immunostaining. Scale bar, 50 µm.
Ube3a-maternal deficient AD mice exhibit progressively increased body weight, accelerated behavioral abnormalities and early death
To further explore the role of Ube3a in AD pathogenesis, we created Ube3a-maternal deficient AD mice by crossing female Ube3a-maternal deficient heterozygous mice with AD males. Because the paternally inherited Ube3a is imprinted in the neuron, Ube3a-maternal deficient heterozygous mice will not express Ube3a in their neurons. Newly generated mice were genotyped and categorized as wild type, Ube3a-maternal deficient, AD and Ube3a-maternal deficient AD (Supplementary Material, Fig. S1A). Immunoblot analysis of the expression of transgenic APP (detected by 6E10 antibody) and PS1 as well as Ube3a further confirmed the definite genotypes (Supplementary Material, Fig. S1B). Mice belong to all four genotypes appeared to be phenotypically similar till their age of 4 months. From the age of 5–6 months, Ube3a-maternal deficient AD mice showed visible increase in body weight compared with other genotypes and at 12 months of age there was about 50% increase in body weight of these mice compared with wild type or AD group (Fig. 3A and B). Ube3a-maternal deficient mice also exhibited significant increase in body weight at 12 months of age when compared with wild type and AD mice group. Interestingly, we noticed that lifespan of Ube3a-maternal deficient AD mice were comparatively shorter with respect to other genotypes (Fig. 3C). Ube3a-maternal deficient AD mice started dying from 9 months onwards and at 20 months, all of them were died. Although 20% of AD mice were died at the age of 20 months. Ube3a-maternal deficient mice survived normally similar like wild type till their age of 20 months.
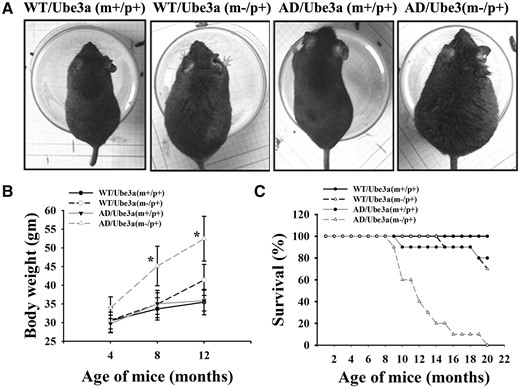
Ube3a-maternal deficient AD mice showed progressive obese phenotype and shorter lifespan. Ube3a-maternal deficient AD mice along with three other genotypes were regularly monitored for their body weight and survival up to 24 months of their age. (A) Snapshot of mice of all four genotypes at their age of 12 months. Note the obese phenotype of Ube3a-maternal deficient AD mice. Comparison of body weight at different ages (B) and survival rate (C) of mice having four different genotypes. Values are mean ± SD of 10 animals in each genotypes. *P < 0.001 when compared with wild type, Ube3a-maternal deficient and AD mice groups (two-way ANOVA followed by Holm-Sidak post-hoc test).
We next compared the cognitive and motor behavioral deficits among these four genotyped mice. The novel object recognition test was used to measure the cognitive function in these groups of mice. This task is based on the rodent‘s usual tendency to explore novel objects and preference for novel object is used as an index to evaluate cognitive function. In this test, Ube3a-maternal deficient AD mice showed significant differences in cognitive dysfunction compared with AD and Ube3a-maternal deficient mice from 8 months onwards. AD mice exhibited significant cognitive deficits from the age of 12 months (Fig. 4A). In rotarod performance test, Ube3a-maternal deficient AD mice also showed accelerated motor co-ordination deficits compared AD and Ube3a-maternal deficient mice (Fig. 4B). Increased body weight of Ube3a-maternal deficient AD mice at higher age (particularly at 12 months of age) might affect rotarod performance. To minimize the interference, we performed the experiment at much slower rotarod speed. Ube3a-maternal deficient mice demonstrated motor deficits from 8 months of age under the present experimental paradigm (15 rpm of rotarod speed); however, at higher speed these abnormalities can be detected from early age. It is also important to note that cognitive and motor deficits are observed in these AD mice from 12 to 18 months of their age, while these abnormalities are detected in Ube3a deficient mice from very early age (30–32). Interestingly, in clasping test, Ube3a-maternal deficient AD mice demonstrated robust abnormalities from 2 months onwards compared with all other mice groups (Fig. 4C and D).
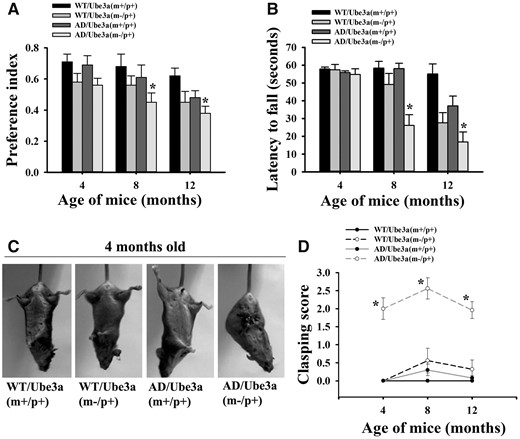
Ube3a-maternal deficient AD mice exhibits exacerbated behavioral abnormalities. Mice of all four genotypes were assessed for cognitive and motor tasks at 4, 8 and 12 months of their ages. (A) Novel object recognition test. Motor performance and coordination as evaluated from rotarod test at 15 rpm (B) and clasping behavior (C, D). All these tests were conducted in a blinded manner. Data represented as mean ± SD; n = 10 in each group. *P < 0.05 (A) and *P < 0.001 (B and D) when compared with wild type, Ube3a-maternal deficient and AD mice groups (two-way ANOVA followed by Holm-Sidak post-hoc test).
Decreased Aβ plaque load in the brain of Ube3a-maternal deficient AD mice
We further attempted to explore the probable cause for aggravated various behavioral deficits in Ube3a-maternal deficient AD mice including the impact of Ube3a deficiency on the APP metabolism and production of Aβ. Mice of all four genotypes were sacrificed at 4, 8 and 12 months of their ages and brain samples were collected and analyzed for various biochemical parameters either by immunohistochemical staining or immunoblot analysis. Notably, the total brain weight was significantly decreased in the case of Ube3a-maternal deficient AD mice compared with all other groups (Supplementary Material, Fig. S2A). The number of GFAP-positive astroglial cell was considerably increased in the cortical region of 12-months old AD mice compared with wild type and Ube3a-maternal deficient mice and this is reported earlier in (33). However, GFAP-positive cells number were significantly lower in the brain of Ube3a-maternal deficient AD mice when compared with AD group (Supplementary Material, Fig. S2B). No TUNEL positive cells were detected in either in AD or in Ube3a-maternal deficient AD mice brain (Supplementary Material, Fig. S2C). These results indicate that inflammation is not a contributing factor in the early death of Ube3a-maternal deficient mice. Next, we compared Aβ plaques load in the brain of AD and Ube3a-maternal deficient AD mice through immunohistochemical staining of Aβ using 6E10 antibody (an antibody that detect soluble monomeric and oligomeric Aβ, Aβ plaques as well as APP). Surprisingly, we observed a significant reduction in the deposition of Aβ plaques in the cortex and hippocampus of Ube3a-maternal deficient AD mice brain with respect to AD mice (Fig. 5 and Supplementary Material, Fig. S3 for lower magnification images). AD mice exhibited fewer Aβ plaques in the cortex and hippocampus at 4 months of age, which was progressively increased over time. Although in Ube3a-maternal deficient AD mice brain, Aβ plaques were detectable from about 8 months of age and increased gradually but was comparatively lower than AD mice. Quantification of amyloid plaques area (6E10 positive) in both cortical and hippocampal regions of 8 and 12 months old mice were shown in Figure 5B and C. At 8 months of age, both cortical and hippocampal regions showed more than 50% reduction of Aβ plaques load in Ube3a-maternal deficient AD mice. Because of the ubiquitin ligase role of Ube3a, we further checked the ubiquitination profile of Aβ plaques in the Ube3a- maternal deficient AD brain along with AD and observed no differences in the staining pattern of ubiquitin. However, ubiquitin immunostaining also showed very clear differences in Aβ plaques load in Ube3a-maternal deficient AD mice brain in comparison with AD group (Fig. 6A). Interestingly, the profile of global ubiquitinated proteins (assessed by immunoblot analysis of ubiquitin) were significantly decreased in Ube3a-deficient cortical samples indicating that Ube3a plays a very important role in the ubiquitination of many neuronal proteins (Fig. 6B and C). In order to understand the cause behind reduced accumulation of Aβ plaques in Ube3a-maternal deficient AD mice brain, we first compared the level of transgenic APP including soluble monomeric and oligomeric Aβ (detected using 6E10 antibody) and PS1 using immunoblot analysis. As shown in Figure 7, total APP and PS1 levels were unaltered in the cortical sample between AD and Ube3a-maternal deficient AD brain. However, monomeric and oligomeric Aβ levels as well as insoluble Aβ (in the gel top) were comparatively decreased in the cortical sample of Ube3a-maternal deficient AD mice. The mRNA level of transgenic APP and PS1 were also unaffected in these mice (Fig. 7E). These results indicate that Ube3a deficiency results in decreased formation of Aβ peptides.
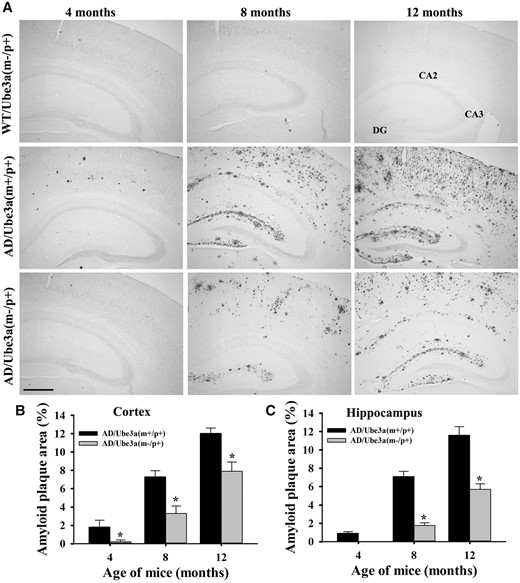
Decreased Aβ plaques load in the brain of Ube3a-maternal deficient AD mice. (A) Representative immunohistochemical staining of Aβ plaques in cortical and hippocampal regions as detected by 6E10 antibody. Brain sections obtained from wild type, AD and Ube3a-maternal deficient AD mice at different ages were placed on same slides and subjected to immunostaining. Brain sections from at least four different mice in each group were analysed. Scale bar, 500 µm. Quantitative analysis of 6E10-positive Aβ plaques area as a percentage of total areas in the cortical (B) and hippocampal (C) region using ImageJ. Values are mean ± SD of 4 animals/genotype/age group. *P < 0.001 compared with AD group (one-way ANOVA).
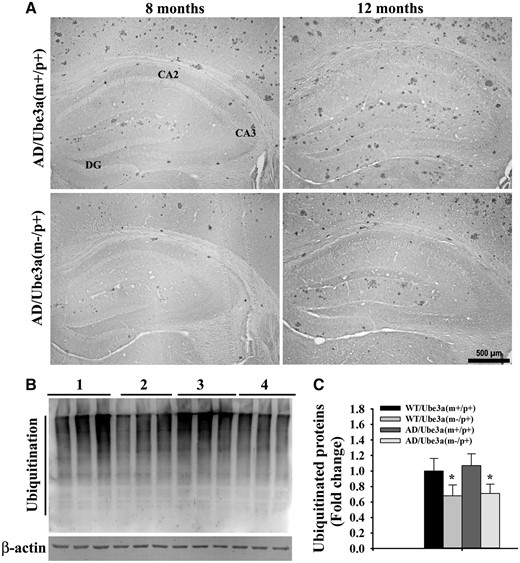
(A) Representative immunohistochemical staining of ubiquitin in the brain section obtained from AD and Ube3a-maternal deficient AD mice of different age groups. Ubiquitin-positive Aβ plaques were considerably lower in Ube3a-maternal deficient AD mice with respect to AD. (B) Immunoblot analysis of ubiquitin in the cortical lysate of 12-months old AD and Ube3a-maternal deficient AD mice. Group 1, wild type; Group 2, Ube3a-maternal deficient; Group 3, AD; Group 4, Ube3a-maternal deficient AD. Each lane of the blot represent sample from different animal. Values are mean ± SD of three animals/genotype/age group. *P < 0.05 compared with AD group (one-way ANOVA).
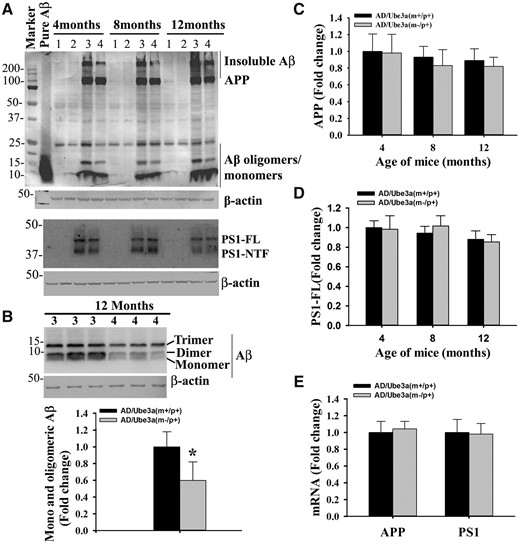
Ube3a deficiency impairs the rate of formation of Aβ peptides. (A, B) Cortical lysates obtained from mice of different genotypes were subjected to immunoblotting to check levels of transgenic human APP, PS1 and monomeric and oligomeric Aβ peptides. Band intensities of monomeric/oligomeric Aβ (B), FL APP (C) and PS1 (D) normalized to β-actin were quantified and plotted as fold change. (E) Quantitative real time RT-PCR analysis of transgenic APP and PS1 mRNA in the cortical sample. Values are mean ± SD of three animals/genotype/age group. Lane 1, wild type; Lane 2, Ube3a-maternal deficient; Lane 3, AD; Lane 4, Ube3a-maternal deficient AD. In B, each gel lanes were loaded with samples from different mice. Note the decreased level of Aβ monomers/oligomers (P < 0.001, Students t-test) and insoluble Aβ in the Ube3a-maternal deficient AD mice.
Ube3a deficiency leads to increased expression of ADAM10 in AD mice brain
Aβ peptide is produced through sequential cleavage of APP by β- and γ-secretases at ectodomain and intramembrane sites, respectively. On the contrary, juxtamembrane cleavage of APP by α-secretase (in between β- and γ-cleavage sites) prevents Aβ generation. Since PS1 (catalytic subunit of γ-secretase) level was unaffected between AD and Ube3a-maternal deficient AD mice brain, we next checked the endogenous expression of BACE1 (β-secretase) and ADAM10 (primary constitutive and inducible α-secretase in the neuron). Figure 8 demonstrated that the deficiency of Ube3a did not affect the expression level of BACE1. However, increased level of BACE1 was detected in AD mice brain, which is reported earlier (34). Interestingly, Ube3a deficiency caused up-regulation of ADAM10 in the brain of Ube3a-maternal deficient and Ube3a-maternal deficient AD mice. Levels of both precursor and matured ADAM10 were significantly increased in the absence of Ube3a (Fig. 8). Quantitative real time RT-PCR analysis of ADAM10 mRNA further revealed that its transcription was significantly increased in the absence of Ube3a (Fig. 9A). Since ADAM10 activity leads to increased formation of sAPPα (α-secretase cleavage N-terminal fragment of APP), we next analyzed the level of sAPPα in the cortical lysate of 12-months old Ube3a-maternal deficient AD mice along with AD. Because of very close molecular weight of full length (FL) APP and sAPPα, we first immunodepleted the FL APP from the sample (using Y188 antibody that detects C-terminal fragments of APP) and then samples were immunoblotted using 6E10 antibody to detect sAPPα. As shown in Figure 9B, immunodepleted samples showed very negligible amounts of FL APP when detected with Y188 antibody. However, sAPPα level was significantly increased in the immunodepleted cortical sample prepared from Ube3a-maternal deficient AD mice with respect to AD. These results indicate that Ube3a is involved in regulating the expression of ADAM10 and up-regulation of ADAM10 in Ube3a-maternal deficient AD mice brain might lead to increased formation of sAPPα. To further understand the mechanism of transcriptional up-regulation of ADAM10 in Ube3a deficient mice, we checked the level of peroxisome proliferator-activated receptor-α (PPARα), because its activity has been shown to be negatively regulated by Ube3a (35). We have detected a significant increase in the level of PPARα in the brain sample of 12-months old Ube3a-maternal deficient as well as Ube3a-maternal deficient AD mice (Fig. 9C). Increased level of PPARα in the Ube3a deficient mice could be due its altered turnover that requires further investigation. Our findings indicate that Ube3a deficiency results in increased activation of PPARα, which in turn causes up-regulation of ADAM10.
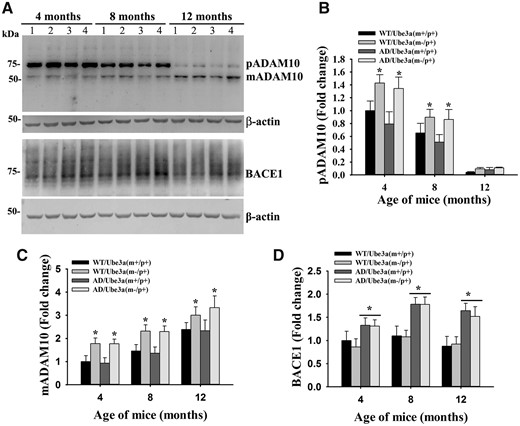
Increased expression of ADAM10 in the cortex of Ube3a-maternal deficient AD mice. (A) Immunoblot analysis of ADAM10 and BACE1 in the cortical lysate prepared from mice of different genotypes at their different ages. (B–D) Band intensities of pADAM10 (precursor ADAM10, mADAM10 (matured ADAM10) and BACE1 were quantified, normalized and plotted as fold change. Values are mean ± SD of three animals/genotype/age group. In (B) and (C), *P < 0.05 compared with either wild type or AD mice and in D, *P < 0.01 in comparison with wild type or Ube3a-maternal deficient mice groups. Two-way ANOVA followed by Holm-Sidak post-hoc test was used to analyze the data. Lane 1, wild type; Lane 2, Ube3a-maternal deficient; Lane 3, AD; Lane 4, Ube3a-maternal deficient AD.
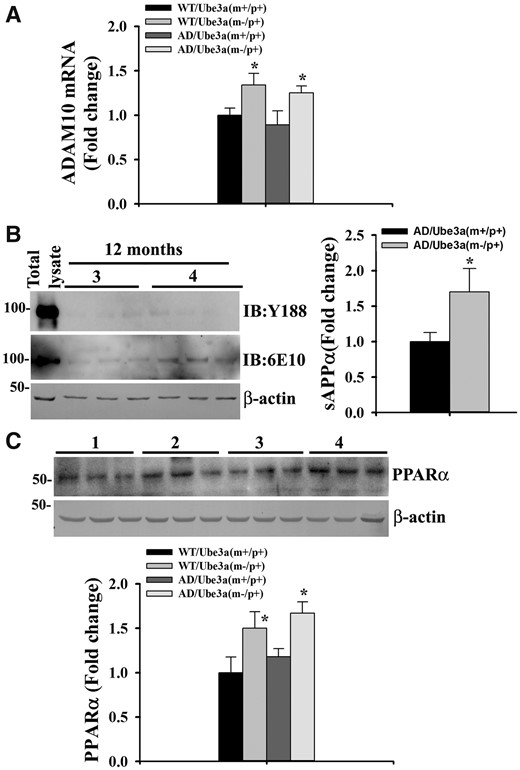
Ube3a deficiency leads to transcriptional up-regulation of ADAM10 and increased formation of sAPPα. (A) Quantitative real time RT-PCR analysis of ADAM10 mRNA in cortical samples of 12-months old mice of different genotypes. Values are mean ± SD; n = 3. *P < 0.05 with regard to either wild type or AD mice groups. (one-way ANOVA). (B) Detection of sAPPα in the cortical lysate of 12 months AD and Ube3a-maternal deficient AD mice. Cortical lysates were immunodepleted with Y188 antibody and then were subjected to immunoblot analysis using Y188 (to check immunodepletion of total APP), 6E10 (to detect sAPPα in immunodepleted samples) and β-actin antibodies. Group 3, AD; Group 4, Ube3a-maternal deficient AD. Right panel showed the quantitation of sAPPα in AD and Ube3a-maternal deficient AD samples. Values are mean ± SD with 3 mice in each group. *P < 0.01 compared with AD mice groups. (Student t-test). (C) Immunoblot analysis of PPARα in the cortical lysate of 12 months old mice of different genotypes. Lane 1, wild type; Lane 2, Ube3a-maternal deficient; Lane 3, AD; Lane 4, Ube3a-maternal deficient AD. Values are mean ± SD with three mice/group. *P < 0.05 with regard to either wild type or AD mice groups. (one-way ANOVA). In (B) and (C), each lane of the blot represent samples from different animals.
Discussion
One of the distinctive features of AD is the extracellular accumulation of insoluble Aβ called senile plaques, which emerge from the processing of APP through sequential action of β- and γ-secretases. In contrast, α-secretase-mediated processing of APP prevents amyloid plaque formation. In AD, activities of β-and γ-secretases overcome the function of α-secretase and exacerbate Aβ production and AD pathogenesis. Therefore, understanding the modulation in the activity of secretases, particularly ADAM10 is hot spot for potential therapeutic approach. In the current study, we revealed that Ube3a regulates the expression of ADAM10 and thereby could be implicated in the pathogenesis of AD. First, we demonstrate that the level of Ube3a is age-dependently decreased in the cortex and hippocampus of AD mice compared with age-matched wild type controls. Second, Ube3a-maternal deficient AD mice exhibit significantly reduced level of Aβ plaques along with exaggerated disease phenotype compared with AD mice. These Ube3a-deficient AD mice were excessively obese and had shorter lifespan. Finally, we show the increased expression of ADAM10 in the brain of Ube3a-maternal deficient AD mice.
Ube3a plays a crucial role in synaptic function and plasticity and Ube3a-maternal deficient mice (model mice for AS) exhibit severe cognitive and motor deficits (25,26,31,36). The level of Ube3a has been shown to be reduced in the normal ageing human brain as well as in the symptomatic HD mice brain (24,37,38). The decrease in soluble level of Ube3a could be due to the oxidative stress that results in oligomerization and aggregation of Ube3a into high molecular weight complex. Oxidative stress is a common event observed across most age-related neurodegenerative disorders including AD and solubility of various ubiquitin ligases including Ube3a are shown to be affected by oxidative stress (39–41). Notably, the progressive age-dependent decline of soluble Ube3a in AD mice indicate that Ube3a might be linked with the cognitive dysfunction in AD mice. There are several evidences indicating the role of enriched environment in improving the cognitive deficit in AD mice and in some cases beneficial effect can be observed irrespective of increased level of Aβ plaques (42–44). Ube3a could play an important mediator in enriched environment-induced improvement in cognitive function, because of its up-regulation during neuronal activity and enriched environment (36). Even, increased level of Aβ plaques during the exposure of enriched environment could also be associated with Ube3a up-regulation. This aspect requires further investigation.
In order to gain further insight about the involvement of Ube3a in AD progression, we made Ube3a-maternal deficient AD mice. Surprisingly, these Ube3a-maternal deficient AD mice exhibited significantly reduced level of Aβ generation and plaque formation in their brain compared with age-matched AD mice. In depth analysis revealed that the deficiency of Ube3a resulted in increased expression of ADAM10 in AD mice brain that could lead to increased α-secretase cleavage of APP and consequently the decreased formation of Aβ. Expression levels of mutant transgenes (APP and PS1) as well as endogenous BACE1 were unaffected in these mice. ADAM10 is one of the major constitutive and inducible APP α-secretase in the neuron (45). Overexpression of ADAM10 suppress the Aβ plaque formation in AD mouse model, while blocking its synaptic trafficking generates a model of sporadic AD (46,47). Activation of PPARα reported to induce the expression of ADAM10 and shift the proteolysis of APP towards α-secretase pathway. Furthermore, disruption of PPARα increases the Aβ plaque load in the brain and reduces the survival rate of 5xFAD mice (48). Interestingly, activities of PPARs are shown to be negatively regulated by Ube3a (35) and we have detected increased level of PPARα in the brain of Ube3a-maternal deficient AD mice. Therefore, it is conceivable that up-regulation of PPARα in the Ube3a-maternal deficient AD mice might be leading increased expression of ADAM10 and subsequent α-secretase cleavage of APP.
Despite decreased Aβ plaque load in their brain, Ube3a-maternal deficient AD mice displayed exacerbated cognitive and motor dysfunction when compared with AD mice. This is not surprising, because, both AD and Ube3a-maternal deficient mice manifest cognitive and motor deficits and therefore, one could expect additive effect of those behaviors in Ube3a-maternal deficient AD mice. Surprisingly, the clasping behavior that is mildly exhibited by older Ube3a-maternal deficient mice is aggravated in Ube3a-maternal deficient AD mice and can be seen from 2 months onwards. The exact reason for abnormal clasping behavior in Ube3a-maternal deficient mice is not fully understood. Dysfunction in basal ganglia circuitry could be linked with clasping behavior as several reports in various mouse model of neurological disorders have indicated (49,50). These observations hinted a possible defect in basal ganglia circuitry in AD mice brain. In fact, AD patients exhibit extrapyramidal motor abnormalities and some of these defects could be attributed due to the dysfunction in basal ganglia (51). Similarly, Ube3a-maternal deficient AD mice are also unusually obese. Excessive obesity might also contribute to increased motor abnormalities and early death of these mice. Obesity is also one of the risk factors for AD (52,53). The reason for increased obesity in Ube3a-maternal deficient AD mice is not clear presently. This could be due to up-regulation of pro-adipogenic factor C/EBPα and well as PPARγ, because both of them are target of Ube3a (35,54). The altered level of C/EBPα and PPARs and their possible link with obese phenotype in Ube3a-maternal deficient mice or AD mice are not known and warrant further investigation. Nonetheless, these exacerbated phenotypes could be helpful to identify altered signaling molecules and pathways that are subtly altered in early stages of AD or Ube3a-maternal deficient mice.
Altogether, our study identified Ube3a as a novel regulator of ADAM10. Aberrant function of Ube3a in AD mice brain might be linked with cognitive and motor deficits as well as α-secretase proteolysis of APP. Therefore, preserving optimal level of Ube3a might be beneficial in AD.
Materials and Methods
Materials
Mouse monoclonal anti-Ube3a (WH0007337M1), mouse monoclonal anti-β-actin (A5316) and rabbit polyclonal anti-PPARα (SAB2101852) were purchased from sigma. Anti-6E10 (SIG-39320), and anti-presenilin1 N-terminal antibody-NT1(SIG-39194) were purchased from Covance. Anti-ADAM10 (AB39177) and anti-APP (Y188, AB32136) were from Abcam, anti-BACE1 (AB5940) was from Millipore, anti-ubiquitin (Z0458) was from Dako and anti-Ube3a (611416) was from BD Bioscience. Alkaline phosphatase (AP), horse radish peroxidase (HRP) and biotin-conjugated secondary antibodies as well as VECTASTAIN-Elite ABC reagent and ImmPact Novared staining kit were purchased from Vector Laboratories.
Animals
B6C3-Tg(APPswe/PSEN1δE9)85Dbo/J transgenic mice (AD mouse model) and heterozygous Ube3a (129-Ube3atm1Alb/J) mice were obtained from the Jackson Laboratory and maintained in animal house facility of the Institute. This AD transgenic mouse line express APPswe (K670N and M671L) mutations in humanized mouse APP cDNA and Exon-9 deleted human PS-1 (PSEN1dE9) cDNA under the regulation of prion gene promoter (55). Male AD transgenic mice were crossed with Ube3a-maternal deficient (m−/p+) females to generate Ube3a-maternal deficient AD mice (AD/Ube3a(m−/p+). Genotyping was carried out as previously described in (31,55). These mixed background mice were used in all experiments. Animals of four different genotypes (wild type, AD, Ube3a-maternal deficient and Ube3a-maternal deficient AD) were subjected to different behavioral tests at 4, 8 and 12 months of their age. Mice were handled strictly according to guidelines proposed by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forestry, Government of India. All animal experiments were approved by the Institutional Animal Ethics Committee (IAEC) of the National Brain Research Centre (Protocol no: NBRC/IAEC/2012/71).
Bodyweight measurement and survival studies
Body weight of mice was measured at 4, 8 and 12 months of age before the beginning of behavioral studies. Life span of mice was assessed once in 15 days. Survival data were analyzed by Kaplan-Meier survival analysis and multiple curves were compared by log rank test.
Behavioral studies
Novel object recognition test
To test cognitive function in mice, novel object recognition test was carried out in an open-field box with dimensions of 45 × 45 × 20 cm. Each mouse was habituated in empty open-field for three sessions (10 min/session) per day for 3 days. Mice were next allowed to explore two identical objects (similar in size, shape and colour) placed in an open field 15 cm away. Exploration of each object in terms of touch and visits was monitored through a camera mounted over the box. Mice were shifted back to their respective home cage just after familiarization of objects. After 2 h, retention test (for short-term memory) was performed. In retention test, one previous object was replaced with novel object, which was quite different in size, shape and colour. Mice were then allowed to explore objects and time spent to explore novel and old objects were measured. No objects have preferential priority of physical complexity and emotional association. To erase possible instinctive odorant cues, the open-field and objects were carefully wiped with 70% alcohol after each session. The amount of time spent in exploring the novel object (retention session) over the total time spent exploring both objects, was used to measure novel object recognition memory.
Tail suspension test
To assess the clasping behavior of limbs, mice were suspended with their tail for 30 s for three times. Severity of clasping was represented on 0–3 scale. Mice showed no clasping were allotted 0 score, mice showed clasping only of two limbs were given score 1, mice clasped their all four limbs with 30 and 5 s given scores 2 and 3 respectively. This test was performed in random manner to eliminate biasness.
Rotarod test
Balance and coordination were assessed through rotarod test. Mice were kept on rotating rod with different speeds (00, 05, 15 and 25 rpm). Mice were equally taught to make balance until wild type mice started staying up to 60 s on rotating rod at each speed. Mice were trained for 3 days and then tested three sessions per day for next 3 days at all three time points (4, 8 and 12 months). All sessions at each time point were averaged to calculate the time mice stayed on rotarod before fall. Maximum time to stay on rod was set to 60 s.
Immunoblotting and dot blot analysis
Mice were sacrificed at their age of 4, 8 and 12 months and different parts of the brain were dissected out and snap frozen in liquid nitrogen. For tissue lysate preparation, sample were homogenized in RIPA lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 10 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 10 mM NaF, 5 mM Na4P2O7, 0.1 mM Na2VO5, complete protease inhibitor cocktail) containing protease and phosphatase inhibitors. Samples were then centrifuged at 15 000×g for 15 min and supernatants were removed, proteins amounts were quantified through BCA methods and then stored at −80 °C in different aliquots until use. One aliquot of the sample was heat-denatured in 2× sample buffer and resolved in different percentage of SDS-PAGE. Protein was transferred onto 0.20 µm nitrocellulose membranes in Towbin buffer (25 mM Tris, 192 mM glycine and 20% (v/v) methanol) under semidry conditions. Membrane was blocked with 5% skimmed milk in TBST (50 mM Tris pH7.4, 150 mM NaCl, 0.1% (v/v) Tween-20) for 2 h and incubated with primary antibody over night at 4 °C under shaking condition. Membrane was washed with TBST and incubated with HRP or AP conjugated secondary antibodies (1:5000 dilutions in TBST) for 2 h at room temperature. Blot was washed and subsequently developed. Band intensities were analyzed using ImageJ software and normalized with β-actin and expressed as fold change.
For the analysis of Ube3a in soluble and insoluble fractions, cortical samples were homogenized in RIPA buffer as above followed by estimation of protein by BCA method. Samples (equal concentrations) were then centrifuged at 15000xg for 15 min and soluble and insoluble fractions were collected. Insoluble fractions were treated with DNase1 at 37 °C for 2 h and then centrifuged at 15 000×g for 10 min. Pellets were collected and re-suspended in 200 µl of 1× sample buffer with 2% SDS, boiled for 10 min and equal volume of different samples were run through SDS-PAGE (along with corresponding total lysates and soluble fraction) followed by immunoblot analysis using Ube3a antibody. For dot blot analysis, collected pellets were re-suspended in 200 µl of 2% SDS, boiled for 10 min and equal volume of different samples were filtered through nitrocellulose membrane followed by probing the membrane with Ube3a and 6E10 antibodies. For APP immunodepletion experiment, soluble fractions (800 µg/200 µl) prepared from cortical lysates of different mice were incubated with 4 µl of Y188 antibody for 16 h at 4 °C. Samples were then mixed with protein G-agarose beads for 4 h at room temperature under shaking conditions followed by centrifugation at 5000g for 10 min. Supernatants were collected and subjected to immunoblot analysis using various antibodies.
Immunohistochemistry
Animals were anesthetized with Xylazine (10 mg/kg body weight) and Ketamine (100 mg/kg body weight) and perfused with PBS followed by 4% PFA in PBS and brains were carefully excised. Collected brains were kept in 4% paraformaldehyde for 24 h and then subjected to 10, 20 and 30% sucrose (in PBS) treatment followed by sectioning in freezing microtome (25 μm thickness). Serial brain sections were kept in PBS with 0.02% sodium azide and stored at 4 °C. For immunohistochemical staining, sections were kept on the glass slides, subjected to antigen retrieval at 70 °C for 30 min, washed, blocked in 2% BSA and then incubated with various primary antibodies for overnight. Anti-Ube3a, anti-6E10 and anti-ubiquitin were used at 1: 200, 1: 1000 and 1: 100 dilutions, respectively. After primary antibody incubation, sections were incubated with appropriate biotinylated secondary antibody at 1:500 s dilution and ABC kit was used to amplify the signal. Staining was developed by using Immpact Novared peroxidase substrate. Stained sections were visualized under Leica DM RXA2 microscope and photographs were assembled using Adobe Photoshop.
Quantitative real-time RT-PCR
RNA was extracted from cortical samples of different mouse lines using Trizol reagent (Sigma) following manufacturer protocol. Quantitative real-time RT-PCR for Ube3a, transgenic APP, PS-1 and ADAM10 was performed using power SYBR Green PCR master mix (Applied Biosystems) after cDNA synthesis from total RNA. PCR reaction was carried out using ViiA7 real time PCR system (Applied Biosystems), and data were subsequently analyzed and expressed as fold change. RT-PCR products were normalized with 18 S RNA as an internal control. Primer sequences for Ube3a, APP, PS-1, ADAM10 and 18 S RNA are as follows: Ube3aF, 5′-CATACCTGAGTCCAGCGAATTA-3′; Ube3aR, 5′-ACGCCAAGTTCGGTTTCT-3′; APPF, 5′-TTCCCGTGAATGGAGAGAGTTC-3′; APPR, 5′-ATGAACTTCATATCCTGAGTCATGTCG-3′; PS1F, 5′-GGTCCACTTCGTATGCTGGT-3′; PS1R, 5′- TTCCCATTCCTCACTGAACC-3′; ADAM10F, 5′-GGAAGATGGTGTTGCCGAC-3′; ADAM10R, 5′-TTTCCATACTGACCTCCCAGC-3’; 18SF, 5′-GAGGGA GCCTGAGAAACGG-3′; 18SR, 5′-GTCGGGAGTGGGTAATTTGC-3′.
Statistical analysis
One- or two-way ANOVA followed by Holm-Sidak past-hoc test were applied to analyze data in SigmaStat software. In some experiments, two-tailed Student‘s t-test was used for comparing the inter group. Data represent mean ± SD and P < 0.05 was considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
We would like to thank Dr Vijayalakshmi Ravindranath (founder Director of National Brain Research Centre) for providing the AD transgenic mice. We would like to thank Dr Ranjit Giri for his suggestions and providing several antibodies and Mr Mahendra Kumar Singh for his technical assistance.
Conflict of Interest statement. None declared.
Funding
This work was financially supported by the core grant of National Brain Research Centre, which is under Department of Biotechnology, Government of India. NRJ is a recipient of TATA Innovation Fellowship from Department of Biotechnology, Government of India (BT/HRD/35/01/03/2013).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}