




Abstract
Heterozygous mutations of the PHOX2B gene account for a broad variety of disorders of the autonomic nervous system, either isolated or combined, including congenital central hypoventilation syndrome (CCHS), tumours of the sympathetic nervous system and Hirschsprung disease. In CCHS, the prevalent mutation is an expansion of a 20-alanine stretch ranging from +5 to +13 alanines, whereas frameshift and missense mutations are found occasionally. To determine the molecular basis of impaired PHOX2B function, we assayed the transactivation and DNA binding properties of wild-type and mutant PHOX2B proteins. Furthermore, we investigated aggregate formation by proteins with polyalanine tract expansions ranging from +5 to +13 alanines using immunofluorescence of transfected cells and gel filtration of in vitro translated proteins. We found that transactivation of the dopamine beta-hydroxylase promoter by PHOX2B proteins with frameshift and missense mutations was abolished or severely curtailed, as was in vitro DNA binding although the proteins localized to the nucleus. The transactivation potential of proteins with polyalanine tract expansions declined with increasing length of the polyalanine stretch, and DNA binding was affected for an expansion of +9 alanines and above. Cytoplasmic aggregation in transfected cells was only observed for the longest expansions, whereas even the short expansion mutants were prone to form multimers in vitro. Such a tendency to protein misfolding could explain loss of transactivation for alanine expansion mutations. However, additional mechanisms such as toxic gain-of-function may play a role in the pathogenic process.
INTRODUCTION
The neuronal circuits of the autonomic nervous system that control vegetative functions have been shown to depend on the Phox2b homeodomain transcription factor as the neurons either fail to form or degenerate in mouse mutants null for Phox2b (paired-like homeobox 2B) (1,2). Subsequently, we showed that heterozygous mutations of PHOX2B are responsible for autonomic dysfunctions in man where congenital central hypoventilation syndrome (CCHS), Hirschsprung disease (HSCR) and tumours of the sympathetic nervous system (TSNS) can be found in various combinations (3–5). Thus far, three types of PHOX2B gene mutations have been described: (i) polyalanine expansions ranging from +5 to +13 alanines of a 20-alanine stretch, C-terminal to the homeodomain, which are the far most frequent mutations (∼95%) (Table 1) (5–7), (ii) frameshift mutations (<5%) (5–8) and (iii) rare missense mutations modifying the PHOX2B homeodomain (4,5).
Some phenotype and genotype correlations have been drawn already. In particular, frameshift mutations predispose to TSNS, as opposed to polyalanine expansions (5). Moreover, a correlation also exists between the length of the polyalanine expansion and the severity of the disease: the respiratory phenotype tends to be more severe in patients with longer expansions, and PHOX2B mutations found in patients with late-onset central hypoventilation syndrome harbour always the smallest expansion (+5 alanines) (5–7,9). Interestingly, the penetrance of the HSCR phenotype is incomplete in CCHS patients harbouring an expansion of +6 to +13 alanines suggesting that the HSCR phenotype depends on modifier gene(s) (5). Finally, a de novo interstitial 4p12 deletion encompassing the PHOX2B gene has been reported in a patient with HSCR, growth failure and psychomotor delay but without respiratory phenotype (10).
To further investigate the phenotype–genotype correlation and to clarify the molecular basis of their pathogenicity, we compared transactivation and DNA binding properties of wild-type and mutant PHOX2B proteins. Furthermore, we explored aggregate formation by mutant proteins with expansions ranging from +5 to +13 alanines both in transfected cells and in vitro. We observed a significant decrease in transactivation potential of all missense and frameshift mutations and all, but the shortest, pathogenic polyalanine expansions. Similarly, all polyalanine-expanded proteins had a propensity to form higher order oligomers in vitro. However, the disease-causing molecular mechanisms may not be the same for all mutations, as impaired DNA binding, nuclear exclusion and aggregate formation in the cytoplasm were observed for some, but not all, mutations.
RESULTS
Impaired transactivation of the dopamine beta-hydroxylase promoter by PHOX2B mutant proteins
Defined enhancer sequences in the promoter region of the gene encoding dopamine beta-hydroxylase (DBH), the key enzyme of noradrenaline synthesis, appear to be direct transcriptional targets of the Phox2a and Phox2b factors (11,12). To address the question of whether PHOX2B mutants are able to transactivate the DBH promoter, we performed co-transfection assays in HeLa cells, which express neither PHOX2B nor DBH. As reporter construct, we used the 978-DBH-luciferase pGL3-basic construct, in which 978 bp upstream of the human DBH promoter (containing the critical enhancer region) is fused to the firefly luciferase reporter gene (12,13). The human PHOX2B mutant alleles studied and the resulting phenotypes are summarized in Table 1. Two additional mutations were generated and also examined: a +4 alanine expansion and a +7 alanine expansion interrupted by a threonine (Ala)11(Thr)(Ala)15 (Fig. 1A). As shown in Figure 1B, forced expression of wild-type PHOX2B induced a 10-fold increase of reporter gene expression when compared with the empty vector. A slight decrease in transactivation activity was first observed for the +5 alanine expansion, which, however, did not reach statistical significance, whereas the +4 alanine expansion (that has never been encountered in patients and controls) clearly stayed in the normal range. Expansions of +6, +7 and +9 alanines showed a significant reduction in luciferase activity, which was almost totally abolished for expansions of +11 or more alanines (Fig. 1B). Interestingly, the transcriptional activity of the (Ala)11(Thr)(Ala)15 mutant remained within the normal range (Fig. 1B). Finally, a plot of relative transactivation versus the number of supernumerary alanine residues fitted an exponential regression curve showing that activation of the DBH promoter declines as a function of the length of the expansion, without any obvious threshold (R2=0.95) (Fig. 1C). Similar to the mutants with the longest expansions, proteins with missense and frameshift mutations were almost as inactive as the empty vector (Fig. 1B), although they localized to the nucleus and were expressed at similar levels as the wild-type protein (Fig. 1D) (data not shown).
DNA binding properties of PHOX2B mutant proteins
As shown previously, PHOX2B activates the DBH promoter through binding to three upstream sequence elements, namely PBD1, PBD2 and PBD3 (Fig. 2A) (12). The ability to bind to the PBD2 domain was tested for the three types of mutations: homeodomain missense mutations, polyalanine expansions and frameshift mutations. We produced wild-type and mutant PHOX2B proteins using coupled in vitro transcription/translation. A protein migrating with a molecular weight (MW) of ∼32 kDa was expressed in all cases at similar levels (data not shown). We performed electrophoretic mobility shift assays (EMSA) using radiolabelled PBD2 oligonucleotide as a probe, because PBD2 was the motif exhibiting the highest binding affinity to Phox2 proteins (Fig. 2A) (12). As expected, the R100L and R141G missense mutations resulted in a complete loss of DNA binding (Fig. 2B). Among the three frameshift mutations studied, the 722del38 mutation showed no DNA binding, whereas the 931del5 and the 936insT frameshift mutants retained some ability to bind DNA although the DNA–protein complex appeared more diffuse, suggestive of abnormal protein conformation (Fig. 2B). Concerning the polyalanine size variations, DNA binding was normal for the −7 alanine contraction, the smallest expansions (+5 and +7 alanines) and the (Ala)11(Thr)(Ala)15 mutation. It was consistently decreased for the +9 alanine expansion and barely detectable for the longest expansions (+12 and +13 alanines) (Fig. 2B). Whenever DNA binding occurred, its specificity was confirmed by competition with cold PBD2 oligonucleotide (data not shown).
PHOX2B alanine expansions lead to multimerization in vitro
We hypothesized that reduced DNA binding by the proteins with longest alanine expansions may be the consequence of spontaneous aggregation of the mutant proteins in vitro. To test this idea, we performed gel-filtration chromatography using in vitro translated and radiolabelled wild-type and mutant PHOX2B proteins (+5, +7, +9 and +13 alanines). To maintain conditions similar to those used in the EMSA experiments, gel filtration was carried out in the same non-denaturing binding buffer. SDS–PAGE followed by phosphoimaging was used to record the elution profiles of radiolabelled PHOX2B proteins. Representative elution profiles are shown in Figure 3A. The wild-type PHOX2B protein was eluted as a single peak at a position corresponding to a MW of ∼75 kDa indicating that native PHOX2B forms homodimers in line with previous results obtained in GST pull-down experiments (Fig. 3) (14). In contrast, all mutant proteins tested (i.e. +5, +7, +9 or +13 alanine expansions) contained labelled species that were eluted at positions corresponding to a higher MW (Fig. 3A and B) (data not shown). As SDS–PAGE revealed a single radiolabelled species of the expected MW (data not shown), the presence of higher MW species is probably due to the formation of homo-multimers. In addition, all mutant proteins contained a second peak that was eluted at or near the position of the wild-type protein (+5, +7 and +9 alanine expansion) or with an intermediate MW (+13 expansion) suggesting that the mutant proteins may exist in different conformational states. In any case, there was a clear tendency for the mutants with longer expansions to form higher order oligomers. Assuming that the multimers have molecular shapes that do not differ too much from that of the wild-type dimers, we estimated that the multimeric species are composed of on average 8 and 12 polypeptide chains for the +5 and the +13 expansions, respectively (Fig. 3B). Oligomer size was substantially more variable for the +5 than for the wild-type, +7 and +13 alanine expanded proteins. This may be interpreted to mean that oligomers formed by the +5 alanine protein are unstable and in dynamic equilibrium with the normal dimeric species. In support of the latter, the lower MW form of the +5 mutant protein eluted as a broad peak slightly ahead of the wild-type and the +7 mutant proteins, as if the oligomers become destabilized under elution conditions.
We also tested whether the proteins with the longest expansion would drive the wild-type chains into higher order oligomers. However, this did not occur because a peak eluting at the position of the wild-type dimer was still observed when a mixture of wild-type and +13 alanine mutant proteins was applied. In addition, the +13 alanine mutant oligomer was now eluted at a lower MW position than when run alone (Fig. 3A and B). This may be taken to mean that wild-type chains do in fact co-aggregate with the mutant polypeptides, but that their presence prevents the formation of higher order aggregates.
Altogether, these results show that PHOX2B proteins with polyalanine expansions spontaneously form oligomers, the size of which correlates with the length of the alanine stretch. Oligomer formation was observed even for the +5 alanine expansion, for which DNA binding remained in the normal range. The presence of substantial amounts of what are probably dimers may explain the essentially normal DNA binding properties of the +5 and +7 mutants and the only modest decrease in binding of the +9 alanine mutant. The fraction of the mutant proteins eluted as dimers or trimers, estimated by integrating the peak areas in Figure 3A, were 72, 87 and 79% for the +5, +7 and +9 mutants, respectively, whereas the +13 mutant never contained labelled species that eluted at position at or near the wild-type protein (Fig. 3A) (data not shown). It is equally possible that oligomers and dimers exist in a dynamic equilibrium, which, according to the law of mass action, is shifted towards the dimers by the DNA binding of the latter.
Cytoplasmic aggregation correlates with the size of the alanine expansion
Several studies showed that proteins with expanded alanine tracts may aggregate either in the cytoplasm or in the nucleus or in both (15–18). To address this issue, we investigated the subcellular distribution of PHOX2B protein in HeLa cells transfected with wild-type or mutant PHOX2B. Expression of wild-type PHOX2B resulted in an exclusively nuclear staining of transfected cells (Fig. 4A). In contrast, the +13 alanine mutant localized almost exclusively to the cytoplasm, indicative of aggregate formation in the cytosol. These aggregates sometimes spread over the whole cytoplasm or concentrated near the nucleus (Fig. 4A and B). Alanine expansions ranging from +9 to +12 were also prone to forming aggregates in the cytosol, although less frequently, and without nuclear exclusion (Fig. 4A). In the latter cases, the number of cells presenting cytoplasmic aggregates increased with the amount of transfected DNA. This was not true for the smaller alanine expansions (+5 to +7 alanines), where cytoplasmic aggregation could not be detected whatever the amount of transfected DNA and even at 48 h after transfection (data not shown). Furthermore, the staining pattern in the nucleus was indistinguishable between the wild-type and mutant proteins with short expansions (Fig. 4A). Nuclear aggregates were not observed using both standard and Apotome Zeiss fluorescence microscopy (data not shown).
One way cells handle misfolded and aggregate-prone proteins is by attempting to refold them using molecular chaperones such as heat shock proteins (HSP). In particular, the role of HSP70 has been studied in polyglutamine and polyalanine protein conformation disorders (15,18–20). HSP70 was found to co-localize with the cytoplasmic aggregates formed by the +13 alanine PHOX2B mutant protein while it remained spread throughout the cytoplasm when tranfected with wild-type PHOX2B (Fig. 4B). Geldanamycin is an antibiotic that activates a heat shock response in mammalian cells (19). When cells expressing the +13 mutant were treated for 48 h with geldanamycin, a major decrease in the number of cells presenting cytoplasmic aggregates was observed (Fig. 4C) (data not shown).
Altogether, these results indicate that PHOX2B mutants with the longest alanine expansion form cytoplasmic aggregates and recruit HSP70, which may counteract aggregate formation as shown by the geldanamycin effect, while proteins with shorter expansions remain nuclear with no obvious signs of aggregate formation.
The effects of co-expressing the +13 mutant on wild-type Phox2 proteins
A dominant-negative effect of mutant proteins with alanine expansions has been proposed for HOXD13 and ZIC2 (21,22). To test this possibility for the PHOX2B mutant proteins, we measured luciferase activity after co-transfecting the wild-type PHOX2B expression construct with increasing amounts of the plasmid coding for the +13 alanine mutant protein. Luciferase activity remained essentially in the normal range upto a 5-fold excess of the mutant construct, significant impairment of transactivation being observed only at a 10-fold excess (Fig. 5A). These results suggest that only a minor fraction of wild-type PHOX2B protein may interact with the misfolded +13 alanine expansion mutant.
Phox2a and Phox2b are paralogous transcription factors that have identical homeodomains, are often expressed together (23) and form heterodimers (14). One may thus hypothesize that PHOX2B aggregates may trap PHOX2A in the cytoplasm resulting in a dominant-negative effect. However, we observed that nuclear localization of Phox2a was preserved in HeLa cells co-transfected with Phox2a and the +13 alanine expansion mutant of PHOX2B (Fig. 5B).
Altogether, these results suggest that a dominant-negative effect of mutant PHOX2B by oligomerization with wild-type PHOX2B or PHOX2A may not be a major pathogenic mechanism.
DISCUSSION
Here, we report a functional study of PHOX2B mutations associated with autonomic nervous system dysfunction. PHOX2B has the longest known alanine tract in mammals (n=20) and one of the shortest disease-causing expansions (+5 alanines) (reviewed in 24,25). Polyalanine tract expansions are by far the most frequent pathogenic mutations in PHOX2B (5). However, missense and frameshift mutations have also been described as in most of the diseases known to result from alanine expansion mutations (21) (reviewed in 25), suggesting similar physiopathologic consequences of all types of mutations. Although polyalanine tracts are frequent in eukaryotic proteins, their normal function(s) remain(s) largely unknown (24). They lie outside of other known functional domains and are regarded as flexible spacer elements essential to protein conformation, protein–protein interactions and/or DNA binding (26,27). Whatever the nature of the repeated amino acid involved (alanine, glutamine, aspartic acid), such homopolymeric tract expansions may lead to protein misfolding and aggregation and result in loss-of-function, gain-of-function or dominant-negative effects including cellular toxicity (28).
In this study, we observed a drastic decrease in the transactivation potential of both missense and frameshift mutations. As for alanine expansions, we observed a correlation between the length of the polyalanine expansion and the decrease in transactivation of the DBH promoter, indicating that the size of the alanine tract influences the level of transactivation. Interestingly, the insertion of a threonine within a 27 alanine tract in the PHOX2B protein appears sufficient to restore promoter activation. Thus, rather than the total number of hydrophobic amino acids, it is the purity of the homopolymeric tract that matters.
We then tested whether reduced transactivation is due to a reduction in DNA binding to the DBH promoter. We observed a drastic decrease of DNA binding for missense and frameshift mutations, but impaired binding for polyalanine expansions only for mutants with a +9 or longer expansions (Fig. 2B). However, in the gel filtration experiments, even the +5 alanine expansion displayed a tendency to form oligomers in vitro. In contrast to the wild-type protein, all polyalanine tract mutants were eluted as two distinct peaks.The mutant proteins most likely exist in two different conformational states that differ in their propensities to aggregate. That mutant proteins with disease-causing homopolymer tracts may exist in two conformations, one properly folded and one aggregate-prone, have been proposed before on theoretical grounds (29). As the size of the PHOX2B–DNA complex detected by EMSA did not vary, the higher order oligomers are unlikely to bind DNA in vitro. The observation that an important fraction of the +5 and +7 alanine mutants elutes during gel filtration at or near the position of the wild-type peak (suggesting that dimers and oligomers exist in a dynamic equilibrium) may explain why DNA binding was in the normal range for the short expansions.
Another explanation for the reduced transactivation could be mislocation of the protein. Indeed, PHOX2B mutants with long, but not those with short expansions, have a marked tendency to form cytoplasmic aggregates in transfected cells. The higher the number of alanine residues, the higher the tendency of the protein to aggregate, which could lead to nuclear exclusion. Recently, Bachetti et al. (30) reported very similar data on the transactivating properties of PHOX2B with polyalanine expansions. In contrast to our results, however, these authors observed intranuclear aggregates for all alanine expansions and, to a small extent, also for the wild-type protein. This discrepancy could be due to the fact that they used a different cell type and a different transfection protocol. Another possibility is that the different results from their use of GFP fusion proteins. Nevertheless, ultastructural studies are needed to definitively rule out formation of intranuclear aggregates in our model.
The aggregates recruit HSP70 and become less prominent when chaperone expression is boosted by treating cells with geldanamycin. One may thus speculate that chaperone proteins promote either refolding or degradation of the expanded proteins at a rate efficiently enough to avoid the accumulation of intracellular aggregates for small expansions. Indeed, although cytoplasmic and/or nuclear aggregations have been observed in cell culture for the shortest disease-causing alanine expansion in some cases (HOXD13, SOX3, RUNX2) (15), this is not the case for PHOX2B (+5 to +7 alanines, this study), nor for ZIC2 (+10 alanines) (22).
Interestingly, the human phenotype resulting from the +5 alanine expansion of PHOX2B is variable, ranging from respiratory distress at birth (CCHS) to later onset variants occurring after months or years during an episode of upper airway infection (Table 1) (5,7,9,31). This may be taken to mean that under conditions of cellular stress, the capacity of the cells' chaperone system becomes unable to cope efficiently with even small expansions, eventually leading to the formation of aggregates. In the case of PHOX2B proteins harbouring longer expansions, chaperone-mediated refolding and the proteasome system may never suffice to cope with the misfolded proteins, thus leading to an early-onset and more severe phenotype. In the case of frameshift and missense mutations, PHOX2B subcellular localization is not impaired as the mutant proteins appear stable and localize to the nucleus.
The question thus arises whether loss of function explains most or all of the phenotypic consequences. In the case of Hoxa13, mice bearing a heterozygous mutation leading to a +10 alanine expansion (Hoxa13+/+10 ala) were phenotypically identical to Hoxa13−/+ mice (17), arguing for pure haploinsufficiency. Protein levels were reduced in the limb bud although transcription, splicing and stability of the Hoxa13 mRNA remained normal. The authors hypothesized a loss-of-function mechanism via degradation of the mutant protein, but protein aggregation and mislocalization have not been tested. In a pure loss-of-function situation, patients with PHOX2B mutations, which are always heterozygous, are expected to have 50% normal activity in the case of null mutations, and somewhat more if the mutant retains residual activity. On the basis of our functional assays, one would argue that mutant proteins with small expansions should retain at least some activity. However, reporter gene transactivation in cultured cells may only incompletely mirror the in vivo situation where functional impairment may also vary according to cell type, as may vary sensitivity to haploinsufficiency, and we do not know at present what happens in the cells involved in the etiopathogeny of the disease. An obvious possibility is that even short alanine expansions perturb the interaction with a co-factor essential for transactivation of some promoters but not of the DBH promoter.
On the other hand, loss-of-function alone may not explain the entire disease spectrum caused by PHOX2B mutations. A heterozygous deletion encompassing the PHOX2B locus, frameshift and missense mutations have been identified in patients without congenital ventilatory phenotype (4,8,10). In contrast, a ventilatory phenotype is always reported in patients harbouring polyalanine-expanded PHOX2B proteins, although it appears later in life in some patients with a +5 alanine expansion. One may thus postulate an alternative disease-causing mechanism for alanine expansions. A dominant-negative effect or a toxic gain-of-function is possibility that has been shown to occur in other polyalanine diseases (28). Bachetti et al. (30) observed some trapping of wild-type PHOX2B by the +13 alanine mutant protein in nuclear aggregates. We observed impaired transactivation by the wild-type protein with a 10-fold excess of the +13 mutant and no co-aggregation with Phox2a, known to form heterodimers with Phox2b, arguing against a dominant-negative effect as a major pathogenic mechanism. Finally, a toxic gain-of-function in vulnerable cell types remains possible. However, while the main cause of polyglutamine tract diseases seems toxic accumulation of protein aggregates that lead to cell death late in life (32–34), formation of potentially toxic aggregates of alanine-expanded proteins has been demonstrated only for ARX (18) and PABP2 (29).
Elucidation of this issue must await the generation of mouse models, in which the cells and tissues relevant for the disease process may be studied directly. It is worth noting in this respect that patients presenting the association of CCHS and HSCR have never been found to harbour a +5 alanine expansion, but present at least a +6 alanine expansion or other types of mutations. These data fit in well with the observation that mutations resulting in short polyalanine expansions lie at the milder end of the phenotypic spectrum of a disease when compared with other types of mutations but also tend to get more severe with longer alanine expansions (5–7,25).
In conclusion, while the phenotypic consequences of frameshift and missense mutations can be explained by loss-of-function, additional mechanisms may be invoked for alanine expansions, of which cellular toxicity is a possibility. All PHOX2B proteins harbouring polyalanine expansions appear to be prone to misfolding, as they spontaneously form oligomers in vitro. However, we never observed signs of aggregate formation in the nucleus and nuclear exclusion resulting from cytoplasmic aggregation only for the longest alanine expansions. Finally, our in vitro studies strengthen phenotype–genotype correlations, as functional impairment indicative of misfolding of PHOX2B proteins correlates with the size of the alanine expansion.
MATERIAL AND METHODS
Plasmids and mutations
Given the 100% degree of sequence identity between human and mouse Phox2b proteins, we introduced the human mutations into mouse cDNA. PHOX2B homeodomain mutations (R100L and R141G) were generated using the quikChange® XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocol. The following oligonucleotides were used: 5′-AACGCAAGCAGCGGCTCATCCGCACCACCT-3′ and 5′-AGGTGGTGCGGATGAGCCGCTGCTTGCGTT-3′ (R100L), 5′-CCTCACCGAGGCGGGAGTCCAGGTGTGGTT-3′ and 5′-GAACCACACCTGGACTCCCGCCTCGGTGAG-3′ (R141G). For all other PHOX2B constructs (−7, +5, +6, +7, +9, +11, +12, +13 alanines, 722del38, 931del5, 936insT), the relevant sequences were PCR amplified using human genomic DNA of patients as templates and the following primers: 5′-GCACTGACCCGGACAGCACT-3′ and 5′-GGGAGCTCCGAGTGGGCGAGCGGGTA-3′ for alanine contraction or expansion, 5′-GCACTGACCCGGACAGCACT-3′ and 5′-GGGAGCTCTGGCTCGCCCGCTGTC-3′ for frameshifts. The PCR fragments were introduced into wild-type mouse cDNA by SacI digestion (for which a site had been generated in the primer) and ligation. Two remaining mutations were accidentally obtained and further studied:+4 alanines and (Ala)11(Thr)(Ala)15. All constructs were validated by DNA sequencing. For in vitro transcription/translation, constructs were subcloned into a pcDNA3.1 vector (Invitrogen) containing a T7 promoter.
We used pCAGGS–IRES2–EGFP as a vector expressing wild-type or recombinant Phox2b driven by a composite chicken beta actin-CMV promoter. pCAGGS–PHOX2B–IRES2–EGFP was constructed by inserting the IRES2–EGFP sequence (Clontech) 3′ to the Phox2b coding sequence into the previously described pCAGGS–Phox2b vector (35). To increase GFP expression, the 3′ end of the untranslated region was deleted by NruI and EcoRI digestion followed by blunt-end ligation. PCAGGS–Phox2a was constructed by introducing the beta-actin intron from pCAGGS (35) as a SpeI–EcoRI fragment 5′ of the Phox2a cDNA in pKS and transferring the insert cut out by SpeI–XhoI into the SpeI–XhoI–cut pCAGGS vector. The IRES2–EGFP sequence isolated by a NotI–NheI digestion was then introduced into pCAGGS–Phox2a cut with the same enzymes. The luciferase reporter construct was prepared using pGL3 basic vector (Promega) containing the firefly luciferase gene under the control of a 978 bp DBH promoter fragment (12,13).
Cell culture and luciferase transfection assays
Plasmids used for luciferase assays were prepared using Qiagen kit. HeLa cells were grown to 95% confluency in Dulbecco's minimum essential medium supplemented with 10% fetal bovine serum in 12-well plates. Cells were transfected with 260 ng of recombinant pCAGGS–PHOX2B–IRES2–EGFP expression vector, 1.2 µg of the firefly luciferase reporter promoter, 100 ng of pRL-CMV Renilla luciferase internal control (Promega) and 2.5 µl of Lipofectamine 2000 in 100 µl of OPTI-MEM (Invitrogen). Cells were harvested and lysed 24–48 h after transfection. Firefly and Renilla luciferase activities were assayed according to the manufacturer's protocol. Luciferase activity of each construct was normalized by the internal control pRL-CMV. Experiments were repeated seven times in duplicate. Co-transfection of wild-type PHOX2B and +13 alanine expansion was assayed in six-well plates. HeLa cells were transfected with 150 ng of pCAGGS–PHOX2B (wild-type)–IRES2–EGFP, 900 ng of reporter plasmid, 40 ng of pRL-CMV and increasing amounts of pCAGGS–PHOX2B +13–IRES2–EGFP (i.e. 150, 300, 750 and 1500 ng). Total plasmid amount was equalized by adding pCAGGS–IRES2–EGFP empty vector. Experiments were repeated five times in duplicate.
In vitro transcription and translation
The TNT® coupled reticulocyte lysate transcription/translation system (Promega) was used to generate in vitro translated proteins according to the manufacturer's protocol. The vectors pcDNA3.1–PHOX2B were opened by EcoRI digestion and translated with or without [35S]methionine. For co-translation of the wild-type and +13 alanine PHOX2B, the amount of each template was reduced by one-half for the same reaction volume. Expressed proteins were analysed by 15% SDS–PAGE and autoradiography and used for EMSA or gel filtration chromatography.
Electrophoretic mobility shift assays
The following double-stranded oligonucleotides (−87)5′-CCGCTAGACAAATGTGATTACCC-3′ (−56) corresponding to the sequence of the PBD2 element in the DBH promoter was end-labelled with [gamma32P]-ATP using T4 polynucleotide kinase. The EMSAs were carried out in 20 µl of the binding reaction buffer (12 mm Hepes–NaOH (pH 7.9), 4 mm Tris–Cl (pH 7.9), 60 mm KCl, 1 mm EDTA, 1 mm DTT and 12% glycerol), 1 µg of poly(dI:dC) as non-specific competitor, labelled probe (20 000–50 000 cpm) and 3 µl of in vitro recombinant translated Phox2b proteins. The reaction mixture was incubated at room temperature for 20 min. Twenty microliters of the DNA–protein complexes were resolved on high ionic strength, non-denaturing 6% polyacrylamide gels. Subsequently, gels were vacuum-dried and autoradiographed at −70°C with an intensifying screen for 6–16 h, depending on the strength of the signals.
Immunocytochemistry
All immunofluorescence experiments were performed on HeLa cells grown to 95% confluency on eight individual chamber culture slides (Falcon®). Subcellular localizations of expansion mutants were analyzed on cells transfected with 150–500 ng of recombinant Phox2b pcDNA3.1 expression vectorsusing lipofectamine 2000. The co-localization of Phox2a and aggregates was checked after co-transfection of 400 ng of +13 alanine recombinant PHOX2B pcDNA3.1 expression vector and 200 ng of pCAGGS–Phox2a wild-type expression vector. Twenty-four or forty-eight hours after transfection, cells were fixed in paraformaldehyde (4% in PBS) for 10 min and then permeabilized with Triton X-100 (0.1% v/v in PBS) for 10 min at room temperature. Non-specific binding sites were blocked with 10% donkey serum (in PBS/BSA 1%/Tween 0.1%). Cells were then exposed to the anti-C-terminal Phox2b rabbit antibody (dilution 1 : 300 in PBS/BSA 1%/Tween 0.1%) (23) for 1 h at room temperature, washed off and subsequently stained with a secondary donkey anti-rabbit Texas red-conjugated antibody (Jackson Corporation, West Grove, PA, USA) for 1 h at room temperature (dilution 1 : 100 in PBS). PHOX2B frameshift mutations were detected using an anti-N-terminal PHOX2B goat antibody (Tebu-bio, dilution 1 : 400) and PHOX2A using anti-N-terminal PHOX2A goat antibody (Tebu-bio, dilution 1 : 200). In both cases, we used Alexa® Fluor 488 donkey anti-goat IgG (Invitrogen, dilution 1 : 400) as secondary antibody. For analysis of HSP70 localization, we used mouse anti-HSP70 (Tebu-bio, dilution 1 : 500) and as secondary antibody Alexa® Fluor 488 donkey anti-mouse IgG diluted (Invitrogen, dilution 1 : 200). The slides were mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories). Images were acquired using either an OlympusX81 fluorescence microscope or an Apotome Zeiss microscope and Axiovision software. Apotome Zeiss system provides an optical slice view reconstructed from fluorescent samples, using a series of ‘grid projection’ acquisitions.
Geldanamycin (InvivoGen) was used as described previously (19) and added to HeLa cells transfected with PolyFect (Qiagen).
Gel filtration
A Sephacryl S-300HR (Amersham) column (total volume 1.1 ml) equilibrated with EMSA buffer and run by gravity flow was used throughout. Ferritin, thyroglobulin, bovine serum albumin, aldolase and ovalbumin were used as MW standards to construct a calibration curve. The in vitro translated [35S]Met-labelled wild-type and mutant PHOX2B proteins (30 µl) were incubated for 20 min at RT in EMSA buffer before being applied to the column and eluted with the same buffer. Fifty-microliter fractions were collected and radioactivity counted. The radioactive high-MW fractions were analysed by SDS–PAGE on 15% gels and the radioactivity in the band corresponding to the PHOX2B polypeptide quantified by phosphoimaging using a FujixBAS1000 phosphoimager. Elution profiles were drawn by Excel and the volume of the peak fractions determined. The partition coefficient Kav (Ve−V0/Vt−V0, where Ve is the elution volume, Vt the total volume of the column and V0 the void volume determined by elution of blue Dextran) was then determined for the peak fractions. The corresponding MWs were read from the calibration curve (Kav versus log MW) constructed with the known MWs of the standards.
ACKNOWLEDGEMENTS
We thank the CCHS families and the referring clinical teams world wide for participation, the Association Française du syndrome d'Ondine for its support and Emmanuelle Perret for technical assistance with the Apotome Zeiss system (PFID, Institut Pasteur). This work was supported by grants from the GIS-Maladies Rares, the EU (QLG2-CT-2001-01467) and from Association Française contre les Myopathies (to C.G.).
Conflict of Interest statement. None declared.
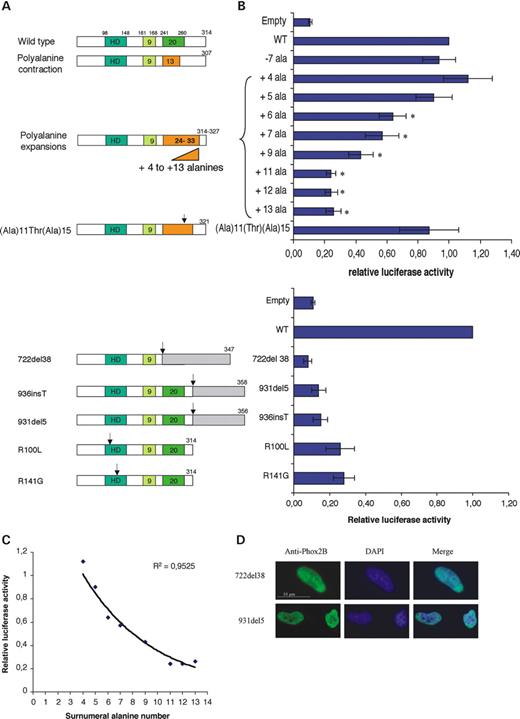
Figure 1. Transactivation of the DBH promoter by wild-type and mutant PHOX2B proteins. (A) Schematic representation of the wild-type and mutant PHOX2B proteins depicting homeodomain and 9- and 20-alanine tracts. The three types of mutations tested (polyalanine tract variations, homeodomain missense and frameshift mutations) are indicated. The final size of each protein is given as the number of amino acid residues. (B) Luciferase activities. The error bars indicate SD based on n=7 duplicate experiments. Each luciferase activity was normalized to an internal control (pRL-CMV, Promega). In each experiment, the activity of the wild-type protein was set to one, and the results are shown as the ratio between the activities of the mutant and wild-type proteins. PCAGGS–IRES2–EGFP was used as negative control (Empty). *P<0.03 (by Wilcoxon test) is significantly different from the wild-type. There is no statistically significant difference between Empty, 722del38, 931del5, 936insT, R100L and R141G groups (ANOVA Kruskal–Wallis). (C) Exponential regression curve. Regression plot of relative luciferase transactivation activity (y) as a function of supernumerary alanine residues (x) for the disease-causing expansions. Luciferase activity declines with the number of additional alanines according to the exponential regression. (D) Transfection of HeLa cells with PHOX2B cDNA carrying either 722del38 or 936insT showing similar nuclear localization as the wild-type. The images were taken at ×1000 magnification on a fluorescence microscope.
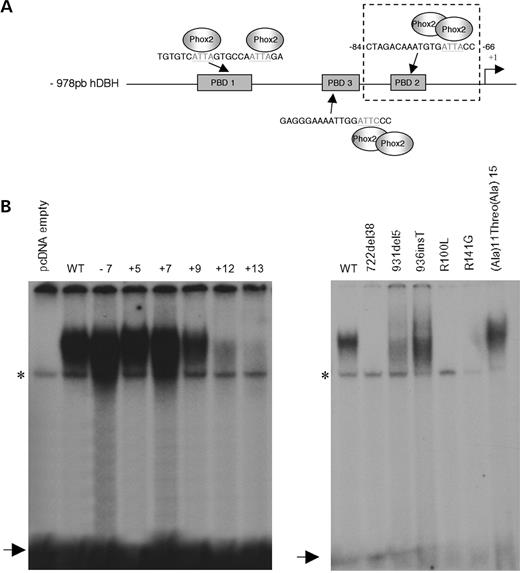
Figure 2. Electromobility shift assay. (A) Diagram of the 978 bp human DBH promoter. The three PHOX2(A/B) binding sites are indicated (PBD1, PBD2, PDB3). We focused on PDB2 previously described as the noradrenergic-cell-specific Phox2a/Phox2b binding site, with which Phox2b interacts as a dimer.(B) Gel shift assays with in vitro translated PHOX2B wild-type (WT) and mutant proteins using the 32P-PBD2-binding site as a probe. (A and B) Two independent gel shift experiments. In all EMSA experiments, in vitro translated pcDNA 3.1 empty vector was used as a control. Free probe is indicated by an arrow and a non-specific band is marked with an asterisk. The mutations are indicated above each lane.
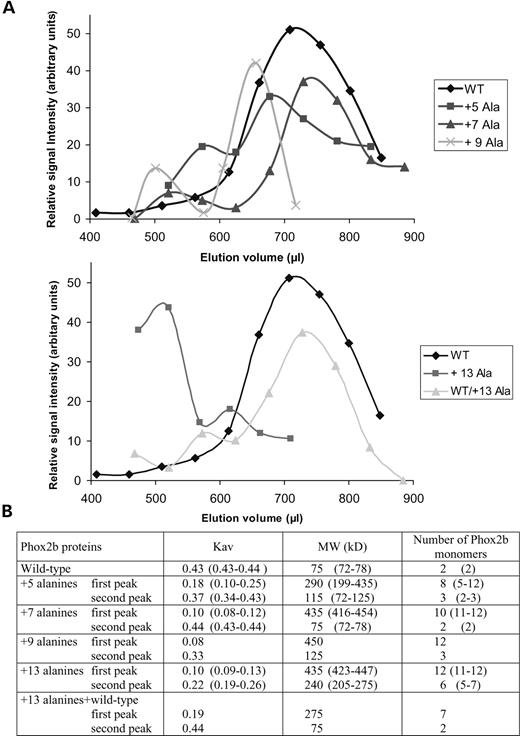
Figure 3. Gel filtration chromatography of wild-type and mutant PHOX2B proteins with polyalanine expansions. (A) Representative elution profiles of in vitro-translated 35S-labelled wild-type and mutant PHOX2B proteins. After SDS–PAGE of the radioactive fractions, the signal intensity of the PHOX2B band as measured by phosphoimaging (see Materials and Methods) was plotted against elution volumes. The areas underneath the peaks were measured to give an estimate of the fraction of the mutant protein found in the higher and lower MW peaks. The fraction of the protein in the lower MW peak was 72% for the +5, 87% for the +7, 79% for the +9 and 23% for the +13 protein. A peak indicative of higher order oligomers was never observed with the wild-type protein, which was always eluted as a single peak. (B) Molecular sizes of Phox2b mutant proteins obtained by gel filtration. The mean and the range of the partition coefficients Kav and of the apparent MWs are given for two to three independent experiments; where one experiment was done, a single value is presented. Wild-type Phox2b (calculated MW 31.6 kDa) elutes as a 75 kDa species assumed to be a dimer of two subunits with an apparent MW of 37.5 kDa each. In the last column, the number of monomers for each molecular species is given that fit best with the mean and the range of the MWs.
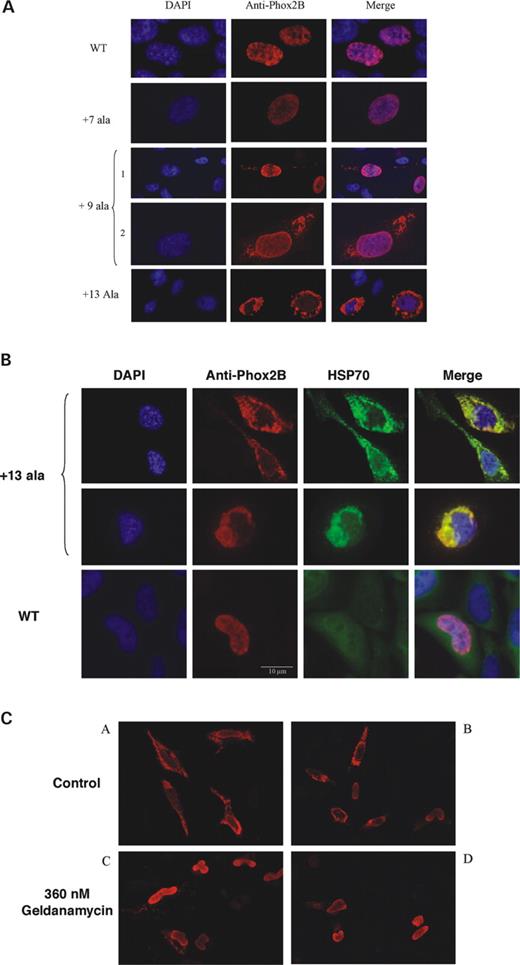
Figure 4. Cellular localization of wt and expanded PHOX2B proteins. (A) Cytoplasmic aggregation correlates with the size of the alanine expansion. The localization of the different proteins was analysed by immunocytochemistry 24 h after transfection using anti-Phox2b antibodies. Wild-type and +7 alanine expansion PHOX2B proteins are exclusively found in the nucleus, whereas cytoplasmic aggregates are visible for the +9 alanine expansion and get larger for the+13 alanine expansion leading to almost complete nuclear exclusion. The images were taken at ×1000 magnification on a fluorescence microscope, except for the +9 (1) and +13 alanine images taken with ×400 magnification. (B) Cytoplasmic aggregates of the +13 alanine mutant protein (red) co-localize with HSP70 heat shock protein (green). When cells are transfected with wild-type PHOX2B, HSP70 is spread throughout the cell. The images were taken on a fluorescence microscope with ×1000 magnification. (C) Geldanamycin reduces the formation of aggregates in transfected cells. HeLa cells were transfected with pcDNA3.1 expressing PHOX2B +13 alanine mutant protein and grown for 48 h in the absence (a and b) or presence (d and c) of geldanamycin (concentration 360 nm). We observed both a decrease in the number of cells with cytoplasmic aggregates and a reduction of their size when aggregates are present in the cytoplasm. The images were taken on a fluorescence microscope with ×200 magnification.
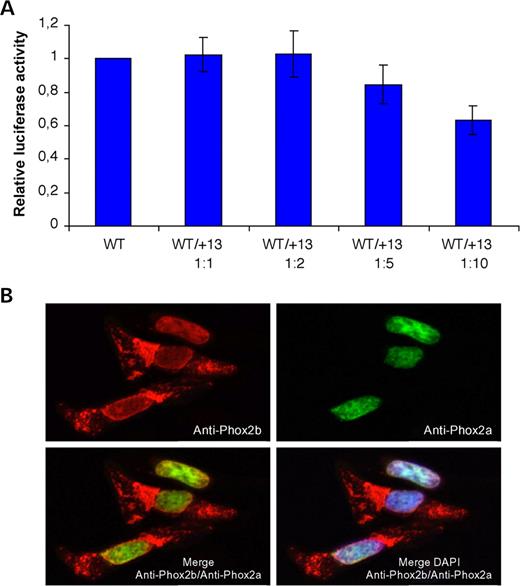
Figure 5. Effects of the +13 alanine expansion on wild-type Phox2b and Phox2a. (A) Co-transfection of the PHOX2B wild-type construct with increasing amounts of the PHOX2B +13 alanine expansion construct. Wild-type/mutant ratios are indicated. The error bars indicate SD based on n=5 duplicate experiments. (B)Co-expression of the PHOX2B +13 alanine mutant with wild-type Phox2a. Despite formation of cytoplasmic aggregates by the +13 alanine mutant, wild-type Phox2a translocates into the nucleus.
PHOX2B mutant alleles and their phenotypic consequences
| PHOX2B nucleotidic variation | Number of patients (n=155) | LO-CHS | CCHS | CCHS+HSCR | CCHS+HSCR+TSNS | CCHS+TSNS | TSNS | HSCR+TSNS |
|---|---|---|---|---|---|---|---|---|
| +5 Alanines | 35 | 7 | 28 | — | — | — | — | — |
| +6 Alanines | 43 | — | 38 | 5 | — | — | — | — |
| +7 Alanines | 55 | — | 38 | 17 | — | — | — | — |
| +9 Alanines | 3 | — | 2 | — | — | 1 | — | — |
| +11 Alanines | 4 | — | 2 | 2 | — | — | — | — |
| +12 Alanines | 3 | — | 1 | 2 | — | — | — | — |
| +13 Alanines | 6 | — | 1 | 4 | — | 1 | — | — |
| 722del38 | 1 | — | — | — | 1 | — | — | — |
| 931del5 | 1 | — | — | — | 1 | — | — | — |
| 936insT | 1 | — | — | — | 1 | — | — | — |
| R100L | 1 | — | — | — | — | — | 1 | — |
| R141G | 1 | — | — | — | — | — | — | 1 |
| PHOX2B nucleotidic variation | Number of patients (n=155) | LO-CHS | CCHS | CCHS+HSCR | CCHS+HSCR+TSNS | CCHS+TSNS | TSNS | HSCR+TSNS |
|---|---|---|---|---|---|---|---|---|
| +5 Alanines | 35 | 7 | 28 | — | — | — | — | — |
| +6 Alanines | 43 | — | 38 | 5 | — | — | — | — |
| +7 Alanines | 55 | — | 38 | 17 | — | — | — | — |
| +9 Alanines | 3 | — | 2 | — | — | 1 | — | — |
| +11 Alanines | 4 | — | 2 | 2 | — | — | — | — |
| +12 Alanines | 3 | — | 1 | 2 | — | — | — | — |
| +13 Alanines | 6 | — | 1 | 4 | — | 1 | — | — |
| 722del38 | 1 | — | — | — | 1 | — | — | — |
| 931del5 | 1 | — | — | — | 1 | — | — | — |
| 936insT | 1 | — | — | — | 1 | — | — | — |
| R100L | 1 | — | — | — | — | — | 1 | — |
| R141G | 1 | — | — | — | — | — | — | 1 |
LO-CHS: late-onset central hypoventilation syndrome, CCHS: congenital central hypoventilation syndrome, HSCR: Hirschsprung disease, TSNS: tumour of the sympathetic nervous system.
PHOX2B mutant alleles and their phenotypic consequences
| PHOX2B nucleotidic variation | Number of patients (n=155) | LO-CHS | CCHS | CCHS+HSCR | CCHS+HSCR+TSNS | CCHS+TSNS | TSNS | HSCR+TSNS |
|---|---|---|---|---|---|---|---|---|
| +5 Alanines | 35 | 7 | 28 | — | — | — | — | — |
| +6 Alanines | 43 | — | 38 | 5 | — | — | — | — |
| +7 Alanines | 55 | — | 38 | 17 | — | — | — | — |
| +9 Alanines | 3 | — | 2 | — | — | 1 | — | — |
| +11 Alanines | 4 | — | 2 | 2 | — | — | — | — |
| +12 Alanines | 3 | — | 1 | 2 | — | — | — | — |
| +13 Alanines | 6 | — | 1 | 4 | — | 1 | — | — |
| 722del38 | 1 | — | — | — | 1 | — | — | — |
| 931del5 | 1 | — | — | — | 1 | — | — | — |
| 936insT | 1 | — | — | — | 1 | — | — | — |
| R100L | 1 | — | — | — | — | — | 1 | — |
| R141G | 1 | — | — | — | — | — | — | 1 |
| PHOX2B nucleotidic variation | Number of patients (n=155) | LO-CHS | CCHS | CCHS+HSCR | CCHS+HSCR+TSNS | CCHS+TSNS | TSNS | HSCR+TSNS |
|---|---|---|---|---|---|---|---|---|
| +5 Alanines | 35 | 7 | 28 | — | — | — | — | — |
| +6 Alanines | 43 | — | 38 | 5 | — | — | — | — |
| +7 Alanines | 55 | — | 38 | 17 | — | — | — | — |
| +9 Alanines | 3 | — | 2 | — | — | 1 | — | — |
| +11 Alanines | 4 | — | 2 | 2 | — | — | — | — |
| +12 Alanines | 3 | — | 1 | 2 | — | — | — | — |
| +13 Alanines | 6 | — | 1 | 4 | — | 1 | — | — |
| 722del38 | 1 | — | — | — | 1 | — | — | — |
| 931del5 | 1 | — | — | — | 1 | — | — | — |
| 936insT | 1 | — | — | — | 1 | — | — | — |
| R100L | 1 | — | — | — | — | — | 1 | — |
| R141G | 1 | — | — | — | — | — | — | 1 |
LO-CHS: late-onset central hypoventilation syndrome, CCHS: congenital central hypoventilation syndrome, HSCR: Hirschsprung disease, TSNS: tumour of the sympathetic nervous system.
References
Pattyn, A., Morin, X., Cremer, H., Goridis, C. and Brunet, J.F. (
Brunet, J.F. and Pattyn, A. (
Amiel, J., Laudier, B., Attie-Bitach, T., Trang, H., de Pontual, L., Gener, B., Trochet, D., Etchevers, H., Ray, P., Simonneau, M. et al. (
Trochet, D., Bourdeaut, F., Janoueix-Lerosey, I., Deville, A., de Pontual, L., Schleiermacher, G., Coze, C., Philip, N., Frebourg, T., Munnich, A. et al. (
Trochet, D., O'Brien, L.M., Gozal, D., Trang, H., Nordenskjold, A., Laudier, B., Svensson, P.J., Uhrig, S., Cole, T., Munnich, A. et al. (
Weese-Mayer, D.E., Berry-Kravis, E.M., Zhou, L., Maher, B.S., Silvestri, J.M., Curran, M.E. and Marazita, M.L. (
Matera, I., Bachetti, T., Puppo, F., Di Duca, M., Morandi, F., Casiraghi, G.M., Cilio, M.R., Hennekam, R., Hofstra, R., Schober, J.G. et al. (
Mosse, Y.P., Laudenslager, M., Khazi, D., Carlisle, A.J., Winter, C.L., Rappaport, E. and Maris, J.M. (
Trang, H., Laudier, B., Trochet, D., Munnich, A., Lyonnet, S., Gaultier, C. and Amiel, J. (
Benailly, H.K., Lapierre, J.M., Laudier, B., Amiel, J., Attie, T., De Blois, M.C., Vekemans, M. and Romana, S.P. (
Kim, H.S., Seo, H., Yang, C., Brunet, J.F. and Kim, K.S. (
Seo, H., Hong, S.J., Guo, S., Kim, H.S., Kim, C.H., Hwang, D.Y., Isacson, O., Rosenthal, A. and Kim, K.S. (
Hong, S.J., Chae, H. and Kim, K.S. (
Adachi, M., Browne, D. and Lewis, E.J. (
Albrecht, A.N., Kornak, U., Boddrich, A., Suring, K., Robinson, P.N., Stiege, A.C., Lurz, R., Stricker, S., Wanker, E.E. and Mundlos, S. (
Caburet, S., Demarez, A., Moumne, L., Fellous, M., De Baere, E. and Veitia, R.A. (
Innis, J.W., Mortlock, D., Chen, Z., Ludwig, M., Williams, M.E., Williams, T.M., Doyle, C.D., Shao, Z., Glynn, M., Mikulic, D. et al. (
Nasrallah, I.M., Minarcik, J.C. and Golden, J.A. (
Sittler, A., Lurz, R., Lueder, G., Priller, J., Lehrach, H., Hayer-Hartl, M.K., Hartl, F.U. and Wanker, E.E. (
Abu-Baker, A., Messaed, C., Laganiere, J., Gaspar, C., Brais, B. and Rouleau, G.A. (
Brown, L.Y. and Brown, S.A. (
Brown, L., Paraso, M., Arkell, R. and Brown, S. (
Pattyn, A., Morin, X., Cremer, H., Goridis, C. and Brunet, J.F. (
Lavoie, H., Debeane, F., Trinh, Q.D., Turcotte, J.F., Corbeil-Girard, L.P., Dicaire, M.J., Saint-Denis, A., Page, M., Rouleau, G.A. and Brais, B. (
Amiel, J., Trochet, D., Clement-Ziza, M., Munnich, A. and Lyonnet, S. (
Karlin, S., Chen, C., Gentles, A.J. and Cleary, M. (
Goodman, F.R., Mundlos, S., Muragaki, Y., Donnai, D., Giovannucci-Uzielli, M.L., Lapi, E., Majewski, F., McGaughran, J., McKeown, C., Reardon, W. et al. (
Albrecht, A. and Mundlos, S. (
Bao, Y.P., Cook, L.J., O'Donovan, D., Uyama, E. and Rubinsztein, D.C. (
Bachetti, T., Matera, I., Borghini, S., Di Duca, M., Ravazzolo, R. and Ceccherini, I. (
Weese-Mayer, D.E., Berry-Kravis, E.M. and Zhou, L. (
Zoghbi, H.Y. and Orr, H.T. (
Michalik, A. and Van Broeckhoven, C. (
Ravikumar, B., Vacher, C., Berger, Z., Davies, J.E., Luo, S., Oroz, L.G., Scaravilli, F., Easton, D.F., Duden, R., O'Kane, C.J. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}