




Abstract
Single nucleotide polymorphisms (SNPs) that alter exon splicing efficiency are an emerging class of functional genetic variants. Since mutations in low-density lipoprotein receptor (LDLR) are a primary cause of familial hypercholesterolemia, we evaluated whether LDLR SNPs may alter splicing efficiency and cholesterol homeostasis. A SNP within LDLR exon 12, rs688, was identified in silico as neutralizing a putative exon splicing enhancer. Studies in human liver samples established that this SNP was associated with significantly decreased LDLR exon 12 splicing efficiency in women in vivo . In vitro minigene splicing studies qualitatively replicated these in vivo results and demonstrated that rs688 specifically modulates splicing efficiency. These effects on splicing may be physiologically relevant because the presence of the rs688 minor allele associates with increased total and LDL-cholesterol in female members of the Framingham Offspring Study. The largest rs688-associated cholesterol differences were observed in pre-menopausal women. In summary, these studies identify an LDLR SNP present in ∼60% of Caucasians that is associated with significant 10% increases in total and LDL-cholesterol in pre-menopausal women.
INTRODUCTION
Single nucleotide polymorphisms (SNPs) that modulate splicing efficiency, often by altering exon splicing enhancers (ESEs), are increasingly recognized as functional genetic variants associated with clinically relevant phenotypes ( 1–5 ). The spliceosome identifies exons based on sequence elements at the intron/exon junction as well as enhancer and silencer elements in the adjoining exons and introns (reviewed in 6 , 7 ). ESEs are recognized by members of the splicing regulatory protein family, e.g. SRp40, that then attract splicing machinery. Within the past few years, algorithms have been developed that predict ESEs within a given RNA sequence ( 8–11 ). Recently, ESE alterations have been linked with altered splicing efficiency and disease. For example, a SNP within the complement component 5 gene was predicted to alter an ESE and was subsequently associated with inefficient splicing of the relevant exon in vitro , and with complement component 5 deficiency in vivo ( 4 ). Similarly, an SNP that neutralized a predicted ESE within the cystic fibrosis transmembrane conductance regulator was associated with inappropriate RNA splicing and an incompletely penetrant cystic fibrosis phenotype ( 5 ). Hence, ESE alterations can have functional impacts on RNA splicing and disease.
Low-density lipoprotein receptor (LDLR) is an excellent gene for candidate SNP studies because LDLR mutations are a primary cause of familial hypercholesterolemia, suggesting that other gene products do not compensate effectively for reduced LDLR function (reviewed in 12–14 ). Although LDLR SNPs and their haplotypes have been associated with cholesterol levels in many studies ( 15–17 ), SNPs that alter LDLR function have not been identified. Here, we report that the minor allele of rs688, predicted in silico to neutralize an LDLR exon 12 ESE, is associated with decreased LDLR splicing efficiency in vivo and increased total and LDL-cholesterol in women. Moreover, in vitro minigene studies demonstrate the functionality of this specific SNP in splicing efficiency.
RESULTS
To discern SNPs that may modulate LDLR ESEs, LDLR SNPs were analyzed relative to ESE matrices ( 18 , 19 ). Candidate SNPs were defined as those wherein only one allele was predicted to generate an ESE capable of binding its cognate regulatory protein. This process identified an exon 12 C/T SNP, rs688, as modulating two putative SRp40 binding sites, i.e. one ESE site is altered from the major allele TGTCAA C to the minor allele TGTCAA T (underline denotes SNP); this alteration decreases the SRp40 affinity score from 3.04 to 1.50, well below the SRp40 binding threshold of 2.67. An overlapping putative SRp40 binding site is altered from TCAA C GG to the minor allelic TCAA T GG, and is predicted to maintain SRp40 binding. Since an LDLR isoform lacking exon 12 has been reported ( 20 ), we interpreted these results as suggesting that rs688 may modulate the splicing efficiency of exon 12 or its flanking exons.
To investigate whether rs688 is associated with LDLR splicing efficiency, we evaluated LDLR splicing efficiency in liver samples obtained at autopsy from 21 female and 22 male age-matched individuals. Splicing was analyzed by using PCR to amplify LDLR cDNA from exon 10 to exon 14, allowing detection and relative quantitation of LDLR isoforms lacking intervening exons. We found that an LDLR isoform lacking exon 12 was readily detectable, while an isoform lacking both exons 11 and 12 was present at lower levels (Fig. 1 ). Parsing the splicing results by rs688 genotype revealed that splicing efficiency tended to decrease in individuals with the minor T allele of rs688 (Fig. 1 ). While the data from the male population did not reach statistical significance, inefficient splicing in women was significantly associated with rs688 ( P = 0.024). The apparent average decrease in splicing efficiency between homozygous rs688C/C and rs688T/T females was 8.6%. Hence, the minor rs688T allele was associated with significant decreases in LDLR splicing efficiency in women in vivo .
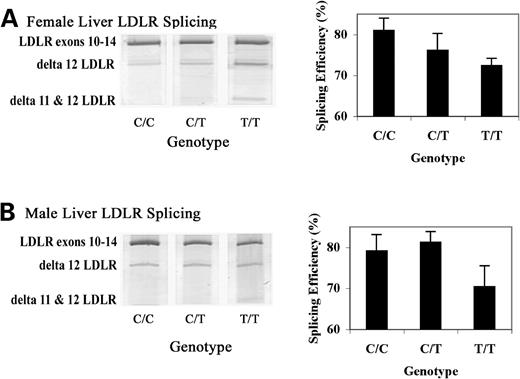
rs688 associates with LDLR splicing efficiency in female human livers in vivo . This figure depicts representative splicing results as well as quantitation for female ( A ) and male ( B ) liver samples. The bands corresponding to LDLR exons 10–14, as well as LDLR exons 10–11–13–14 (delta 12 LDLR), and LDLR exons 10–13–14 (delta 11 & 12 LDLR) are labeled. Sequencing established that the faint and inconsistent band above delta 12 LDLR represents delta 11 LDLR, which was not included in the quantitative analysis. rs688 was significantly associated with decreased splicing efficiency in females, i.e. P = 0.024 by non-parametric analysis. rs688 was not significantly associated with splicing efficiency in males ( P = 0.114).
To evaluate whether rs688 itself is functional, or only linked to functional SNP(s), we evaluated rs688 specifically by using a minigene, transfected cell in vitro approach. We began this process by cloning LDLR exons 9–14, along with the intervening introns, from the genomic DNA of two rs688C/C individuals and two rs688T/T individuals. These LDLR minigenes were transfected into HepG2 cells, a transformed liver cell line. RNA was then isolated 24 h later, and splicing efficiency evaluated (Fig. 2 ). This analysis revealed that the splicing efficiency differences observed in vivo were qualitatively replicated in vitro , i.e. the proportion of LDLR containing exons 11–12 was greater in the two rs688C minigenes than in the two rs688T minigenes (Fig. 2 ). The apparent splicing efficiency difference was ∼12%. Hence, although the in vitro splicing assay was not as efficient as that in vivo , the in vitro assay results support the in vivo results in that the rs688T allele is associated with less efficient LDLR splicing.
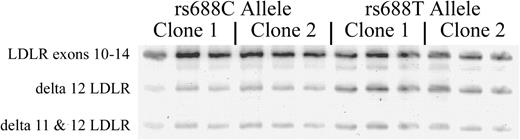
rs688 in vivo splicing patterns are replicated in vitro . Minigenes containing genomic LDLR exons 9–14 were cloned from two rs688C/C individuals and two rs688T/T individuals. One clone from each person was transfected into HepG2 cells and splicing patterns analyzed in triplicate beginning with cell transfections. The two rs688C genotype clones were associated with a higher splicing efficiency, i.e. 56 ± 3.8 and 53.4 ± 0.4 (%, mean ± SD, triplicate analysis) than the two rs699T genotype clones, i.e. 42.6 ± 0.8 and 40.4 ± 1.3. Similar results were observed in two separate experiments. Hence, relative differences in splicing efficiency in vivo are replicated in vitro .
Direct sequencing of the minigenes revealed several additional SNPs that differentiated the rs688C and rs688T alleles (Table 1 ), suggesting the possibility that rs688 may not be functional but only linked with a functional SNP. Therefore, we evaluated the effects of rs688 specifically by using site-directed mutagenesis to swap only rs688 within the minigenes, i.e. minigenes containing rs688C (rs688C) were converted to the minor T allele (rs688C→t) and rs688T minigenes to rs688C (rs688T→c). Sequencing of the resultant clones demonstrated the specificity of the mutagenesis. When these minigenes were evaluated for splicing efficiency, we found that converting rs688C to the minor T allele (rs688C→t) reduced exon 12 splicing efficiency, i.e. the LDLR isoform containing exon 12 was decreased relative to isoforms lacking exon 12 (Fig. 3 A and C). In parallel, converting rs688T to the major C allele (rs688T→c) increased the efficiency of exon 12 inclusion (Fig. 3 B and C). Hence, although additional SNPs define the rs688C and rs688T haplotypes, the major and minor rs688 alleles are sufficient to account for the differences in splicing efficiency. The magnitude of the rs688 effect on splicing is not atypical given that multiple regulatory elements contribute to splicing efficiency, including sequences at the intron/exon boundary as well as exonic and intronic enhancers and silencers ( 6 , 7 ). In summation, rs688 is a functional SNP on the basis of its association with splicing efficiency in vivo and its direct modulation of LDLR splicing in vitro .
![rs688 specifically modulates LDLR splicing efficiency in vitro . For each of the rs688C and rs688T minigenes, we used site-directed mutagenesis to convert the C allele to the minor T allele, or vice versa. The eight constructs were then transfected into HepG2 cells in parallel and RNA isolated for analyses 24 h later. These results depict representative images ( A and B ) and quantitation [ C , mean ± SD ( n = 3)]. Splicing efficiency was increased by the rs688C allele, regardless of haplotypic background ( P < 0.001).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/14/10.1093_hmg_ddm124/2/m_ddm12403.jpeg?Expires=1716624285&Signature=QKpRgizzervOKJutsELtYYZS1DraCuM7HTEdrWYNG9h6nnq~KYS4zacbzvpcjn1e97kx2d7g5ZnsDoIqouI4~dpurDvJ23KGz3jRJxMLZgposNpegnrwTR-SBZG1zc~eFUlFxBhfx9-vGi8iChZXPuE6KVoo2s~gGRuRVS5dTk7x0eUDPe4Wp5gCbcpprzvJD7myJEawhzE8E1W2cQTzRgrwW0~arQXv0cHcujJzsbS2YVWUtsI~czo4v738dX1cA1iGWh7eOHAOIKQUmQ2GgPtguYLLDoxU~vC53pPubb~S4VWA8igujjIht1bzSL0rcJ~3mQZMd~VUPBWtQJfCLQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
rs688 specifically modulates LDLR splicing efficiency in vitro . For each of the rs688C and rs688T minigenes, we used site-directed mutagenesis to convert the C allele to the minor T allele, or vice versa. The eight constructs were then transfected into HepG2 cells in parallel and RNA isolated for analyses 24 h later. These results depict representative images ( A and B ) and quantitation [ C , mean ± SD ( n = 3)]. Splicing efficiency was increased by the rs688C allele, regardless of haplotypic background ( P < 0.001).
Six additional SNPs near rs688 are differential within the LDLR minigenes
| SNP | Exon/intron | Frequency | rs688T | rs688C |
|---|---|---|---|---|
| rs1003723 | Intron 9 | C:0.40 | T | C |
| rs5930 | Exon 10 | A:0.36 | G | A |
| rs2738445 | Intron 11 | C:0.43 | T | C |
| rs2738446 | Intron 11 | G:0.29 | G | C |
| rs2738447 | Intron 11 | A:0.28 | C | A |
| rs5925 | Exon 13 | C:0.48 | C | T |
| SNP | Exon/intron | Frequency | rs688T | rs688C |
|---|---|---|---|---|
| rs1003723 | Intron 9 | C:0.40 | T | C |
| rs5930 | Exon 10 | A:0.36 | G | A |
| rs2738445 | Intron 11 | C:0.43 | T | C |
| rs2738446 | Intron 11 | G:0.29 | G | C |
| rs2738447 | Intron 11 | A:0.28 | C | A |
| rs5925 | Exon 13 | C:0.48 | C | T |
Three SNPs are within the 646 bp intron 11 with rs2738447 being the closest to exon 12 at 54 bp 5′ to exon 12. These intronic SNPs are in addition to the exon 10, exon 12 (rs688) and exon 13 SNPs that also distinguish these haplotypes.
Six additional SNPs near rs688 are differential within the LDLR minigenes
| SNP | Exon/intron | Frequency | rs688T | rs688C |
|---|---|---|---|---|
| rs1003723 | Intron 9 | C:0.40 | T | C |
| rs5930 | Exon 10 | A:0.36 | G | A |
| rs2738445 | Intron 11 | C:0.43 | T | C |
| rs2738446 | Intron 11 | G:0.29 | G | C |
| rs2738447 | Intron 11 | A:0.28 | C | A |
| rs5925 | Exon 13 | C:0.48 | C | T |
| SNP | Exon/intron | Frequency | rs688T | rs688C |
|---|---|---|---|---|
| rs1003723 | Intron 9 | C:0.40 | T | C |
| rs5930 | Exon 10 | A:0.36 | G | A |
| rs2738445 | Intron 11 | C:0.43 | T | C |
| rs2738446 | Intron 11 | G:0.29 | G | C |
| rs2738447 | Intron 11 | A:0.28 | C | A |
| rs5925 | Exon 13 | C:0.48 | C | T |
Three SNPs are within the 646 bp intron 11 with rs2738447 being the closest to exon 12 at 54 bp 5′ to exon 12. These intronic SNPs are in addition to the exon 10, exon 12 (rs688) and exon 13 SNPs that also distinguish these haplotypes.
Since loss of a single LDLR allele causes an approximate doubling of LDL-cholesterol ( 12 , 14 ), we hypothesized that these rs688 effects on splicing efficiency may be sufficient for rs688 to be associated with increased total and/or LDL-cholesterol. We evaluated this hypothesis in members of the Framingham Offspring Study (FOS), which is a longitudinal cohort study with medical exams about every 4 years beginning in 1971. We genotyped rs688 in DNA from a total of 1314 subjects for association with lipid profiles. For initial analyses, we used lipid profile data determined at FOS Exam 1 in 1971. These individuals were fasting and not taking cholesterol-lowering drugs at the time of study. Multiple linear regression analyses revealed that rs688 was not associated with significant differences in HDL-cholesterol, consistent with past observations regarding LDLR familial mutations and cholesterol homeostasis ( 12 , 14 ). However, rs688 was associated with significantly increased total cholesterol and LDL-cholesterol in women (Table 2 ). Similar increases were not observed in men (Table 2 ). Interestingly, the association in women suggests a possible dominant mechanism because cholesterol values for rs688 C/T and T/T individuals were both significantly higher than C/C individuals, but not different from each other. The increases in cholesterol of 6–7 mg/dl may be physiologically relevant because they are similar in magnitude to those reported for the well-characterized apoE4 relative to apoE3 ( 21 ).
rs688 is associated with increased total and LDL-cholesterol in women. The values are mean ± SE. All lipoprotein levels were adjusted for age, BMI and apoE genotype
| Women | Men | |||||
|---|---|---|---|---|---|---|
| rs688C/C | rs688C/T | rs688T/T | rs688C/C | rs688C/T | rs688T/T | |
| N | 224 | 316 | 120 | 185 | 339 | 135 |
| Total Cholesterol (mg/dl) | 186 ± 2 | 193 ± 2** | 193 ± 3* | 201 ± 3 | 197 ± 2 | 199 ± 3 |
| LDL (mg/dl) | 113 ± 2 | 118 ± 2* | 118 ± 2 | 129 ± 3 | 127 ± 2 | 129 ± 3 |
| HDL (mg/dl) | 57 ± 1 | 58 ± 1 | 59 ± 1 | 44 ± 1 | 44 ± 1 | 44 ± 1 |
| Women | Men | |||||
|---|---|---|---|---|---|---|
| rs688C/C | rs688C/T | rs688T/T | rs688C/C | rs688C/T | rs688T/T | |
| N | 224 | 316 | 120 | 185 | 339 | 135 |
| Total Cholesterol (mg/dl) | 186 ± 2 | 193 ± 2** | 193 ± 3* | 201 ± 3 | 197 ± 2 | 199 ± 3 |
| LDL (mg/dl) | 113 ± 2 | 118 ± 2* | 118 ± 2 | 129 ± 3 | 127 ± 2 | 129 ± 3 |
| HDL (mg/dl) | 57 ± 1 | 58 ± 1 | 59 ± 1 | 44 ± 1 | 44 ± 1 | 44 ± 1 |
* P ≤ 0.05, ** P < 0.01 significantly different compared with rs688 C/C genotype.
rs688 is associated with increased total and LDL-cholesterol in women. The values are mean ± SE. All lipoprotein levels were adjusted for age, BMI and apoE genotype
| Women | Men | |||||
|---|---|---|---|---|---|---|
| rs688C/C | rs688C/T | rs688T/T | rs688C/C | rs688C/T | rs688T/T | |
| N | 224 | 316 | 120 | 185 | 339 | 135 |
| Total Cholesterol (mg/dl) | 186 ± 2 | 193 ± 2** | 193 ± 3* | 201 ± 3 | 197 ± 2 | 199 ± 3 |
| LDL (mg/dl) | 113 ± 2 | 118 ± 2* | 118 ± 2 | 129 ± 3 | 127 ± 2 | 129 ± 3 |
| HDL (mg/dl) | 57 ± 1 | 58 ± 1 | 59 ± 1 | 44 ± 1 | 44 ± 1 | 44 ± 1 |
| Women | Men | |||||
|---|---|---|---|---|---|---|
| rs688C/C | rs688C/T | rs688T/T | rs688C/C | rs688C/T | rs688T/T | |
| N | 224 | 316 | 120 | 185 | 339 | 135 |
| Total Cholesterol (mg/dl) | 186 ± 2 | 193 ± 2** | 193 ± 3* | 201 ± 3 | 197 ± 2 | 199 ± 3 |
| LDL (mg/dl) | 113 ± 2 | 118 ± 2* | 118 ± 2 | 129 ± 3 | 127 ± 2 | 129 ± 3 |
| HDL (mg/dl) | 57 ± 1 | 58 ± 1 | 59 ± 1 | 44 ± 1 | 44 ± 1 | 44 ± 1 |
* P ≤ 0.05, ** P < 0.01 significantly different compared with rs688 C/C genotype.
Since apoE4 has a more robust association with total and LDL-cholesterol in post-menopausal women than pre-menopausal women ( 21 ), we also evaluated the rs688 association with cholesterol in these two subgroups. For this evaluation, we used data from FOS Exam 3 because ∼50% of the women were post-menopausal at the time of Exam 3 (average age of 48 years), while only ∼10% of the women were post-menopausal at the time of Exam 1 (average age of 36 years). We also pooled the rs688C/T and rs688T/T individuals because rs688T appears to have a dominant effect (Table 2 ). This approach yielded two findings. First, this stratified analysis found statistical evidence of interaction between rs688 and menopause status with both total and LDL-cholesterol ( P = 0.02 and P = 0.0042, respectively). Secondly, rs688 was associated significantly with total and LDL-cholesterol in pre-menopausal women but not post-menopausal women (Table 3 ). In pre-menopausal women, the presence of the rs688 minor allele was associated with an ∼10% higher level of both total and LDL-cholesterol.
rs688 is associated with increased total and LDL-cholesterol in pre-menopausal women
| Pre-menopausal | Post-menopausal | |||||||
|---|---|---|---|---|---|---|---|---|
| rs688 genotype | C/C | C/T or T/T | C/C | C/T or T/T | ||||
| n | Mean ± SE | Mean ± SE | P -value | n | Mean ± SE | Mean ± SE | P -value | |
| LDL (mg/dl) | 218 | 109 ± 4 | 122 ± 3 | 0.011 | 218 | 147 ± 4 | 145 ± 3 | 0.647 |
| Total cholesterol (mg/dl) | 218 | 183 ± 5 | 199 ± 4 | 0.004 | 218 | 232 ± 4 | 234 ± 3 | 0.599 |
| Pre-menopausal | Post-menopausal | |||||||
|---|---|---|---|---|---|---|---|---|
| rs688 genotype | C/C | C/T or T/T | C/C | C/T or T/T | ||||
| n | Mean ± SE | Mean ± SE | P -value | n | Mean ± SE | Mean ± SE | P -value | |
| LDL (mg/dl) | 218 | 109 ± 4 | 122 ± 3 | 0.011 | 218 | 147 ± 4 | 145 ± 3 | 0.647 |
| Total cholesterol (mg/dl) | 218 | 183 ± 5 | 199 ± 4 | 0.004 | 218 | 232 ± 4 | 234 ± 3 | 0.599 |
These lipoprotein values and their resultant P -values are from the FOS Exam 3 and have been adjusted for age, BMI and apoE genotype. Although estrogen treatment was considered initially as a covariate because of its relevance to menopause, only 31 of the 436 women were on estrogen therapy. We did not see a significant association between estrogen treatment and total or LDL-cholesterol. Moreover, a significant interaction between estrogen and rs688 was not detected. Therefore, estrogen treatment was not included as a covariate.
rs688 is associated with increased total and LDL-cholesterol in pre-menopausal women
| Pre-menopausal | Post-menopausal | |||||||
|---|---|---|---|---|---|---|---|---|
| rs688 genotype | C/C | C/T or T/T | C/C | C/T or T/T | ||||
| n | Mean ± SE | Mean ± SE | P -value | n | Mean ± SE | Mean ± SE | P -value | |
| LDL (mg/dl) | 218 | 109 ± 4 | 122 ± 3 | 0.011 | 218 | 147 ± 4 | 145 ± 3 | 0.647 |
| Total cholesterol (mg/dl) | 218 | 183 ± 5 | 199 ± 4 | 0.004 | 218 | 232 ± 4 | 234 ± 3 | 0.599 |
| Pre-menopausal | Post-menopausal | |||||||
|---|---|---|---|---|---|---|---|---|
| rs688 genotype | C/C | C/T or T/T | C/C | C/T or T/T | ||||
| n | Mean ± SE | Mean ± SE | P -value | n | Mean ± SE | Mean ± SE | P -value | |
| LDL (mg/dl) | 218 | 109 ± 4 | 122 ± 3 | 0.011 | 218 | 147 ± 4 | 145 ± 3 | 0.647 |
| Total cholesterol (mg/dl) | 218 | 183 ± 5 | 199 ± 4 | 0.004 | 218 | 232 ± 4 | 234 ± 3 | 0.599 |
These lipoprotein values and their resultant P -values are from the FOS Exam 3 and have been adjusted for age, BMI and apoE genotype. Although estrogen treatment was considered initially as a covariate because of its relevance to menopause, only 31 of the 436 women were on estrogen therapy. We did not see a significant association between estrogen treatment and total or LDL-cholesterol. Moreover, a significant interaction between estrogen and rs688 was not detected. Therefore, estrogen treatment was not included as a covariate.
DISCUSSION
The primary findings of this report include (i) rs688 is associated with significant differences in LDLR splicing efficiency in the female human liver in vivo , (ii) rs688 is a functional SNP as demonstrated by in vitro minigene splicing assays and (iii) rs688 is associated with increased total and LDL-cholesterol, especially in pre-menopausal women. Considered together, these findings provide mutually supporting evidence for the overall hypothesis that rs688 alters LDLR splicing efficiency, and thereby, cholesterol homeostasis in women. Although LDLR mutations have been recognized as causing hypercholesterolemia since the Nobel-prize winning work of Brown, Goldstein and collaborators, this is the first report of a common functional LDLR SNP. As such, these results provide insights into the genetic basis of cholesterol homeostasis and may lead to insights in cholesterol-associated diseases.
As a common SNP within a well-studied gene, rs688 has been evaluated in prior association studies. Discovered initially by Leitersdorf and Hobbs ( 22 ) in 1988, this SNP has been referred to as C1773T or as the Hin cII LDLR restriction fragment length polymorphism. Rs688 is a common SNP within European Caucasians, with minor allele carriers, i.e. C/T or T/T individuals, representing 60–65% of the population (dbSNP, build 126). The frequency of rs688 carriers in other races varies from 0–17% in African populations to 17–34% in different Asian populations (dbSNP, build 126). In prior reports, rs688 or tightly linked SNPs like rs5925, which is 3279 bp away in exon 13, have been inconsistently associated with increased LDL-cholesterol. For example, rs688 was associated with increased LDL-cholesterol in a gender-independent fashion in an Alberta Hutterite population ( 15 ). Rs688 was also associated with increased LDL-cholesterol in a normotensive Japanese population, and with hypertension in Japanese ( 23 ). However, rs5925 was not associated with increased cholesterol by others ( 15–17 ), which may reflect differences in age and/or gender of the study population. The identification of rs688 as a functional SNP will hopefully encourage direct evaluation of the SNP in additional populations.
We interpret the observation that rs688C/T and rs688T/T individuals associate with cholesterol similarly as suggestive that rs688 may act in a dominant fashion. Trends towards a dominant action were also observed in the Hutterite and Japanese population studies ( 15 , 23 ). In considering the underlying mechanism, we note that the rs688T allele increases the proportion of LDLR lacking exon 12 or exons 11 and 12. The loss of these exons shifts the LDLR reading frame, leading to premature stop codons in exon 13 and 14 for exon-12 deficient LDLR and exon 11 and 12 deficient LDLR, respectively (Fig. 4 A). Hence, translation of these LDLR isoforms are predicted to produce proteins that retain the LDL binding domain encoded by exons 1–7, but lack the transmembrane domain encoded by exons 16–17. Therefore, we propose a model (Fig. 4 B) wherein these soluble LDL ‘receptors’ act in a dominant negative fashion by binding LDL and thereby inhibiting its uptake by full length, cell surface LDLR. This scenario is supported by reports of secreted LDLR fragments that are encoded by exons 1–7 and capable of binding ligand ( 24 ). This model is supported further by findings that a soluble apoER2 protein acts as a dominant negative receptor; this soluble receptor is generated by inclusion of an alternatively spliced exon encoding a furin cleavage site ( 25 ). Experimentation is underway to evaluate whether the soluble LDLR fragments encoded by exon 12 deficient LDLR are secreted. We note that this is not a ‘given’ because certain LDLR mutations associated with hypercholesterolemia produce forms of LDLR that are improperly folded and degraded within the endoplasmic reticulum. For example, the ‘Lebanese’ familial hypercholesterolemia LDLR mutation, which consists of a nonsense mutation in exon 13, is retained within the endoplasmic reticulum ( 26 ). As an alternative model, we note that this intracellular LDLR protein could act in a dominant fashion by binding LDL within the cell, especially since LDLR proteins lacking the propeller domain do not release LDL-cholesterol at endosomal pH ( 27 , 28 ). In summary, while experimentation to discern among these possibilities is underway, our current hypothesis is that ‘soluble’ LDLR protein encoded by exon 12-deficient LDLR isoforms acts as a dominant negative receptor either outside the cell or, possibly, within the endosomal compartment.
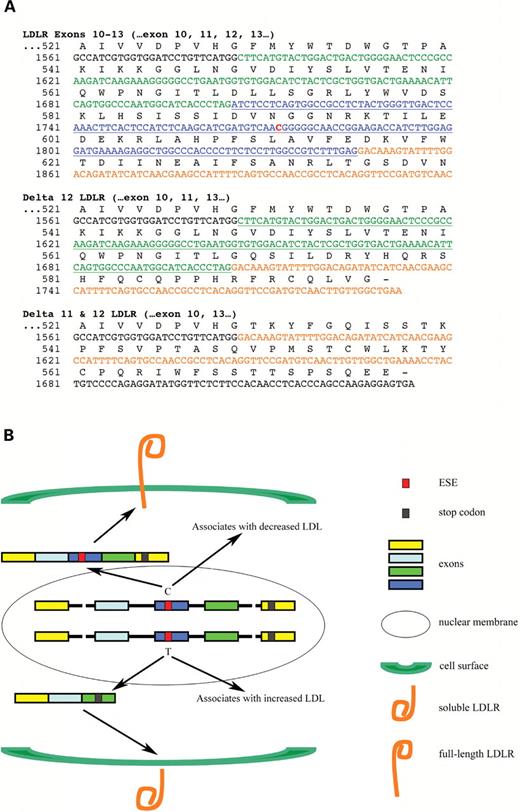
Inefficient LDLR exon splicing: primary sequence and LDLR function. ( A ) The sequence whereby the lack of exon 12 or exons 11 and 12 alters the LDLR reading frame, leading to premature stop codons. Exons 10 are 14 are presented in black font while exons 11, 12 and 13 are presented in green, blue and orange font, respectively. rs688 is in red font. The corresponding amino acids are above the nucleotide sequence. ( B ) The opposing effects of the rs688C and rs688T alleles on (i) the LDLR exon 12 ESE, (ii) the proportion of LDLR mRNA lacking exon 12, (iii) the generation of predicted soluble forms of LDLR and (iv) cholesterol homeostasis. In addition to reflecting the findings of this report, this model proposes the testable hypothesis that the soluble LDLR isoforms antagonize LDL uptake; such actions have been reported previously for recombinant soluble LDLR isoforms and for a soluble form of ApoER2 ( 27–29 ).
We have considered whether the rs688 association with splicing is likely to account for the rs688 association with cholesterol. Consistent with this possibility, the association of rs688 with splicing and cholesterol were both significant only within women. Moreover, the liver samples used in the splicing study were from women that were generally pre-menopausal, i.e. their average age at death was 27 ± 9 years (mean ± SD, range of 15–44, n = 21). Hence, the in vivo splicing results and cholesterol associations were derived from similar populations. Regarding the magnitude of the cholesterol differences with rs688, we note loss of one allele of LDLR is sufficient for an ∼100% increase in LDL-cholesterol ( 12 , 14 ). Hence, the 10% increase in LDL-cholesterol associated with rs688, which acts to decrease splicing efficiency, may be reasonable. Regarding alternative interpretations, the possibility that rs688 alters function by causing an amino acid change is not applicable because rs688 is the third base in the codon encoding Asn591, and does not alter this amino acid within LDLR (Fig. 4 A). The formal possibility exists that rs688 is linked with another functional SNP that actually causes the cholesterol differences. An rs688- linked functional SNP that alters exon 12 splicing efficiency is unlikely as demonstrated by our in vitro splicing studies wherein we used site-directed mutagenesis to implicate rs688 alone in splicing efficiency. While other functional SNPs linked to rs688 may be possible, the parsimonious explanation is that the known functional SNP, rs688, accounts for the cholesterol differences. In summation, the parallels between gender and menopausal status of the rs688 association with splicing and with cholesterol, and the absence of robust alternative interpretations, suggests that the rs688 association with cholesterol is likely due to rs688 actions on splicing efficiency.
We are unclear as to the mechanism(s) whereby the rs688 association with splicing efficiency was restricted to liver tissue from female individuals. Close evaluation of the splicing results indicates that splicing efficiency in the rs688C/C versus rs688T/T men showed similar trends to the women, although these results were not sufficiently consistent to reach statistical significance. A possible explanation for the male/female differences in rs688 association with splicing involves SRp40, which is the splicing factor predicted to bind the ESE neutralized by rs688. SRp40 is regulated by physiologic changes, e.g. SRp40 expression and activity decreases in the myometrium during pregnancy, and this change occurs in parallel with changes in relevant exon splicing efficiency ( 29 ). Overall, the regulation of SRp40 is complex and occurs at the levels of transcription, RNA splicing and SRp40 phosphorylation (reviewed in 30 ). Another clear difference regarding LDLR and cholesterol in males versus females is that LDLR transcription is positively regulated by estrogen ( 31 ); estrogen differences between men and women, and the decline in estrogen associated with menopause, is thought to contribute to pre-menopausal women having lower cholesterol levels than age-matched men or post-menopausal women. We note that the splicing patterns in the minigene studies were performed in HepG2 cells, which were derived from a male individual, but are maintained in media containing estrogen as well as estrogenic compounds like phenol red. Estrogen could cause the splicing patterns of HepG2 cell cultures to resemble those of human females. Hence, some interplay between estrogen actions on LDLR expression and on SRp40 activity may modulate the rs688 association with splicing and with cholesterol. Studies to evaluate this possibility are currently underway.
In summary, rs688 is a functional LDLR SNP that modulates LDLR exon splicing efficiency in vitro and is associated with decreased LDLR splicing efficiency in vivo as well as increased total and LDL-cholesterol in vivo in women. The magnitude of increased total and LDL-cholesterol is ∼10% in pre-menopausal women. Although somewhat modest by itself, this rs688-associated increase may modulate cholesterol-associated disease susceptibility, especially in combination with other cholesterol-modulating factors. As such, these results build on prior work demonstrating that LDLR mutations cause familial hypercholesterolemia ( 12–14 ). Overall, we anticipate these studies may prove useful to understanding the genetic aspects of cholesterol homeostasis and associated diseases, and as a model approach for evaluating genetic polymorphisms that modulate splicing efficiency.
MATERIALS AND METHODS
Evaluation of LDLR splicing in vivo
Human liver samples were obtained from the Brain and Tissue Bank for Developmental Disorders (Baltimore, MD). The samples were from deceased individuals with an average age at death for women of 27 ± 9 years (mean ± SD, range of 15–44, n = 21) and for men of 26 ± 9 (range 13–46, n = 22). The average post-mortem interval (PMI) for women was 13 ± 5 h (mean ± SD, range 4–19, n = 21) while the PMI for men was 10 ± 3 h (range 3–14, n = 22). Total RNA was prepared and converted to cDNA in one microgram aliquots (SuperScript II, Invitrogen) as we described previously ( 32–34 ). Primers corresponding to sequences within LDLR exons 10 and 14 were used to PCR-amplify (Platinum Taq , Invitrogen) cDNAs corresponding to LDLR exons 10–14, as well as isoforms lacking exons 11 and/or 12; primer sequences were 5′-CCTGGCCAGCAGCATGCCGTC and 5′- CATCGTGGTGGATCCTGTTC. PCR profiles consisted of pre-incubation at 94°C for 60 s, followed by cycles of denaturation at 94°C for 30 s, annealing at 60°C for 45 s and extension at 72°C for 90 s (Perkin Elmer Cetus, GeneAmp PCR System 9600). The minimal number of PCR cycles necessary to discern products were performed, i.e. 30 cycles. PCR products were separated by PAGE and visualized by SYBR-gold fluorescence on a fluorescence imager (Fuji FLA-2000). The identities of the PCR products were determined by gel purification and direct sequencing (Davis Sequencing). The amounts of full length and alternatively spliced LDLR isoforms were quantified by fluorescence intensity. For each sample, fluorescence values were corrected for background and normalized for length differences among amplicons. The splicing efficiency was then quantified in each sample as the amount of LDLR containing exons 10–14 divided by the total LDLR PCR product for that sample. PCR amplification efficiencies among the PCR amplicons were not significantly different. Data from men and women were analyzed separately, with statistical differences in splicing efficiency analyzed by one-tailed, non-parametric tests for three ordered groups, i.e. Jonckheere–Terpstra tests.
Evaluation of LDLR minigene splicing in vitro
LDLR minigenes were generated from the genomic DNA of two rs688C/C individuals and two rs688T/T individuals. Overlapping LDLR genomic fragments containing exons 9–12 and exons 12–14, as well as the internal introns, were amplified with high fidelity polymerase (Herculase, Stratagene) and primers LDLR Exon9F 5′ GCTCCATCGCCT ACCTCT TCTTC, LDLR Exon12R 5′ CTCAAAGACGGCCAAGGA GAAGG (3679 bp product) and LDLR Exon12F 5′ATCTCC TCAGTGGCCGCCTCTA and LDLR Exon14R 5′ CTGTG AGGCAGCTC CTCATGTC (3664 bp), respectively. PCR fragments were cloned into pCR 2.1 TOPO T/A cloning vector (Invitrogen). Exon 9–12 clones were digested ( Hin dIII/ Bsp DI) and the fragment gel purified. Exon 12–14 clones were also digested ( Hin dIII/ Bsp DI) and the exon 9–12 fragments ligated into the 12–14 vectors. Full length exon 9–14 clones in pCR 2.1 were digested ( Hin dIII and Not I) to release the exon 9–14 insert, which was then gel purified and cloned into pCDNA3.1/V5-His-TOPO (Invitrogen) prepared by digestion ( Hin dIII/ Not I). Identities of the exon 9–14 minigenes were confirmed by sequencing.
The rs688 SNP was interchanged between the rs688C and rs688T genomic LDLR backbones by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit, Stratagene). A small portion of the full-length clones containing rs688 was subcloned into Litmus 28i vector (New England Biolab), producing a small vector with the pertinent LDLR sequence that could be well-utilized by the QuikChange Multi-Site-Directed Mutagenesis Kit (Stratagene). Briefly, the rs688C and rs688T clones were digested (AvrII/BsiWI) to release a fragment from the end of exon 11 to the beginning of intron 12. The fragments were gel purified and ligated into AvrII/BsiWI cut Litmus 28i. Mutagenesis was performed with the following oligos: 5′ pAAGCATCGATGT CAA T GGGGGCAACCGGAAG (underline denotes SNP), to convert the major allele to the minor allele, and 5′pAAGCATCGATGT CAA C GGGG GCAACCGGAAG, to convert the minor allele to the major allele. Mutagenesis was performed according to manufacturers' instructions. Positive clones were confirmed by sequencing. Mutated fragments were then cut from Litmus 28i (AvrII/BsiWI) and cloned back into AvrII/BsiWI digested LDLR vector, and positive clones again confirmed by sequencing.
To evaluate splicing in the minigene constructs, the clones were transfected into HepG2 cells by using FuGene as directed by the manufacturer (Roche). Twenty-four hours post-transfection, RNA was isolated and analyzed for LDLR splicing patterns by RT–PCR as described above except that only vector-specific LDLR was analyzed by substituting a sense PCR oligo corresponding to vector sequences at the beginning of the transcription product (5′ACTAGTCCAGT GTGGTGGAATTGCC 3′). PCR conditions were 94° for 30 s, 60° for 30 s and 72° for 60 s.
Genetic association studies
DNA samples from members of the FOS were genotyped for rs688 by using unlabeled PCR primers and TaqMan FAM or VIC dye-labeled MGB probes (Assays-by-Design, Applied Biosystems) on an ABI-7000 (Applied Biosystems). We excluded from the analysis individuals that were not fasting at the time of lipid profile determination, or that were taking cholesterol-lowering drugs. Hence, we report on a total to 1314 subjects (655 women and 659 men). Lipid profiles and apoE status on this population were provided by the FOS ( 21 ). Hardy–Weinberg equilibrium was evaluated by using SNPalyze (Dynacom). Rs688 association with lipid profiles was evaluated for significance by using a General Linear Model (SAS), relating multiple continuous or discrete independent variables, i.e. age, rs688, body mass index and apoE genotype, to a dependent variable, e.g. LDL-cholesterol. ApoE genotype categories were reduced to 3: 3/3, 2/2 or 2/3 and 3/4 or 4/4; the rare apoE 2/4 subjects were excluded ( 21 ). Age was modeled as a continuous variable. Women and men were analyzed separately. Menopause was defined as the absence of menses for 1 year. Written informed consent for the FOS subjects overall was obtained from FOS participants with a study protocol approved by the Boston Medical Center Institutional Review Board. This specific work was approved by the FOS review committee and the University of Kentucky Institutional Review Board.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the Framingham Heart Study of the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine. This work was supported by the National Heart, Lung and Blood Institute's Framingham Heart Study (Contract No. N01-HC-25195) as well as the National Institute on Aging (R01AG026147 and T32AG000242).
Conflict of Interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}