




Abstract
α-synuclein is a neuronal protein implicated genetically in Parkinson's disease. α-synuclein localizes to the nucleus and presynaptic nerve terminals. Here we show that α-synuclein mediates neurotoxicity in the nucleus. Targeting of α-synuclein to the nucleus promotes toxicity, whereas cytoplasmic sequestration is protective in both cell culture and transgenic Drosophila . Toxicity of α-synuclein can be rescued by administration of histone deacetylase inhibitors in both cell culture and transgenic flies. α-synuclein binds directly to histones, reduces the level of acetylated histone H3 in cultured cells and inhibits acetylation in histone acetyltransferase assays. α-synuclein mutations that cause familial Parkinson's disease, A30P and A53T, exhibit increased nuclear targeting in cell culture. These findings implicate nuclear α-synuclein in promoting nigrostriatal degeneration in Parkinson's disease and encourage exploration of histone deacetylase inhibitors as potential therapies for the disorder.
INTRODUCTION
Parkinson's disease is a neurodegenerative condition that affects 1% of the population over 65 ( 1 ) and is characterized by a relatively selective depletion of dopaminergic neurons in the substantia nigra pars compacta. Currently, the cause of most cases of the disease is unknown.
Though the majority of Parkinson's disease cases are sporadic in origin, mutations in several proteins, including α-synuclein, have been linked to Parkinson's disease ( 1–3 ). As its name implies, α-synuclein was initially localized to presynaptic nerve terminals and the nucleus ( 4 ). Subsequent studies have focused on α-synuclein's role in the synapse, although nuclear localization of the protein has been noted in a variety of experimental systems, including the α-synuclein transgenic Drosophila developed by our laboratory ( 5 ), mice ( 6 , 7 ) and cultured cells ( 8 , 9 ). Perhaps more importantly, intranuclear inclusions formed from α-synuclein are present in patients with multiple system atrophy, a clinically distinct synucleinopathy ( 10 , 11 ). Thus, accumulation of nuclear α-synuclein can be seen not only in experimental animal models, but in human patients as well. The function of α-synuclein in the nucleus has been unclear. α-synuclein can associate with histones in vitro ( 7 ). α-synuclein also translocates to the nucleus of mice injected with the herbicide, paraquat ( 7 ). These results suggest that the formation of histone-α-synuclein complexes may be relevant to neurotoxicity in Parkinson's disease.
Several proteins implicated in human neurodegenerative diseases exert their pathogenic effects in the nucleus ( 12–15 ). We decided to investigate whether α-synuclein could act in the nucleus to promote neurotoxicity. To examine the ability of α-synuclein to promote cell death in the nucleus and the cytoplasm, we created constructs of α-synuclein tagged with either a nuclear localization sequence (NLS) or a nuclear export sequence (NES). We used these constructs to transiently transfect SH-SY5Ys, a dopaminergic cell line. We also created transgenic flies expressing either construct. By assaying for cell viability, we find that α-synuclein promotes neurotoxicity when targeted to the nucleus. Conversely, sequestering α-synuclein in the cytoplasm does not yield significant toxicity. Furthermore, we show that α-synuclein associates with histones and acts to inhibit their acetylation. Finally, familial mutations linked to Parkinson's disease demonstrate increased propensity to localize to the nucleus in cell culture. These findings support the hypothesis that α-synuclein acts in the nucleus to promote the degeneration of dopaminergic neurons in Parkinson's disease.
RESULTS
Nuclear localization of α-synuclein promotes cell death, whereas cytoplasmic localization is protective
To address whether altering the subcellular localization of α-synuclein can influence neurotoxicity, SH-SY5Y neuroblastoma cells were transfected with the following constructs: β-galactosidase, syn WT (wild-type human full-length α-synuclein), syn NLS (human full-length α-synuclein with a C-terminal NLS) and syn NES (human full-length α-synuclein with a C-terminal NES). The localization of the α-synuclein constructs expressed in SH-SY5Y cells was assessed by immunofluorescence with an antibody that recognizes α-synuclein. Syn WT showed diffuse localization to both nuclear and cytoplasmic compartments (Fig. 1 A). To ensure that the observed nuclear localization of wild-type α-synuclein was not an artifact of non-specific antibody reactivity, several antibodies against α-synuclein were tested (Supplementary Material, Fig. S1). Syn NLS targeted primarily to the nucleus, whereas syn NES localized predominantly to the cytoplasm (Fig. 1 A). Thus, the constructs localized to appropriate subcellular compartments. Viability of cells expressing the various α-synuclein constructs was examined by immunostaining for activated caspase-3 48 h after transfection. Compared with control cells expressing β-gal, cells expressing syn WT exhibited significantly increased toxicity (Fig. 1 B and Supplementary Material, Fig. S2). Cells expressing syn NLS showed increased toxicity relative to cells expressing syn WT (Fig. 1 B and Supplementary Material, Fig. S2). Sequestration of α-synuclein in the cytoplasm with the syn NES construct prevented α-synuclein-associated neurotoxicity. Similar results were observed using TUNEL staining as an apoptotic marker (Supplementary Material, Fig. S3).
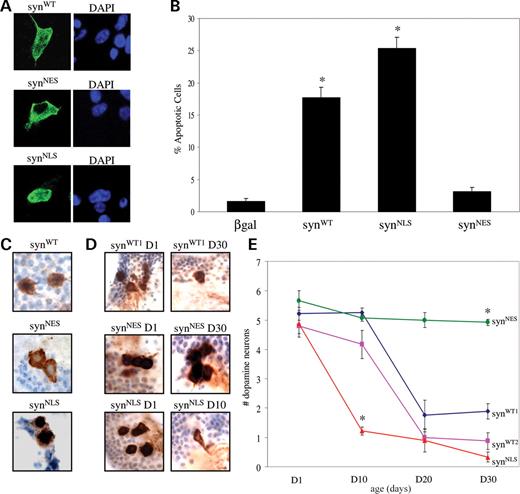
Nuclear targeting of α-synuclein promotes toxicity, whereas cytoplasmic sequestration is protective. ( A ) The subcellular localization of α-synuclein is altered by the addition of NLS or NES tags in SH-SY5Y cells. α-synuclein appears in green. Nuclei are stained blue with DAPI. ( B ) Cell loss in SH-SY5Y cells associated with syn WT expression is increased with the NLS tag ( P <0.01) and decreased with the NES tag ( P <0.01). ( C ) In dopaminergic neurons of transgenic flies, syn WT localizes to both the cytoplasm and nucleus. Syn NLS localizes predominantly to the nucleus, whereas syn NES is found mostly in the cytoplasm. α-synuclein, detected by LB509 antibody, appears brown. Nuclei are stained blue with hematoxylin. ( D ) Tyrosine hydroxylase immunostaining reveals a normal number of dorsomedial dopamine neurons in 1-day-old elav-GAL4 /+; UAS - synWT1 /+ flies, whereas toxicity to dopamine neurons was observed in 30-day-old α-synuclein transgenic flies. Accelerated loss of dopamine neurons was seen in 10-day-old elav-GAL4 /+; UAS - synNLS /+ transgenic flies. In contrast, no neuronal loss is seen in 30-day-old elav-GAL4 /+; UAS - synNES /+ transgenic flies. ( E ) Quantitative analysis of dorsomedial dopamine neurons over time in transgenic flies. Values represent mean ±SEM. Asterisk indicates that the difference between dopamine neurons in 10-day-old elav-GAL4 /+; UAS - synNLS /+ and 10-day-old elav-GAL4 /+; UAS - synWT1 /+ is statistically significant ( P <0.01). Similarly, the difference between dopamine neurons in 30-day-old elav-GAL4 /+; UAS - synNES /+ and 30-day-old elav-GAL4 /+; UAS - synWT1 /+ is statistically significant ( P <0.01).
We next tested the role of nuclear α-synuclein in an animal model of Parkinson's disease ( 16 ). We created multiple transgenic lines carrying the syn NLS and syn NES complementary DNA constructs under the control of upstream activating sequence (UAS) for the yeast transcription factor GAL4 and used the GAL4-UAS bipartite system to direct α-synuclein expression to the brain, using the pan-neuronal driver, elav-GAL4 . Using western blot analysis on fly heads, we identified syn NLS and syn NES lines, which expressed equivalent levels of protein (when syn NLS levels are normalized to 1, syn NES levels average 1.03±0.04). Levels of syn NLS and syn NES were intermediate between two lines of wild-type α-synuclein transgenic flies (syn WT1 and syn WT2 ) previously characterized in the laboratory (Supplementary Material, Fig. S4). To confirm that α-synuclein constructs localize to appropriate subcellular localizations, we stained tissue sections through the brain of the adult fly with an antibody against α-synuclein. Examination of dopamine neurons ( Ddc-GAL4 driver) revealed that syn WT localized to both the nucleus and the cytoplasm (Fig. 1 C). Syn NLS protein localized predominantly to the nucleus, whereas syn NES protein was found mostly in the cytoplasm (Fig. 1 C). To address whether nuclear localization promotes toxicity in our transgenic flies, we immunostained brains from flies 1, 10, 20 and 30 days after eclosion with an antibody against tyrosine hydroxylase, which specifically identifies dopaminergic neurons. We focused our analysis on the dorsomedial group of dopaminergic neurons because these neurons are preferentially sensitive to α-synuclein toxicity ( 16–18 ). In non-transgenic flies, these cells are represented throughout normal lifespan by four or five robustly staining tyrosine hydroxylase positive cells. Similarly, in young adult flies expressing wild-type α-synuclein, the dorsomedial cluster consists of the normal number of dopaminergic neurons (Fig. 1 E). Thirty-day-old adult flies expressing wild-type α-synuclein demonstrate a marked loss of tyrosine-hydroxylase immunostaining in cells in this cluster (Fig. 1 E). These findings are consistent with a neurotoxic effect of α-synuclein ( 16–21 ) most likely representing loss of these neurons ( 16 ). In contrast, aged transgenic flies expressing syn NES showed no loss of dopaminergic neurons (Fig. 1 E). No significant difference in the number of dopaminergic neurons in flies expressing syn NES was observed at 30 days compared with 1 day (Fig. 1 D and E). Thus, sequestering α-synuclein in the cytoplasm is neuroprotective. Next, we examined whether increasing the amount of α-synuclein in the nucleus accelerated toxicity in our transgenic flies. In 1-day-old transgenic flies expressing syn NLS , the number of dorsomedial dopaminergic neurons did not differ significantly from the number in young flies expressing wild-type α-synuclein (Fig. 1 D and E). Therefore, expression of nuclear α-synuclein, like expression of wild-type α-synuclein, does not disrupt the development of dopaminergic neurons. However, the neurodegenerative process was significantly accelerated in flies expressing syn NLS . On average, one dopaminergic neuron was observed in 10-day-old syn NLS flies, whereas dorsomedial dopaminergic neurons were relatively well preserved in both wild-type α-synuclein transgenic lines at the same time point (Fig. 1 D and E). Thus, localization of α-synuclein to the nucleus enhances neurotoxicity of dopaminergic neurons.
Administration of histone deacetylase (HDAC) inhibitors protects against α-synuclein-dependent neurotoxicity
Since α-synuclein can associate with histones in vitro ( 7 ) and because histone acetylation pathways are critical in polyglutamine toxicity and perhaps other types of neuronal cell death as well ( 22 , 23 ), we investigated whether altering histone acetylation levels could affect the neurotoxicity of α-synuclein. SH-SY5Y cells transfected with β-gal, syn WT , syn NLS or syn NES constructs were treated with either of two HDAC inhibitors, sodium butyrate or SAHA, or drug-free solvent. Apoptotic cells were identified by staining for activated caspase-3 and by counting TUNEL-positive nuclei. As previously observed, both syn WT and syn NLS constructs exhibited increased toxicity relative to β-gal and syn NES (Fig. 2 A–D). Administering either sodium butyrate (Fig. 2 A and C) or SAHA (Fig. 2 B and D) diminished toxicity to baseline levels.
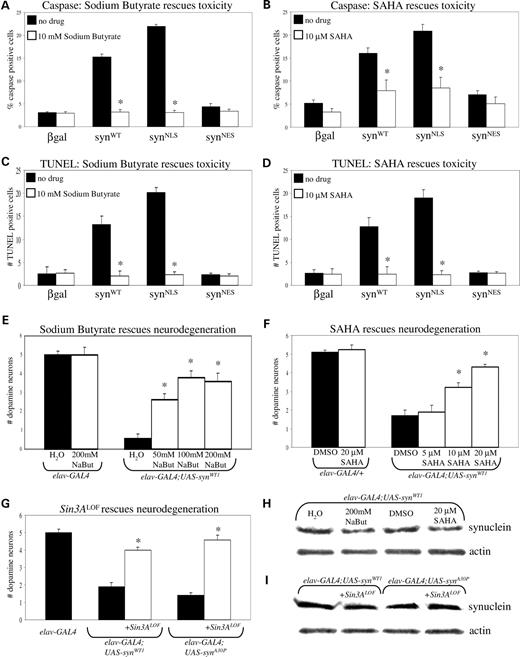
Inhibiting HDAC activity rescues α-synuclein toxicity in transiently transfected SH-SY5Y cells and transgenic flies. ( A ) Administration of 10 m m sodium butyrate rescues toxicity associated with syn WT and syn NLS constructs in SH-SY5Y cells ( P <0.01). Apoptotic cells were identified by staining for activated caspase-3. ( B ) Administration of 10 µ m SAHA rescues toxicity associated with syn WT and syn NLS constructs in SH-SY5Y cells ( P <0.01). Apoptotic cells were identified by staining for activated caspase-3. ( C ) Administration of 10 m m sodium butyrate rescues toxicity associated with syn WT and syn NLS constructs in SH-SY5Y cells ( P <0.01). Apoptotic cells were identified by TUNEL staining. ( D ) Administration of 10 µ m SAHA rescues toxicity associated with syn WT and syn NLS constructs in SH-SY5Y cells ( P <0.01). Apoptotic cells were identified by TUNEL staining. ( E ) Administration of sodium butyrate rescues toxicity to dorsomedial dopamine neurons of 20-day-old wild-type α-synuclein transgenic flies in a dose-dependent fashion ( P <0.01). Animals were fed 50, 100 and 200 m m sodium butyrate. ( F ) Administration of SAHA rescues toxicity to dorsomedial dopamine neurons of 20-day-old wild-type α-synuclein transgenic flies in a dose-dependent fashion ( P <0.01). Animals were fed 5, 10 and 20 µ m SAHA. ( G ) Genetically reducing deacetylase function in 20-day-old α-synuclein transgenics rescues toxicity to dorsomedial dopamine neurons. Flies expressing wild-type or mutant (A30P) α-synuclein heterozygous for a Sin3A mutation were compared with wild-type or mutant α-synuclein flies lacking the Sin3A mutation ( P <0.01). ( H ) Expression of wild-type α-synuclein transgene is not significantly altered following administration of either sodium butyrate or SAHA. A western blot of equivalent amounts of adult brain homogenate expressing wild-type α-synuclein pan-neuronally and treated with either solvent alone or solvent with sodium butyrate or SAHA was probed with Clone 42 α-synuclein antibody. ( I ) Expression of wild-type α-synuclein transgene is not significantly altered following reduction of Sin3A activity.
We observed a similar effect of HDAC inhibitors on our transgenic flies. Flies expressing α-synuclein pan-neuronally were fed various doses of sodium butyrate (Fig. 2 E) or SAHA (Fig. 2 F) for the first 20 days of adult life. Non-transgenic control flies ( elav-GAL4/+ ) fed the maximum dose of sodium butyrate or SAHA had the normal amount of dopaminergic neurons in the dorsomedial cluster. Transgenic α-synuclein flies fed water or DMSO showed loss of neurons in the dorsomedial cluster. Transgenic α-synuclein flies fed increasing concentration of either sodium butyrate or SAHA showed rescue of dorsomedial neurons in a dose-dependent fashion. To test the significance of histone acetylation in mediating α-synuclein toxicity genetically, we altered HDAC enzyme levels and examined cell loss. The Drosophila Sin3A locus encodes a transcriptional-repressor protein that is a component of HDAC complexes. We observed that decreasing the levels of active HDAC complex by a partial loss of function mutant, Sin3A08269 , in heterozygous α-synuclein transgenics rescued the loss of dorsomedial neurons (Fig. 2 G). Taken together, both pharmacological and genetic manipulation of HDAC activity modifies α-synuclein toxicity.
To determine whether the decrease in toxicity observed in sodium butyrate- and SAHA-fed flies was due to a down-regulation of α-synuclein protein expression, we compared α-synuclein transgene expression by western blot in solvent and drug-fed animals. Steady-state levels of α-synuclein were unaffected by the administration of HDAC inhibitors (Fig. 2 H). Similarly, we examined whether transgene expression was altered in response to the Sin3A mutation. We found no difference in levels between flies expressing α-synuclein pan-neuronally with or without reduced Sin3A activity (Fig. 2 I).
To address the possibility that the decrease in toxicity observed in sodium butyrate- and SAHA-fed flies is due to a general anti-apoptotic mechanism, we fed flies expressing the familial tauopathy-associated R406W mutant tau ( elav-GAL4 /+; UAS - tauR406W ) the maximum dose of sodium butyrate, SAHA or solvent for 10 days. Expression of mutant tau produces apoptosis at 10 days ( 24 , 25 ). Apoptotic cells were identified by TUNEL stain. Tau flies showed a significant increase in TUNEL staining relative to non-transgenic controls (Supplementary Material, Fig. S5). Administering HDAC inhibitors to tau transgenic flies did not decrease the number of TUNEL-positive cells, indicating that HDAC inhibitors do not act as neuroprotective agents in all neurodegenerative disease models.
α-synuclein associates with histones and inhibits histone acetylation
To confirm the reported finding that α-synuclein associates with histones, we conducted GST-pull-down experiments with bacterially purified GST-α-synuclein (and GST as a control) and histones extracted from SH-SY5Y cells. GST-synuclein associated with histones, whereas GST alone did not (Fig. 3 A). To determine whether endogenous α-synuclein binds histones in vivo , we performed co-immunoprecipitation experiments with nuclear extracts from mouse brain. α-synuclein and histone H3 co-immunoprecipitated (Fig. 3 B). To further evaluate syn NLS and histone H3 co-localization in vivo , we over-expressed UAS-syn NLS in the salivary glands of Drosophila larvae ( da-GAL4 driver) and immunostained polytene chromosomes with antibodies against α-synuclein and histone H3. syn NLS and histone H3 co-localized on salivary gland polytene chromosomes (Fig. 3 C). These results together support an in vivo interaction between histones and α-synuclein.
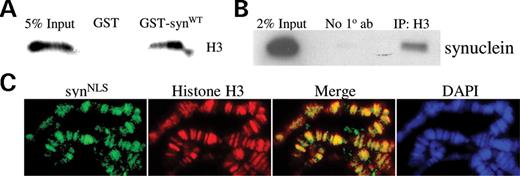
α-synuclein associates with histones in vitro and in vivo . ( A ) Purified GST and GST-synuclein proteins were incubated with histones extracted from SH-SY5Y cells. GST-synuclein directly interacts with histones, whereas GST does not. ( B ) Co-immunoprecipitation of histone H3 and endogenous α-synuclein from wild-type mouse brain. Nuclear extracts of SV129 mouse brain were immunoprecipitated with a polyclonal H3 antibody and probed for α-synuclein. ( C ) Polytene chromosomes were prepared from UAS - synNLS /+; da-GAL4 /+ flies and stained with DAPI and antibodies against α-synuclein and histone H3. syn NLS and histone H3 co-localize and closely mirror DAPI staining.
Since inhibition of deacetylation rescued α-synuclein toxicity, we examined the acetylation levels of histone H3 in SH-SY5Y stable cell lines transfected with syn NLS and syn NES . Expression of syn NLS reduced acetylation of histone H3 by ∼50% relative to untransfected and syn NES -transfected cells. Total histone levels were equivalent, as determined by histone H3 antibody (Fig. 4 A and B).
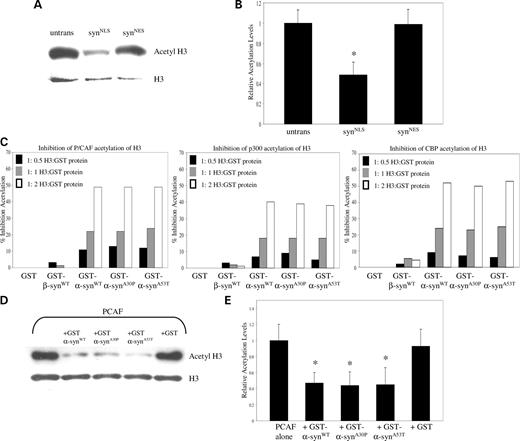
α-synuclein inhibits histone acetylation in stably transfected cells and in in vitro histone acetyltransferase assays. (A) SH-SY5Y cells stably expressing syn NLS show a reduction in the acetylation level of histone H3, relative to expression-matched syn NES stable transfectants. (B) Quantification of western blot results ( n =3). SH-SY5Y cells stably transfected with syn NLS exhibit a significant reduction in histone H3 acetylation ( P <0.05) relative to untransfected and syn NES samples. (C) GST, GST-β-syn WT , GST-α-syn WT , GST-α-syn A30P or GST-α-syn A53T were pre-incubated with histone H3 and subsequently added to an in vitro histone acetylation assay containing purified P/CAF, p300 or CBP. All three α-synuclein constructs strongly inhibit histone acetylation to a similar extent. GST and GST-β-syn WT do not significantly inhibit histone acetylation. (D) GST, GST-α-syn WT , GST-α-syn A30P or GST-α-syn A53T were pre-incubated with chromatin and subsequently added to an in vitro histone acetylation assay containing purified P/CAF. Western blot analysis reveals that all three α-synuclein constructs strongly inhibit histone acetylation to a similar extent. (E) Quantification of western blot results ( n =6). Chromatin pre-incubated with GST-α-syn WT , GST-α-syn A30P or GST-α-syn A53T exhibits a significant reduction in histone H3 acetylation upon completion of an in vitro histone acetylation assay ( P <0.01).
Previous reports have suggested that regulatory proteins may bind to histones and sterically hinder their lysine residues from serving as acetyltransferase substrates ( 26 ). To test whether wild-type and mutant (A30P and A53T) α-synuclein binding can alter histone acetylation, we conducted in vitro histone acetyltransferase assays (Fig. 4 C). We measured the ability of α-synuclein to inhibit histone H3 acetylation by several neuronally expressed acetyltransferase enzymes: CBP, p300 and P/CAF. Equimolar amounts of GST, GST-β-syn WT , GST-α-syn WT , GST-α-syn A30P and GST-α-syn A53T were pre-incubated with histone H3 peptide to promote protein–protein interactions. Following incubation, the remaining reaction components were added, and acetylation of the histone peptides was assessed. All three α-synuclein proteins inhibited acetylation of histone H3 in a dose-dependent fashion, whereas GST and GST-β-syn WT did not (Fig. 4 C). α-synuclein, but not the related protein β-synuclein, has been implicated in the pathogenesis of Parkinson's disease and other synucleinopathies. β-synuclein did not block histone acetylation. There was no significant difference in the ability of mutant α-synuclein to inhibit acetylation when compared with wild-type, suggesting that mutant and wild-type α-synuclein bind equally well to histones. Furthermore, histone acetylation was inhibited to a similar degree in the presence of each individual histone acetyltransferase enzyme (Fig. 4 C). We were unable to observe α-synuclein-dependent inhibition of acetylation in the absence of pre-incubation with histones (data not shown). Since we were also unable to detect direct binding of α-synuclein to histone acetyltransferase enzymes (data not shown), we suggest that α-synuclein inhibits histone acetylation through direct association with histones. To validate these results with a more physiological histone acetyltransferase enzyme substrate ( 27 ), we performed histone acetyltransferase reactions on a chromatin template. All three α-synuclein proteins exhibited a 2-fold inhibition of histone H3 acetylation (Fig. 4 D and E), suggesting that mutant and wild-type synuclein interact with chromatin in a similar fashion.
Increased levels of nuclear α-synuclein are associated with disease-linked mutations of α-synuclein
Since α-synuclein proteins with familial Parkinson's disease mutations did not display an enhanced ability to inhibit histone acetylation in our in vitro assays, we wondered whether an increased propensity to localize to the nucleus might explain their toxicity in patients. We transfected SH-SY5Y cells with syn WT , syn A30P and syn A53T , and using quantitative confocal analysis, we determined nuclear/cytoplasmic ratios corresponding to the relative intensity of α-synuclein staining in nuclear versus cytoplasmic compartments. In contrast to wild-type α-synuclein, both familial Parkinson's disease mutants demonstrated a modest but significant increase nuclear localization (Fig. 5 A and B). These findings were further corroborated by western blot of nuclear and cytoplasmic fractions (Fig. 5 C). We conclude that familial Parkinson's disease mutations may confer increased toxicity through increased nuclear accumulation.
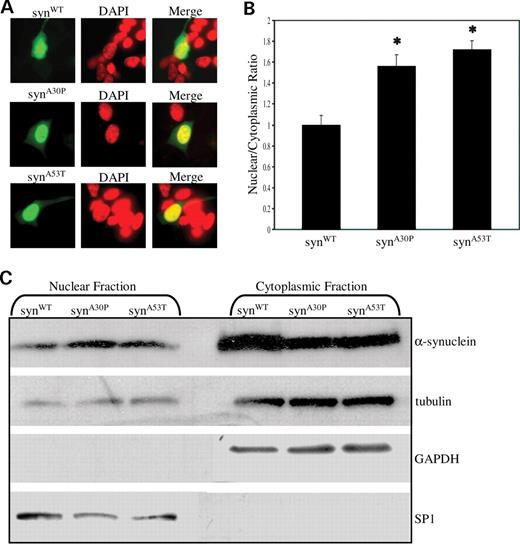
Increased levels of nuclear α-synuclein are associated with disease-linked mutations of α-synuclein. ( A ) Familial Parkinson's disease mutant proteins preferentially accumulate in the nucleus of transfected SH-SY5Y cells compared with wild-type α-synuclein. syn WT localizes diffusely to both nuclear and cytoplasmic compartments. Syn A30P and syn A53T localize predominantly to the nucleus. ( B ) Quantification of nuclear and cytoplasmic intensities by confocal microscopy reveals increased nuclear targeting of α-synuclein mutants relative to wild-type ( P <0.01). For each construct, a nuclear/cytoplasmic ratio was determined, corresponding to the ratio of mean nuclear and cytoplasmic fluorescent intensities. Values were normalized to syn WT . For each construct, at least 50 randomly selected cells positive for α-synuclein staining were analyzed. ( C ) Cytoplasmic and nuclear fractions of syn WT , syn A30P and syn A53T transfected Cos-1 cells were separated by SDS–PAGE and probed for α-synuclein. Cytoplasmic marker GAPDH appears in the cytoplasmic fraction, whereas nuclear marker SP1 appears in the nuclear fraction.
Localization of α-synuclein in wild-type mouse brain
Since the degree of α-synuclein overexpression is likely modest in most patients who do not carry causative duplication or triplication alleles, we examined nuclear α-synuclein localization in an endogenous expression system, wild-type mouse brain. We detected endogenous α-synuclein in the nuclei of dopaminergic neurons in wild-type mice (arrowheads, Fig. 6 C and D). No nuclear α-synuclein immunoreactivity was detected in the negative control (Fig. 6 A) or in α-synuclein knockout animals (Fig. 6 B). Thus, under physiological conditions, detectable amounts of α-synuclein are present in the nuclei of dopaminergic neurons.
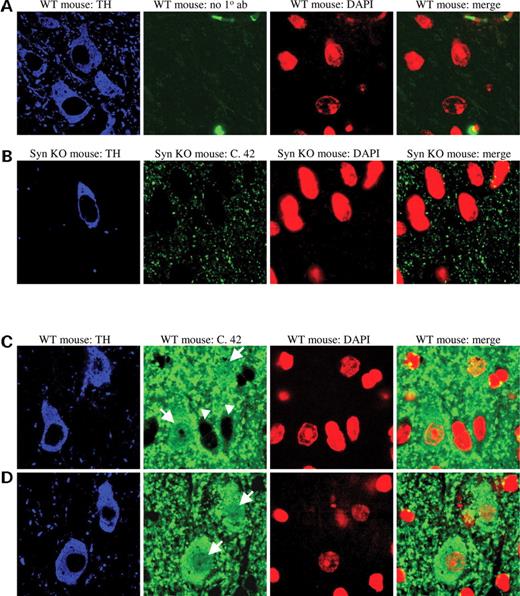
α-synuclein immunoreactivity in wild-type and α-synuclein knockout mouse brain. ( A ) Wild-type mouse brain immunostained for tyrosine hydroxylase (blue) to identify dopamine neurons and DAPI (red) to identify nuclei. α-synuclein antibody was not included as a control. ( B ) α-synuclein knockout mouse brain immunostained for tyrosine hydroxylase (blue), α-synuclein (green) and DAPI (red). α-synuclein antibody does not detect non-specific staining in the nucleus. ( C and D ) Wild-type mouse brain immunostained for tyrosine hydroxylase (blue), α-synuclein (green) and DAPI (red). Arrows point to dopaminergic neurons with nuclear α-synuclein staining. Arrowheads indicate cells with glial morphology lacking nuclear α-synuclein staining, further indicating the specificity of the immunostain.
DISCUSSION
α-synuclein was first identified as a neuronal protein localized to the nucleus and the synapse ( 4 ). Since then, most work has focused on possible roles for α-synuclein in the nerve terminal, although a definitive function for the protein has yet to be defined ( 28 ). However, a number of studies have also documented α-synuclein in the nucleus ( 5–11 ). We have observed α-synuclein immunoreactivity in the nuclei of transiently transfected SH-SY5Y cells (Fig. 1 and Supplementary Material, Fig. S1). We have also found endogenous α-synuclein in the nuclei of dopaminergic neurons in wild-type mice (Fig. 6 ). These findings do not appear to reflect non-specificity of our immunological reagents because we do not detect α-synuclein immunoreactivity in α-synuclein knockout animals (Fig. 6 ). In addition, α-synuclein-containing aggregates are present in the nucleus in multiple system atrophy, an α-synucleinopathy related clinically and pathologically to Parkinson's disease.
We present multiple lines of evidence to suggest that nuclear α-synuclein is toxic to neurons. Targeting α-synuclein to the nucleus in both transiently transfected SH-SY5Y neuroblastoma cells and transgenic flies increased α-synuclein-mediated toxicity. Conversely, sequestering α-synuclein in the cytoplasm through the addition of an NES tag protected for α-synuclein neurotoxicity in both of our model systems. Together with the mechanistic experiments discussed in what follows, these findings support an important role for α-synuclein in mediating neurotoxicity in the nucleus.
To investigate the mechanism by which nuclear α-synuclein promotes neurotoxicity, we began by evaluating histone acetylation because histone acetylation pathways have been implicated in polyglutamine toxicity and possibly other types of neurodegeneration ( 22 , 23 ). Inhibition of histone acetylation was observed in our in vitro histone acetyltransferase assays (Fig. 4 ). We confirmed the in vitro binding of α-synuclein to histones, reported by Goers et al . ( 7 ), and demonstrated an association in vivo as well (Fig. 3 ). Our data are most consistent with a model in which α-synuclein acts to inhibit histone acetylation by directly associating with histones and shielding residues involved in acetylation. Such histone ‘masking’ is one of the mechanisms by which ataxin-3 inhibits transcription ( 29 ). Further investigation should reveal whether α-synuclein represses histone acetylation through alternative mechanisms.
If α-synuclein promotes neurotoxicity by decreasing histone acetylation, then increasing histone acetylation should ameliorate α-synuclein toxicity. In a healthy neuron, levels of histone acetylation are determined by the equilibrium between the forward rate of acetylation by histone acetyltransferases and the reverse rate of deacetylation by histone deacetyltransferases. We hypothesized that nuclear α-synuclein mediates neurotoxicity by inhibiting histone acetylation. We reasoned that if this hypothesis is correct, then the administration of HDAC inhibitors should restore the acetylation state to that found in healthy cells and rescue α-synuclein toxicity. Both SAHA and sodium butyrate diminished apoptotic death observed in response to syn WT and syn NLS expression (Fig. 2 A–D). SAHA and sodium butyrate also rescued dopaminergic neuron degeneration in α-synuclein transgenic flies (Fig. 2 E and F). HDAC inhibitors are known to have a wide array of targets, including transcription factors and cell cycle molecules. For this reason, we supported our HDAC inhibitor feeding results with genetic analysis. Reducing the activity of a component of the HDAC machinery, the Sin3A corepressor, in our α-synuclein transgenic flies resulted in a 2-fold rescue of dopaminergic neuron degeneration, suggesting that the therapeutic effects of the HDAC inhibitors were most likely through acetylation of histone residues (Fig. 4 C).
HDAC inhibitors have been administered successfully in a number of disease models, including Drosophila and mouse models of Huntington's disease, and mouse models of amyotrophic lateral schlerosis and spinal and bulbar muscular atrophy ( 22 , 30–35 ). We wondered whether the HDAC inhibitors were acting to inhibit cell loss through a universal anti-apoptotic mechanism. To test this hypothesis, we fed SAHA and sodium butyrate to transgenic flies expressing the familial tauopathy-associated R406W mutant tau. Expression of mutant tau produces apoptosis at 10 days ( 24 , 25 ). Administration of HDAC inhibitors did not significantly protect against tau-mediated cell death (Supplementary Material, Fig. S5). To probe this issue further, we also used a transgenic fly line expressing the apoptotic gene, reaper , in the eye, using the gmr-GAL4 driver. These flies exhibit a rough eye phenotype, and genetically reducing the activity of the Sin3A co-repressor did not suppress this eye toxicity (data not shown). Taken together, these findings indicate that HDAC inhibitors do not act as general anti-apoptotic agents and suggest that their therapeutic benefit in our α-synuclein models is a result of α-synuclein's direct interaction with histones.
Missense mutations in the α-synuclein gene are associated with rare forms of autosomal dominant Parkinson's disease ( 36–38 ). The course of inherited Parkinson's disease is often more aggressive than sporadic forms of the disease. We chose to examine two well-characterized mutations, A30P and A53T, in our in vitro histone acetyltransferase assay system. We hypothesized that these mutations would inhibit histone acetylation to a greater degree than wild-type α-synuclein, yet found rather that both mutants behaved similarly to wild-type. Thus all three forms of α-synuclein appear to associate to a similar extent with histones. We then wondered whether an increased tendency to localize to the nucleus might explain the increased toxicity of these α-synuclein mutations in patients. We over-expressed all three forms of α-synuclein in SH-SY5Y cells, and through quantification, using confocal microscopy, we were able to observe increased nuclear targeting of A30P and A53T. Similar results were seen with SDS–PAGE analysis of nuclear and cytoplasmic fractions of cells expressing each form of α-synuclein. These findings suggest that familial Parkinson's disease mutations may confer increased toxicity through increased nuclear localization. Although the increase in nuclear localization seen with the mutant forms of α-synuclein is modest, the statistically significant change may have important consequences for the viability of neurons over the long lifespan of a postmitotic neuron.
The mechanism by which α-synuclein enters the nucleus is unclear. There are no canonical NLSs embedded within the primary amino acid code to suggest active transport through the nuclear pores. It is possible that some unknown protein with an NLS sequence could bind α-synuclein and shuttle the protein into the nucleus. Alternatively, α-synuclein is a relatively small protein (14 kDa) and may diffuse passively through the nuclear pores. Recent developments in the Parkinson's disease field indicate that the dosage of wild-type α-synuclein is positively correlated with the severity of Parkinson's disease phenotypes. Several families with early-onset Parkinson's disease have been shown to harbor duplications and triplications of the α-synuclein locus ( 39–43 ). Xu et al . ( 44 ) have previously demonstrated an increase in detergent-soluble and -insoluble forms of α-synuclein in the substantia nigra of Parkinson's disease patients relative to age-matched controls. Interestingly, this difference is not seen in dopaminergic regions of the midbrain unaffected in Parkinson's disease. Additionally, Lee et al . ( 45 ) have recently documented increased α-synuclein levels in the plasma of Parkinson's disease patients relative to controls. Overall, these findings suggest that elevated levels of α-synuclein may underlie dopaminergic neurodegeneration in Parkinson's disease. Increased total levels of α-synuclein in Parkinson's disease could lead to increased α-synuclein accumulation in the nucleus through either passive diffusion or active transport. Our data suggest that elevated levels of nuclear α-synuclein then decrease histone acetylation, leading to aberrations in transcriptional control and, ultimately, cell death.
A number of properties of α-synuclein may be important in disease pathogenesis, including abnormal aggregation of the protein ( 46 ), phosphorylation ( 5 , 47 , 48 ), ubiquitination ( 49 ), phospholipid binding ( 50 ) and tyrosine hydroxylase regulation ( 51 ). It will be interesting to determine whether the transcriptional effects we observe act in concert with these other mechanisms to influence neurotoxicity or whether the pathways operate independently.
Although our findings have clear implications for disease pathogenesis and treatment, they also raise intriguing possibilities regarding the normal function of α-synuclein. The precise function of α-synuclein is not yet clear, but data from multiple sources suggest a role in regulating neurotransmitter release at the synapse ( 52–54 ). Specifically, α-synuclein may regulate transmitter release by facilitating proper folding of synaptic proteins critical for neurotransmitter release ( 55 ). Although α-synuclein may influence neurotransmitter release directly, recent work suggests that α-synuclein diffuses away from the synaptic bouton into the axonal cytoplasm in response to neuronal depolarization and thus may have important targets outside the nerve terminal as well ( 56 ). Our work suggests that these targets may be nuclear. It would be interesting to determine whether the synaptic pool of α-synuclein ultimately translocates to the nucleus. If α-synuclein does act as a messenger between the synapse and nucleus, a certain level of α-synuclein in the nucleus may be critical for maintaining normal neuronal function. However, in Parkinson's disease, through mutations or other mechanism, excessive elevation of nuclear α-synuclein levels might tip the balance toward inappropriate transcriptional silencing of downstream targets required for the viability of substantia nigra dopaminergic neurons. If, as our results suggest, increased levels of nuclear α-synuclein in Parkinson's disease lead to decreased histone acetylation and neurotoxicity, then treatments that raise levels of neuronal acetylation may be attractive therapeutic strategies in Parkinson's disease.
MATERIALS AND METHODS
Cell viability assays
Transfected SH-SY5Y cells in 12 well-plates (Lipofectamine 2000, Invitrogen) were fixed in formalin 48 h after transfection and stained for α-synuclein (1:500, LB509, Zymed) and activated caspase-3 (1:500, Cell Signaling). β-galactosidase-transfected cells served as controls for transfection-related toxicity and were stained with anti-β-gal antibody (1:500, Promega) and activated caspase-3 (1:500, Cell Signaling). For each construct, toxicity was measured as the percentage of α-synuclein-transfected cells staining positively for activated caspase-3. For each construct, 10 random fields were counted at 40× magnification. TUNEL analysis was performed in a similar fashion. For TUNEL analysis, toxicity was measured as the percentage of α-synuclein-transfected cells staining positively for TUNEL (TdT FragE1, Oncogene). One-way ANOVA and Newman–Keuls test were used for statistical analysis.
Histological and immunostaining analysis
Adult flies were fixed in formalin, embedded in paraffin and sectioned into serial 4 µm sections. Immunostaining on paraffin sections were performed using an avadin–biotin–peroxidase complex method as described ( 16 ). For evaluation of dopamingeric neurons, the number of tyrosine hydroxylase-immunoreactive cells (detected using rabbit polyclonal antibody to tyrosine hydroxylase, Chemicon; 1:500) at the level of the giant fiber commissure were counted in well-oriented frontal sections.
Plasmid constructs
Full-length human wild-type, A30P, and A53T α-synuclein and β-galactosidase were cloned into pcDNA3.1. Insertion of SV40 large T antigen NLS and mitogen-activated kinase kinase NES at the 3′ end of human wild-type α-synuclein was performed by standard subcloning techniques. For transgenic Drosophila lines, syn NLS and syn NES constructs were subcloned into pUAST.
Drosophila stocks, crosses, transgene expression, drug feedings, polytene chromosomes
Drosophila were grown on standard cornmeal medium at 25°C. Mutant α-synuclein cDNAs were cloned into the GAL4 responsive pUAST expression vector, and transgenic strains ( UAS - synNLS and UAS - synNES ) were created by embryo injection. At least four independent transgenic lines were derived for each construct. Expression of all α-synuclein transgenes was driven by the UAS/GAL4 bipartite expression system. The HDAC inhibitor concentration ranges tested were based on prior Drosophila experiments (SAHA, Biomol and sodium butyrate, Alfa Aesar). For testing the effect of Sin3A on the α-synuclein neurodegeneration phenotype, Sin3A 08269 males were crossed to elav-GAL4 ; UAS - synWT1 and elav-GAL4 ; UAS - synA30P virgin females. For western anaylsis of transgene expression, one adult head from each UAS-construct expressed under the elav-GAL4 driver was ground in 2× Laemmli sample buffer and loaded per lane. All western blots were performed in triplicate or quadruplicate, normalized to tubulin and quantified using Image J software. For polytene chromosome experiments, salivary glands of UAS - synNLS /+; da-GAL4 /+ larvae were dissected in PBS, fixed in 50% acetic acid for 30 s on a glass slide, cover-slipped and squashed to flatten chromosomes. Slides were frozen in liquid nitrogen and coverslips were removed. Slides were fixed in formalin and microwaved in 10 m m sodium citrate, pH 6.6. Slides were stained for α-synuclein (1:100, Clone 42, Transduction Laboratories), α-histone H3 (1:5000, anti-H3, Abcam) and DAPI (Vector).
Histological and immunostaining analysis
Neuronal apoptosis was detected with the TUNEL assay using a commercially available kit (TdT FragE1, Oncogene). Neurodegeneration was quantified by counting the number of TUNEL-positive cells per hemibrain. At least eight hemibrains were examined per genotype. Multivariate ANOVA and Newman–Keuls test were used for statistical analysis. α-Synuclein staining on human and mouse nigral tissue was performed with Clone 42 (1:100) and a tyramide signal amplification kit (Molecular Probes). All other immunofluorescence detection of α-synuclein was performed using secondary antibodies coupled to Alexa Fluor 488 or Alexa Fluor 555.
Histone acetylation analysis of SH-SY5Y cells
Histones were extracted from expression-matched SH-SY5Y stable lines expressing pcDNA, pcDNA/syn NLS or pcDNA/syn NES according to a protocol described by Stein and Mitchell ( 57 ). Histone concentrations were measured by Bradford. Equivalent amounts of histones were separated by SDS–PAGE and probed for anti-acetyl H3 (1:20 000, Upstate) and anti-H3 (1:5000, Upstate). Western blot quantification ( n =3) was performed using Image J software. One-way ANOVA and Newman–Keuls test were used for statistical analysis.
GST-pull down, immunoprecipitation and chromatin fractionation
GST-pull down assays using purified GST-synuclein (wild-type) or GST (negative control) and histones extracted from SH-SY5Y cells were performed as described previously ( 58 ). Immunoprecipitation experiments were performed using wild-type mouse brains (strain SV129). Isolated nuclei were digested with 25 U micrococcal nuclease for 6 min at 37°C. Nuclease digestion was terminated using 0.5 m m EDTA, and nuclei were extracted using the N-PER kit (Pierce). Nuclear extracts were incubated with anti-H3 antibody (Abcam) bound to Dynal beads (Dynal). Beads were washed extensively with PBS+0.5% NP40+complete protease inhibitor tablet (Roche). α-synuclein was detected with Clone 42 antibody (Transduction Laboratories). Chromatin fractionations on SH-SY5Y stable lines were performed as described in Ward and Chen ( 59 ).
Histone acetyltransferase assays
Full-length wild-type, A30P, A53T α-synuclein, wild-type β-synuclein (cDNA was a generous gift from E. Masliah) and CBP-HAT were cloned into pGEX-4T-3 (Amersham Biosciences) and purified by standard methods. Active P/CAF and p300 enzymes were purchased from Upstate. The effect of α-synuclein proteins on histone acetylation was assayed in vitro by a modified histone acetyltransferase assay (Upstate). GST fusion proteins were incubated with histone H3 peptide (1:2, 1:1 or 1:0.5 pmol ratio histone:GST-protein) and histone acetyltransferase buffer at 37°C with gentle agitation for 4 h. Following incubation, P/CAF and acetyl CoA were added to a final volume of 50 ml, and the mixture was incubated for 20 min with gentle agitation at 30°C. Histones were probed with anti-acetyl-lysine antibody (1:250, Upstate) and goat anti-rabbit IgG, HRP conjugate (1:10 000, Upstate). Acetylation was detected by addition of tetramethylbenzidine reagent and subsequent ELISA plate reading at 415 nm. For each condition, the experiment was performed in triplicate, and resulting ELISA values were averaged. Similar results were seen in at least three separate experiments. For chromatin HAT assays, recombinant human histone octamers ( 60 ) were assembled into chromatin, as described previously ( 61 ). Histone acetyltransferase assays were performed as detailed earlier, with a 1:20 pmol ratio (chromatin:GST-protein), run on western blot and probed for anti-acetyl H3 (1:20 000, Upstate) and anti-H3 (1:10 000, Upstate).
Confocal microscopy
All confocal images were taken with a Zeiss LSM META 510 upright confocal microscope (Harvard Center of Neurodegeneration and Repair, Optical Imaging Facility). Wild-type, A30P and A53T pcDNA3.1 constructs were transfected into SH-SY5Y cells and stained for α-synuclein (1:500, Clone 42, Transduction Laboratories) 48 h after transfection. For each construct, at least 50 images were obtained of representative cells. For each image, average nuclear and cytoplasmic fluorescent intensities were measured using Image J software. A corresponding nuclear/cytoplasmic ratio was determined and normalized to the wild-type α-synuclein construct.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Kami Ahmad, Welcome Bender, Stephen Blacklow, Matthew Frosch and Anthony Philippakis for critical comments. Bruce Horwitz provided SV129 mouse brains. We thank William Dauer for his generous contribution of synuclein knockout mouse brains. Michael Schlossmacher kindly contributed the 9661 polyclonal α-synuclein antibody. This work was supported by NIH grant NS41536 to M.B.F. and NIH grant NS049869 to E.K. Fly stocks were obtained from the Bloomington Stock Center. Fly injection services were provided by Douglas Rennie at the Cutaneous Biology Research Center at Massachusetts General Hospital.
Conflict of Interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}