





Abstract
Gene regulation change has long been recognized as an important mechanism for phenotypic evolution. We used the evolution of yeast aerobic fermentation as a model to explore how gene regulation has evolved and how this process has contributed to phenotypic evolution and adaptation. Most eukaryotes fully oxidize glucose to CO2 and H2O in mitochondria to maximize energy yield, whereas some yeasts, such as Saccharomyces cerevisiae and its relatives, predominantly ferment glucose into ethanol even in the presence of oxygen, a phenomenon known as aerobic fermentation. We examined the genome-wide gene expression levels among 12 different yeasts and found that a group of genes involved in the mitochondrial respiration process showed the largest reduction in gene expression level during the evolution of aerobic fermentation. Our analysis revealed that the downregulation of these genes was significantly associated with massive loss of binding motifs of Cbf1p in the fermentative yeasts. Our experimental assays confirmed the binding of Cbf1p to the predicted motif and the activator role of Cbf1p. In summary, our study laid a foundation to unravel the long-time mystery about the genetic basis of evolution of aerobic fermentation, providing new insights into understanding the role of cis-regulatory changes in phenotypic evolution.
Introduction
Glucose is the primary energy source in nearly all eukaryotic organisms. In the presence of oxygen, most eukaryotes fully oxidize glucose into CO2 and H2O to maximize energy yield, but the budding yeast Saccharomyces cerevisiae and its relatives predominantly degrade glucose into ethanol; this phenomenon is known as aerobic fermentation or the Crabtree effect (De Deken 1966). Aerobic fermentation evolved during the Cretaceous period when fruit-bearing angiosperms began to explode and glucose became abundant in the environment (Bakker 1978; Benner et al. 2002). The emergence of aerobic fermentation enabled these yeasts to rapidly consume surrounding glucose by transforming it into ethanol. Because ethanol can be used as an energy source by budding yeast after glucose depletion but is poisonous to many other species, aerobic fermentation might have provided a selective advantage (Thomson et al. 2005; Piskur et al. 2006). Many kinds of mammalian tumor cells also undergo fermentation, so aerobic fermentation has been used as a diagnostic criterion for tumor cells (Mandelkern and Raines 2002). Furthermore, fermented ethanol is by far the most common type of biofuel produced, accounting for more than 90% of all ethanol production (source: DOE Energy Efficiency and Renewable Energy). A better understanding of the genetic basis of the evolution of aerobic fermentation is of great biological interest and may provide a boost for developing therapeutic interventions and biofuel production.
The fermentation and respiration pathways diverge after glucose is degraded to pyruvate via glycolysis in cytoplasm (Pronk et al. 1996). In respiratory species, such as the dairy yeast Kluyveromyces lactis and the filamentous fungus Ashbya gossypii, pyruvate is fully oxidized to CO2 and H2O in mitochondria through the tricarboxylic acid (TCA) cycle in the presence of oxygen, and the free energy released in this process is stored in ATPs by oxidative phosphorylation. In contrast, in S. cerevisiae and other fermentative yeasts, pyruvate is converted into ethanol and H2O even under the aerobic condition. Genes encoding mitochondrial proteins have been reported to be expressed at high levels in K. lactis. For example, the CYC1 gene, which encodes the electron carrier protein cytochrome c, shows a higher expression level in K. lactis than in S. cerevisiae (Freire-Picos et al. 1994). Additionally, QCR7 and QCR8, which encode subunits VII and VIII of the mitochondrial bc1 complex, are constitutively expressed at high levels in K. lactis (Freire-Picos et al. 1995). In contrast, in S. cerevisiae, transcription of nuclear genes encoding mitochondrial respiration chain proteins is downregulated during growth on glucose (Forsburg and Guarente 1989), whereas genes involved in converting pyruvate to ethanol, such as PDC1 and ADH1, are strongly expressed (Holland MJ and Holland JP 1978; Schmitt et al. 1983). Heterologous DNA arrays also revealed large expression differences in genes related to carbohydrate metabolism and respiratory functions between S. cerevisiae and K. lactis growing in a complete medium (Becerra et al. 2004). Recent studies based on large collections of microarray data further suggested that gene expression divergence was associated with the evolution of aerobic fermentation (Ihmels et al. 2005; Field et al. 2009; Lin and Li 2011a). All these studies indicated that the changes in the regulation of genes involved in glucose metabolism is probably the major factor for their different glucose metabolic styles.
It was proposed that the expression divergence of mitochondrial ribosomal genes was associated with the loss of the “AATTTT” element in their promoters in fermentative yeasts (Ihmels et al. 2005). However, it remains unclear about the genetic basis for the expression divergence of genes involved in major processes of glucose metabolism, such as genes responsible for oxidative phosphorylation, the TCA cycle, or fermentation. Field et al. (2009) found that the transition from nucleosome-depleted promoter to nucleosome-occupied promoter might have contributed to the expression divergence of respiration-related genes and the evolution of aerobic fermentation. However, whether the nucleosome reorganization was the leading or a minor cause for gene expression divergence is subject to debate (Tirosh et al. 2010; Lin and Li 2011a). A recent study also suggested that the elongation of 5′-untranslated region (5′-UTR) of respiration-related genes in fermentative species was linked to the gene expression reprogramming and the evolution of aerobic fermentation (Lin and Li 2012). These studies notwithstanding, the genetic basis of the gene regulation divergence underlying the evolution of yeast aerobic fermentation remains unclear.
Given that the most significant difference between aerobic fermentative and respiratory yeasts is how glucose is metabolized under the aerobic condition (Entian and Barnett 1992; Gancedo 1998), the best way to learn which genes have experienced expression change during the evolution of aerobic fermentation is to compare the gene expression levels between the two types of species. The genome-wide gene expression levels under the same rich medium in 12 completely sequenced yeasts have been recently measured using tiling array approaches, and these yeasts include six aerobic fermentative species and six respiratory species (Tsankov et al. 2010). These data offer us an unprecedented opportunity to identify the genes that have experienced most significant expression change and the genetic variations that have contributed to the gene expression reprogramming underlying the evolution of aerobic fermentation. In this study, we compared the expression differences in 82 transcriptional modules (Ihmels et al. 2002) and found that genes involved in mitochondrial respiration have experienced most significant changes during the evolution of aerobic fermentation. Moreover, our computational and experimental studies on the promoter sequences of these genes suggested that massive loss of cis-regulatory elements was associated with the gene expression divergence event, indicating an important role of cis-regulation change in gene expression divergence and phenotypic evolution.
Materials and Methods
Gene Expression Data Analysis
The genome-wide gene expression data for the 12 hemiascomycete yeasts were obtained from literature (Tsankov et al. 2010). All the 12 species were grown in the same in-house rich medium to mitigate differences in growth rates between species (1.5% yeast extract, 1% peptone, 2% dextrose, 2 g/l SC amino acid mix, mg/l adenine 100, 100 mg/l tryptophan, and 100 mg/l uracil) (Tsankov et al. 2010). The gene expression values were measured during the same midlog phase, using species-specific microarrays. In each species, three to five biological replicates of Cy3-labeled RNA samples were mixed with a reference Cy5-labeled genomic DNA sample and hybridized on two-color Agilent 55- or 60-mer oligoarrays. The expression value for each gene was calculated as the median of the log 2 of the Cy3 to Cy5 ratios across all probes (Tsankov et al. 2010). To compare the gene expression levels between different species, we have normalized the expression value in each data set by subtracting its median values from the expression value for each gene. The orthologous genes for the 12 species were obtained from Fungal Orthogroups Repository (http://www.broad.mit.edu/regev/orthogroups/, supplementary table S1, Supplementary Material online). Based on their glucose metabolism style, the 12 species were assigned to two groups: the fermentative and the respiratory yeast group. The fermentative yeast group included six species: S. cerevisiae, S. paradoxus, S. mikatae, S. bayanus, Candida glabrata, and S. castellii. The six species in the respiratory group were S. kluyveri (Lachancea kluyveri), K. lactis, K. waltii, C. albicans, Debaryomyces hansenii, and Yarrowia lipolytica. The gene lists of the 86 transcriptional modules in S. cerevisiae were retrieved from Ihmels et al. (2002). We only examined the modules with at least 10 genes. The expression level differences between aerobic fermentative species and respiratory species were tested by the two sample Kolmogorov–Smirnov test (K-S test). The K-S statistic D is defined as the maximum absolute difference between the cumulative distribution functions (CDFs) of the two samples. For each transcriptional module, we calculated the D value for the two sample sets, which were defined as the normalized gene expression levels of the six fermentative yeasts and of the six respiratory yeasts. Therefore, the D value of the K-S test quantifies the differences in the gene expression levels of a given transcriptional module between the two types of yeasts.
To determine whether the D value of one module is statistically different from that of the other module, we conducted bootstrap analysis with 1,000 pseudoreplicates on each module. The bootstrap analysis is similar to what is commonly used in phylogenetic analysis. In each transcription module, the total number of gene expression values in fermentative yeasts is m and the total number of gene expression values in respiratory yeasts is n. In each bootstrap replicate, m and n gene expression values were randomly chosen with replacements from the data sets of fermentative and respiratory yeasts, respectively, to constitute two new pseudoreplicate data sets. The D values were calculated between the m and n gene values. One thousand D values were obtained in 1,000 bootstrap replicates. Student’s t-test (one tail) was conducted to determine whether the means of D values are significantly different between two modules.
Promoter Analysis
To study cis-regulatory element changes, we analyzed the promoter sequences for our target gene set. We examined 15 hemiascomycete yeast species. That is, in addition to the 12 species mentioned above, we included three more respiratory yeasts: Zygosaccharomyces rouxii, K. thermotolerans, and A. gossypii. The one-to-one gene orthologs of the additional species were retrieved from the Yeast Gene Order Browser database, in which gene orthology was based on conserved synteny structure (Byrne and Wolfe 2005). Because 99% of known yeast TFBSs are found in the 800 bp upstream of translation start codon (source: TRANSFAC), we retrieved 800-bp sequences upstream of the translation start site for each gene. We used the motif discovery tool Multiple EM for Motif Elicitation (MEME) (Bailey and Elkan 1994) to identify a set of over-represented “seed motifs" for the target gene sets from each species. We ran MEME on the 800-bp promoter sequences with the following parameters: “-mod zoops -revcomp -dna.” Motif width was allowed to range between 7 bp and 10 bp, and both strands of the promoters were searched. For computationally predicted binding sites, occurrences were taken to be those listed in the MEME output, and the “letter-probability-matrix" was used as the position weight matrix.
These motifs were then used to identify other members of the putative regulon by searching in all promoter sequences using the MEME counterpart Find Individual Motif Occurrences (FIMO). As a result, new members were added to each group and some original ones were removed, so that a more specific set of motifs was obtained, again using MEME. This cycle of locating over-represented motifs (with MEME) followed by searching for new genes containing the motifs (with FIMO) was repeated until no new members were found. The resulting “refined motifs" are our candidate regulatory elements. The log-odds matrix of the motif(s) was used to search against the database of known motifs using TOMTOM with default parameters. We input the core motif MEME identified into WebLogo (Schneider and Stephens 1990) to generate logograms of target gene promoters.
Phylogenetic Analysis of CBF1 Genes
The amino acid sequence of S. cerevisiae CBF1 gene (YJR060W) was used as a query to run Blast search against the protein database or genomic database of the 15 hemiascomycete yeasts with E value < 10−10. A complete list of the CBF1 genes from the 15 hemiascomycete species is included in supplementary table S2, Supplementary Material online. Preliminary multiple sequence alignments of all Cbf1 protein sequences under study were carried out using MUSCLE version 3.52 with default parameter settings (Edgar 2004). These alignments were manually inspected and corrected using GeneDoc version 2.6.002 (Nicholas 1997). We constructed phylogenetic trees using both the maximum likelihood (ML) and neighbor-joining (NJ) methods. We used ProtTest 1.4 (Abascal et al. 2005) to identify the most appropriate model and parameters (JTT + I + G + F model) for the Cbf1 protein alignment. These model and parameters were used in our ML tree reconstruction using Phyml 2.4 (Guindon and Gascuel 2003) with 100 bootstrap replicates. The proportion of invariable sites and the α parameter of β-law distribution were optimized according to the data. NJ trees were constructed using MEGA 4.0 (Tamura et al. 2007). The confidence of internal branches of a NJ tree was assessed with 1,000 bootstrap pseudoreplicates using “pairwise deletion option” of amino acid sequences with the Poisson correction and the Jones–Taylor–Thornton model.
Yeast Strains Manipulation and Motif Mutation Construction
The yeast strain, K. lactis KB101 (MATa ade trpl ura3gal80-1), is a gift from Dr Zhenglong Gu’s laboratory (table 1). The predicted binding motif CACGTG in the promoters of candidate genes was deleted using the in vivo site-directed mutagenesis method (Storici et al. 2001). The construction was done by polymerase chain reaction (PCR)-based mutagenesis involving two sequential steps (Gray et al. 2004). The promoter fragments were amplified from the K. lactis KB101 gnomic DNA and cloned into pGEM-T (easy) Vector (Promega), followed by QuikChange Site-Directed Mutagenesis Kit (Stratagene) to delete the Cbf1 binding sites (CACGTG). The plasmid DNA from the positive clones was purified by the Qiagen Miniprep kit and sequenced by ABI 3700 automated sequencer (Applied Biosystems Inc.). The amplification primers and deletion mutant primers were listed in supplementary table S3, Supplementary Material online.
Plasmid and Strain Information
| Name | Information (Phenotype, Genotype, etc.) | Reference |
|---|---|---|
| Plasmid/strain | ||
| pET32a | pET-32 series plasmid | Kit |
| pET32a-KlCBF1 | pET32a plasmid with CBF1 gene of K. lactis | This study |
| KB101 | wild-type strain of K. lactis (MATa ade trpl ura3 gal80-1) | ATCC 96265 |
| Promoter disruption of KB101 | ||
| Klhap4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 hap4pr::KlURA3-kanMX4 | This study |
| Klcox4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 cox4pr::KlURA3-kanMX4 | This study |
| Klqcr7pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 qcr7pr::KlURA3-kanMX4 | This study |
| Cbf1 binding motifs (CACGTG) deletion of KB101 | ||
| Klhap4pr-Δ6 | MATa ade trpl ura3 gal80-1 hap4pr-Δ6 | This study |
| Klcox4pr-Δ6 | MATa ade trpl ura3 gal80-1 cox4pr-Δ6 | This study |
| Klqcr7pr-Δ6 | MATa ade trpl ura3 gal80-1 qcr7pr-Δ6 | This study |
| Name | Information (Phenotype, Genotype, etc.) | Reference |
|---|---|---|
| Plasmid/strain | ||
| pET32a | pET-32 series plasmid | Kit |
| pET32a-KlCBF1 | pET32a plasmid with CBF1 gene of K. lactis | This study |
| KB101 | wild-type strain of K. lactis (MATa ade trpl ura3 gal80-1) | ATCC 96265 |
| Promoter disruption of KB101 | ||
| Klhap4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 hap4pr::KlURA3-kanMX4 | This study |
| Klcox4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 cox4pr::KlURA3-kanMX4 | This study |
| Klqcr7pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 qcr7pr::KlURA3-kanMX4 | This study |
| Cbf1 binding motifs (CACGTG) deletion of KB101 | ||
| Klhap4pr-Δ6 | MATa ade trpl ura3 gal80-1 hap4pr-Δ6 | This study |
| Klcox4pr-Δ6 | MATa ade trpl ura3 gal80-1 cox4pr-Δ6 | This study |
| Klqcr7pr-Δ6 | MATa ade trpl ura3 gal80-1 qcr7pr-Δ6 | This study |
Plasmid and Strain Information
| Name | Information (Phenotype, Genotype, etc.) | Reference |
|---|---|---|
| Plasmid/strain | ||
| pET32a | pET-32 series plasmid | Kit |
| pET32a-KlCBF1 | pET32a plasmid with CBF1 gene of K. lactis | This study |
| KB101 | wild-type strain of K. lactis (MATa ade trpl ura3 gal80-1) | ATCC 96265 |
| Promoter disruption of KB101 | ||
| Klhap4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 hap4pr::KlURA3-kanMX4 | This study |
| Klcox4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 cox4pr::KlURA3-kanMX4 | This study |
| Klqcr7pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 qcr7pr::KlURA3-kanMX4 | This study |
| Cbf1 binding motifs (CACGTG) deletion of KB101 | ||
| Klhap4pr-Δ6 | MATa ade trpl ura3 gal80-1 hap4pr-Δ6 | This study |
| Klcox4pr-Δ6 | MATa ade trpl ura3 gal80-1 cox4pr-Δ6 | This study |
| Klqcr7pr-Δ6 | MATa ade trpl ura3 gal80-1 qcr7pr-Δ6 | This study |
| Name | Information (Phenotype, Genotype, etc.) | Reference |
|---|---|---|
| Plasmid/strain | ||
| pET32a | pET-32 series plasmid | Kit |
| pET32a-KlCBF1 | pET32a plasmid with CBF1 gene of K. lactis | This study |
| KB101 | wild-type strain of K. lactis (MATa ade trpl ura3 gal80-1) | ATCC 96265 |
| Promoter disruption of KB101 | ||
| Klhap4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 hap4pr::KlURA3-kanMX4 | This study |
| Klcox4pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 cox4pr::KlURA3-kanMX4 | This study |
| Klqcr7pr::KlURA3-kanMX4 | MATa ade trpl ura3 gal80-1 qcr7pr::KlURA3-kanMX4 | This study |
| Cbf1 binding motifs (CACGTG) deletion of KB101 | ||
| Klhap4pr-Δ6 | MATa ade trpl ura3 gal80-1 hap4pr-Δ6 | This study |
| Klcox4pr-Δ6 | MATa ade trpl ura3 gal80-1 cox4pr-Δ6 | This study |
| Klqcr7pr-Δ6 | MATa ade trpl ura3 gal80-1 qcr7pr-Δ6 | This study |
Cloning of the KlCBF1 Gene and Cbf1 Protein Purification
The coding region of CBF1 was amplified from the K. lactis KB101 strain genomic DNA and cloned into the pET32 plasmid (named as pET32a-KlCBF1) with XhoI (New England Biolabs) and EcoRI (New England Biolabs) sites. The pET32a-KlCBF1 was then transformed into Escherichia coli NovaBlue (DE3) competent cell (Novagen) for protein induction. The overnight culture was diluted 1:100 in 500 ml of Luria broth and was grown to an OD600 of 0.6. IPTG was added to the culture to the final concentration of 1 mM, and the induced culture was grown for additional 4 h. Cells were then centrifuged to harvest and resuspended in 25 ml binding buffer (20 mM Tris-HCl, 300 mM NaCl, pH 7.5), with protease inhibitor (protease Inhibitor cocktail set I, Calbiochem) to prevent protein degradation. The resuspended cells were disrupted by Microfluidizer, and the supernatants were harvested by centrifuging at 4,000 × g for 60 min in 4 °C. The Cbf1 proteins were purified by the affinity chromatography (Ni sepharose 6 Fast Flow, GEHealthcare). The Ni2+ column was first balanced with binding buffer before sample loading and then sequentially washed with binding buffer containing 0 mM, 30 mM, 40 mM, and 70 mM imidazole. The Cbf1 proteins were collected in elusion buffer (20 mM Tris-HCl, 300 mM NaCl, 90 mM imidazole, pH 7.5) and then dialyzed by Viva spin 20 (GE Healthcare) with binding buffer.
Electrophoretic Mobility Shift Assay
To validate the Cbf1 binding sites predicted, the electrophoretic mobility shift assay (EMSA) was carried out according to the manufacturer’s procedure (Invitrogen; EMSA Kit, E33075). Primers for the DNA-substrate amplicons were listed in supplementary table S3, Supplementary Material online. Around 110-bp length promoters with/without Cbf1 binding motifs (CACGTG) were amplified from previous constructed plasmids. The purified amplicons (120 ng) were incubated with serial amounts of purified Cbf1 proteins (120 ng up to 1 µg) in binding buffer (50 mM Tris-HCl, 250 mM KCl, 0.1 mM dithiothreitol and 0.1 mM ethylenediaminetetraacetic acid, pH 7.4) for assay optimization (data not shown). The optimized DNA:protein ratio is the purified amplicons (120 ng) incubated with 300 ng purified Cbf1 proteins. After incubation in 30 °C for 30 min, the samples were electrophoresed on a 6% nondenaturing polyacrylamide gel at 400 mA, 250 V, 4 °C in TBE buffer. DNA and protein were stained using the SYBR Green and SYPRO Ruby dye (Invitrogen; EMSA Kit) and detected by 300 nm UV transillumination.
Swapping of Cbf1 Binding Site Deletion Promoter in the KB101 Strain
We select 10 genes of Module 5 from K. lactis (KLLA0A06754g, KLLA0C00825g, KLLA0C10384g, KLLA0D05082g, KLLA0D12782g, KLLA0D18095g, KLLA0E23639g, KLLA0F03641g, KLLA0E05654g, and KLLA0F25960g) for testing the function of Cbf1 binding motif and successfully obtained three mutant strains (KLLA0D05082g, KLLA0C-00825g, and KLLA0F13838g). The motif of interest was first replaced by an KlURA3 + kanMX4 cassette with about 45 bp flanking homologous regions to the motif of interest at both ends (Storici et al. 2003). The transformation is conducted by electroporation with Bio-Rad Gene Pulser and Pulse Controller devices (Sanchez et al. 1993), and transformants were selected as Ura+ and G418R colonies. Electroporation modified from Meilhoc et al. (1990) and Sanchez et al. (1993) was performed in a 0.2-cm cuvette; the final volume was always between 50 µl and 55 µl, the voltage was 1,000 V, the capacitance was 25 µF, and the resistance was 400. The transformants were selected as Ura+ and G418R colonies. The insertion of KlURA3 + kanMX4 cassette at the targeted site was confirmed by diagnostic PCR and sequencing. The inserted cassette was further replaced by a second transformation with the appropriate fragment into the URA3-inserted strain. The transformants were selected by 5-FOA counter selection. Only the strains that carried the desired sequence will survive and form colonies on the media with 5-FOA (1 μg/ml). The constructs at the targeted site were confirmed by diagnostic PCR. We also sequenced the entire promoter region to confirm no other mutations within the promoter. The strains used in this study are listed in table 1. We preserved at least four individual transformants for each strain.
Growth Pattern Analysis
The yeast cells were precultivated at 30 °C in YPAD medium for 24 h. Overnight yeast cultures were used to prepare the starting cultures with OD600 = 0.1 and were grown in YPAD media at 30 °C with 200 rpm shaking. Aliquots (0.5–1.0 ml) were taken from the cultures at 2-h intervals for analysis of cell OD600, glucose consumption, and ethanol production. The glucose consumption of each sampling time point was measured by a glucose assay kit (SIGMA). The ethanol concentration was determined by an ethanol assay kit (R-Biopharm, South Marshall, MI).
Expression Level Analysis of Respiration-Related Genes by Quantitative Real-Time PCR
To monitor the effect on the expression of deletion of a Cbf1 motif in a gene promoter, the yeast cells were harvested and the total RNA was extracted by the EPICENTRE MasterPure Yeast RNA Purification Kit following the manufacturer’s instructions. An aliquot of 5 μg total RNA from each sample was used for cDNA synthesis (the final volume was 100 μl), and the reverse transcription was carried out with oligo-dT primers following the manufacturer’s instructions of the Super-script II kit (Invitrogen). Real-time PCR analyses were performed in 25 μl reaction volumes containing 1× Power SYBR Green PCR Master Mix (Foster City, CA), 2 μl cDNA, 1 μl each of gene-specific forward and reverse primers (5 μM) with 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primers were designed by using the Primer Express software from Applied Biosystems (Foster City, CA). The expression levels of target genes in each strain were measured by eight replicates (four biological replicates from four individual transformants of each strain were used for RNA isolation, and two technical replicates were conducted for each biological replicate). The relative expression level of each gene was normalized to that of the Act1 gene (ΔCt) and quantified with the ΔΔCt relative quantification method, and the relative expression ratio was determined following ABI’s guideline. The amplification efficiency of each primer pair was tested by using 2-fold serial dilutions of the templates. The Q-PCR relative expression ratio was determined by the ΔΔCt value using the formula, the relative expression ratio of mutated/wild type = 2[−ΔΔCt], as suggested by Applied Biosystems, and the amplification efficiency of the target gene and the reference gene was approximately equal.
Results
Expression Evolution of Genes Involved in Mitochondrial Respiration
We obtained the genome-wide gene expression values in six aerobic fermentative yeasts and six respiratory yeasts from Tsankov et al. (2010). Because the mean/median values are substantially different among the 12 species (the mean/median values ranging from −2.83/−2.80 in S. cerevisiae to −1.98/−1.93 in D. hansenii), we normalized the gene expression values in each series by subtracting the median value across all genes from the original values, so that the expression values in each series is centered at 0 and are comparable among species (see Materials and Methods, supplementary fig. S1, Supplementary Material online). Because these expression values were measured by microarray, which lacked individual measurement accuracy, we used previously defined 86 “transcriptional modules” (supplementary table S4, Supplementary Material online) (Ihmels et al. 2002) as units to compare the expression level differences between the two types of yeasts. The “transcriptional modules” were inferred based on a large collections of expression data in S. cerevisiae (Ihmels et al. 2002). The genes in each transcriptional module are believed to be coregulated and to share common cis-regulatory elements (Ihmels et al. 2002). To reduce the potential bias caused by a small sample size, we excluded those transcriptional modules with less than 10 genes and selected 82 transcriptional modules for subsequent analyses.
Because the expression values in over 90% modules do not have a normal distribution (the Shapiro–Wilk normality test), we used the nonparametric two-sample K-S test to estimate the differences in the normalized expression levels of each transcriptional module between the two types of yeasts. In a K-S test, the D value, ranging from 0 to 1, denotes the maximum absolute difference between the two cumulative distributions. Specifically, a larger D value indicates a greater difference between the two distributions. Among the 82 transcriptional modules, the D values range from 0.043 to 0.56, and 63% of the D values are less than 0.2 (supplementary table S5, Supplementary Material online). Module 46 has the smallest D value (D = 0.0428, P = 0.15), and, according to the Gene Ontology (GO) annotation, most genes in this module are involved in the protein catabolic process (supplementary table S5, Supplementary Material online), indicating that the expression of genes involved in the protein catabolic process have been maintained at very a stable level during evolution of hemiascomycete yeasts (fig. 1A). Ihmels et al. (2005) suggested that mitochondrial ribosomal protein (RP) genes have experienced transcriptional modifications based on the analysis of large collections of gene expression data in S. cerevisiae and the respiratory yeast C. albicans. The MPR genes are grouped in Module 12, and it has indeed a large D value, 0.4327, P < 1 × 10−16 (fig. 1B). However, it is the sixth largest D value among the 82 modules (supplementary table S5, Supplementary Material online). In contrast, Module 5 has the largest D value, 0.56 (P < 1 × 10−16, fig. 1C). To determine the statistical significance of the D difference between Module 5 and Module 12, we conducted bootstrap analysis with 1,000 pseudoreplicates (see Materials and Methods), and our results revealed that the D value of Module 5 is significant higher than that of Module 12 (Student's t-test, P = 0, supplementary fig. S2, Supplementary Material online). We also calculated the D values for each transcriptional module using un-normalized expression values and obtained the same results (supplementary table S5, Supplementary Material online).
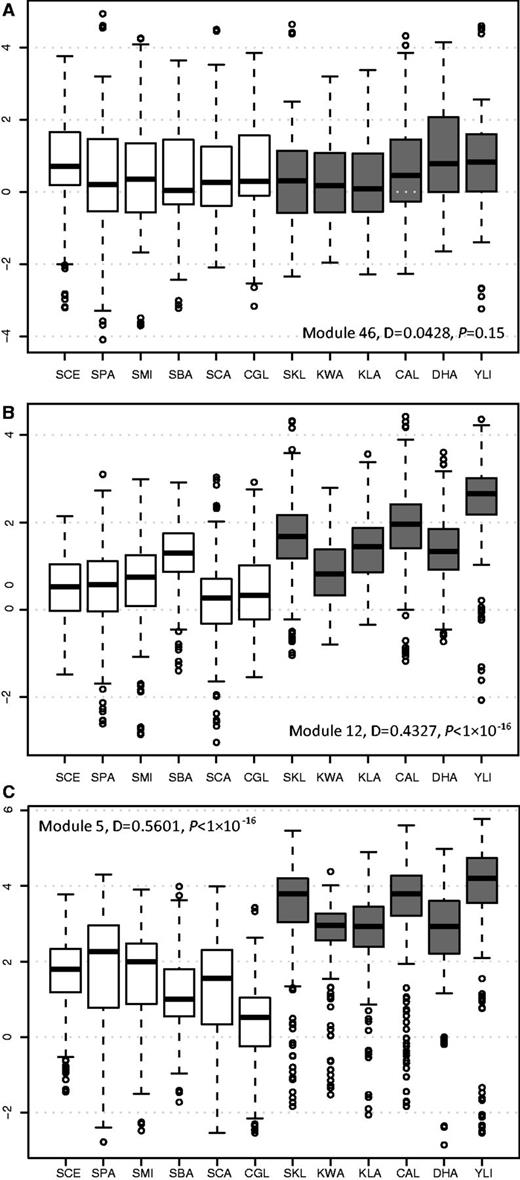
Comparison of gene expression levels of transcriptional modules between aerobic fermentative species and respiratory species. Box plots representing normalized gene expression values of Module 46 (A), Module 12 (B), and Module 5 (C) in the six respiratory yeasts (in gray) and six aerobic fermentative yeasts (in white). The species names are abbreviated as follows: Saccharomyces cerevisiae, SCE; S. paradoxus, SPA; S. mikatae, SMI; S. bayanus, SBA; Candida glabrata, CGL; S. castellii, SCA; S. kluyveri, SKL; Kluyveromyces lactis, KLA; K. waltii, KWA; C. albicans, CAL; Debaryomyces hansenii, DHA; and Yarrowia lipolytica, YLI.
As shown in figure 1C, the expression levels of Module 5 genes are consistently lower in the six aerobic fermentative yeasts than in the six respiratory species growing on glucose-rich media, indicating that the expression of these genes has been significantly downregulated in fermentative species during the evolution of aerobic fermentation. Module 5 genes are mainly involved in mitochondrial energy generation and phosphorylation oxidation and are regulated by the HAP complex (Ihmels et al. 2002). In addition to Module 5, two other modules (Modules 9 and 72) with a D value more than 0.5 are also mainly involved in energy production (supplementary table S5, Supplementary Material online). The common ancestor of aerobic fermentative yeasts in the Hemiascomycete lineage had experienced a whole-genome duplication (WGD) event, and retained gene pairs generated by the WGD are enriched in genes involved in sugar metabolism (Wolfe and Shields 1997; Kellis et al. 2004; Conant and Wolfe 2007). Theoretically, if both copies of a WGD gene pair have been retained, the expression level of either duplicated gene can be reduced as it would be compensated by the other copy. To test whether the presence of WGD pairs contributed to the gene expression reduction, we removed the members with WGD genes in Modules 55, 9, and 12 and recalculated their R values. The R values for the Modules 5, 9, and 72 without WGD genes are 0.55, 0.50, and 0.58, respectively. These values are basically the same as the modules with WGD genes (0.56, 0.50, and 0.52). Therefore, it is not likely that the reduction of expression levels in these modules is due to the presence of WGD pairs. As the main difference between aerobic fermentative and respiratory yeasts is how glucose is metabolized in rich media in the presence of oxygen, the expression data obtained under this condition in the 12 species provide a more accurate measurement than those studies based on other conditions (Ihmels et al. 2005). Therefore, the Module 5 genes, which are involved in mitochondrial energy production, have experienced the most significant transcriptional downregulation and had most likely contributed to the evolution of aerobic fermentation. This hypothesis is also supported by other studies based on individual genes or genome-wide gene expression analysis (Mulder et al. 1995a, 1995b; Field et al. 2009).
Enrichment of HAP Binding Motifs in Module 5 Promoters in Both Types of Yeasts
The regulatory divergence of Module 5 genes during the evolution of aerobic fermentation could be due to changes in trans-acting factors or in cis-regulatory elements. To answer this question, we first compared the over-represented sequence motifs in the promoters of Module 5 genes between the two types of yeasts. The promoter sequences (from the start codon of an open reading frame to 800 bp upstream) of Module 5 genes were retrieved from the 15 hemiascomycete yeasts, including six aerobic fermentative species and nine respiratory species (see Materials and Methods). We used MEME to infer over-represented motifs in the promoters of Module 5 genes in each species. In S. cerevisiae, a motif with a core consensus sequence of ATTGG is present in 75.6% (37/49) promoters of Module 5 genes (fig. 2A and B, supplementary table S6, Supplementary Material online). The motif matrix was submitted to TOMTOM for comparison against a database of known yeast motifs (Gupta et al. 2007), and it is most similar to that of the HAP complex in S. cerevisiae (P value = 2.78 × 10−6). Previous ChIP-chip and computational studies have shown that the Module 5 genes were controlled by the HAP complex in S. cerevisiae (Ihmels et al. 2002; Harbison et al. 2004; MacIsaac et al. 2006). Therefore, the predicted over-represented motif by MEME is consistent with the previous studies. The HAP complex is composed of a DNA-binding heteromer Hap2p/Hap3p/Hap5p and a regulated activation subunit Hap4p (Buschlen et al. 2003). The complex is known to regulate the transcription of genes involved in respiratory metabolism in response to carbon source (Buschlen et al. 2003; Harbison et al. 2004). Deletion of the consensus HAP complex binding sequence led to significantly reduced expression of QCR8, a Module 5 gene, and caused severely impaired growth on the respiration-only medium (de Winde and Grivell 1992).
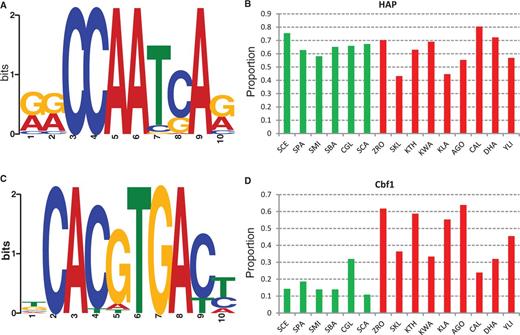
Presence of the HAP complex and Cbf1 binding motifs in the promoters of Module 5 genes. (A) The sequence logo of the HAP binding motifs in Saccharomyces cerevisiae. (B) The proportion of Module 5 gene promoters with the presence of the HAP binding motif in aerobic fermentative yeasts (in green) and respiratory yeasts (in red). (C) The sequence logo of the Cbf1 binding motifs in Kluyveromyces lactis. (D) The proportion of Module 5 gene promoters with the presence of the Cbf1 binding motif in aerobic fermentative yeasts (in green) and respiratory yeasts (in red). The species names are abbreviated as in figure 1: Zygosaccharomyces rouxii, ZRO; Kluyveromyces thermotolerans, KTH; and Ashbya gossypii, AGO.
The HAP complex motif is highly enriched in the promoters of Module 5 genes not only in S. cerevisiae but also in the other five fermentative yeasts (fig. 2B). Interestingly, this motif is also frequently found in the promoters of Module 5 genes in the nine respiratory species examined (fig. 2B). In terms of the proportion of genes with the HAP complex motifs in the promoters, no significant difference was detected between the two types of yeasts (P = 0.27, Fisher’s exact test, two tails). It has been shown that the distributions of binding motifs tend to be highly enriched in a specific region of promoters instead of a random distribution (Lin et al. 2010; Wu 2011). As shown in supplementary figure S3, Supplementary Material online, the distribution of predicted HAP complex binding motifs forms a sharp peak in the promoters in 11 of the 12 species examined, supporting the functionality of these predicted motifs. These observations reveal that the binding sequences of the HAP complex and its target genes have been conserved during the evolution of aerobic fermentation. In addition, the HAP complex members have been functionally conserved between S. cerevisiae and respiratory yeast K. lactis (Mulder, Scholten, de Boer, et al. 1994; McNabb et al. 1995; Nguyen et al. 1995; Bourgarel et al. 1999). Therefore, with respect to the HAP complex, no significant changes in the trans-acting factor or cis-regulatory elements were associated with the expression divergence of Module 5 genes during the evolution of aerobic fermentation.
Scarcity of Cbf1 Binding Motifs in the Promoters of Module 5 Genes in Fermentative Yeasts
In the respiratory yeasts, a motif with the core consensus sequence of CACGTGA is prevalent in the promoters of Module 5 genes (fig. 2C, supplementary table S6, Supplementary Material online). From a TOMTOM motif search, this motif is highly similar to that of Cbf1p (Centromere binding factor 1) in S. cerevisiae (P value = 6.84 ×10−8). Thus, we called this predicted motif the Cbf1 motif. The Cbf1 motif is highly enriched in the promoters of Module 5 genes in respiratory yeasts. For example, 63.8% of Module 5 genes in A. gossypii contain at least one Cbf1 motif in their promoters, and it is 55.3% in K. lactis and 61.7% in the salt-tolerant yeast Z. rouxii (fig. 2D). In comparison, a much lower frequency of the Cbf1 motif is found in the aerobic fermentation species: 14.3% in S. cerevisiae, 10.8% in S. bayanus, and 10.8% in S. castellii (fig. 2D). Therefore, the presence of the Cbf1 motif in the promoters of Module 5 genes is significantly more frequent in respiratory yeasts than in fermentative yeasts (P < 0.0001, Fisher’s exact test, two tails, fig. 2D).
To evaluate the sensitivity and accuracy of the predicted binding targets of Cbf1p by MEME, we compared our results with two previous ChIP-chip studies in S. cerevisiae (Harbison et al. 2004; Lavoie et al. 2010). The promoters of eight and six Module 5 genes were inferred to be bound by Cbf1p in the two ChIP-chip analyses (supplementary table S7, Supplementary Material online). Among them, four genes were listed as Cbf1 targets in both studies. Our analysis showed that a Cbf1 motif is present in seven Module 5 genes, five of which overlap with the results by Harbison et al. (2004), even higher than the overlap between the two experimental studies. The binding locations of Cbf1p in the human pathogen C. albicans were also inferred in one of the two studies (Lavoie et al. 2010). We predicted 11 Cbf1 target genes in C. albicans, which included 66.7% (6/7) predicted by ChIP-chip study (Lavoie et al. 2010) (supplementary table S8, Supplementary Material online). These observations support the high sensitivity and accuracy of our prediction of Cbf1 targets in K. lactis (or among different yeast species).
Evolution of the CBF1 Gene Family and Its Target Genes in Hemiascomycete Yeasts
The S. cerevisiae CBF1 gene encodes a transcription factor containing a basic helix-loop-helix (bHLH) protein domain (Cai and Davis 1990). The homolog of S. cerevisiae CBF1 has been characterized in K. lactis and the Cbf1 proteins from the two species are functionally interchangeable (Mulder et al. 1994). In most yeasts, the orthologous gene of CBF1 has not been identified or functionally characterized. To provide a better understanding of the evolution of CBF1 and its role in the evolution of aerobic fermentation, we searched for the homologs of CBF1 in the 15 species under study and reconstructed their evolutionary history (see Materials and Methods, and supplementary table S2, Supplementary Material online). The bHLH domain is highly conserved among these CBF1 homologs, though the remaining parts are much more divergent. A single CBF1 orthologous gene was identified in each of the 15 species examined (fig. 3). Because the common ancestor of the six aerobic fermentation species had experienced a WGD (Wolfe and Shields 1997; Kellis et al. 2004), theoretically two CBF1 genes are expected in each of these fermentative species. Therefore, one copy of the CBF1 duplicate genes produced by WGD has been lost in all the six fermentative yeasts. The topology of the NJ tree of CBF1 genes is consistent with their species tree except for C. glabrata (Wapinski et al. 2007), probably due to an accelerated evolutionary rate at the whole genome level after its speciation (Jiang et al. 2008). Thus, as a transcription factor, the copy number and its core domain of Cbf1p has been highly conserved during the evolution of aerobic fermentation in the hemiascomycete yeasts.
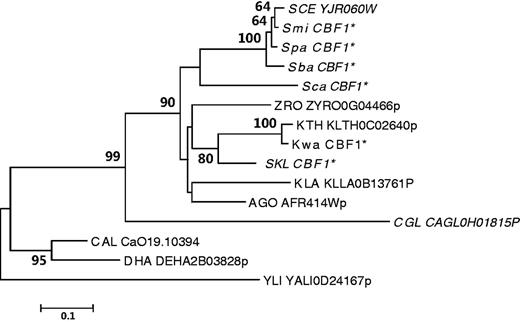
The evolutionary history of CBF1 genes in hemiascomycete yeasts. One CBF1 gene is found in each of the 15 yeasts examined. The phylogenetic tree was constructed based on the sequences of the highly conserved HLH domain. The NJ and ML consensus trees were topologically congruent. Only the NJ tree is shown, and the NJ tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The species names are abbreviated as in figure 2. *Predicted CBF1 coding sequences in this study. Fermentative species are shown in italics.
Because of the conservation of CBF1 in yeasts, the significant downregulation of Module 5 genes in fermentative yeasts was more likely achieved through loss of Cbf1 binding motifs. To obtain a better understanding of how the Cbf1 motif in the promoters of Module 5 genes became rare during the evolution of aerobic fermentation, we traced the binding pattern of the Cbf1 motif across the 15 species. Because the presence of the Cbf1 motif in a promoter was predicted by in silico analysis, incorporation of the phylogenetic conservation information can reduce the possibility of false positive. We thus defined the presence of the Cbf1 motif in an orthologous group of Module 5 genes in each type of yeasts if the motif is found in at least three species. Under these criteria, the Cbf1 motif is present in 75% (37/49) of orthologous groups in respiratory yeasts but in only 12% (6/49) of orthologous groups in fermentative yeasts (supplementary table S9, Supplementary Material online). Because the six aerobic fermentative species descended from a common ancestor after its divergence from Z. rouxi, it appears that most of the Cbf1 motifs in the promoters of Module 5 genes had been lost at the early stage of aerobic fermentation evolution, according to the presence and absence patterns of the Cbf1 motif (supplementary table S9, Supplementary Material online). In some orthologous groups of Module 5 genes, the Cbf1 binding motif is absent in all fermentative yeasts but is present in all respiratory yeasts. One good example is the ATP4 (YPL078C) gene, which encodes a subunit of the mitochondrial ATP synthase (Velours et al. 1988). Genes with this pattern of cis-regulatory changes can be used as good candidates to experimentally validate whether loss of the Cbf1 binding motif has contributed to their different expression patterns.
EMSA Analysis of Cbf1 Protein Binding to Predicted Cbf1 Binding Sites
To determine whether the Cbf1 protein binds to our predicted motif in the promoters of Module 5 genes, we conducted an EMSA using wild-type promoter sequences containing the predicted Cbf1 binding site(s) and mutant promoters with deletion of the 6-bp Cbf1 core binding sites (CACGTG). We selected the respiratory yeast K. lactis as our experimental system. The coding sequence of K. lactis CBF1 was transformed into E. coli, and the K. lactis Cbf1 protein was purified from the E. coli culture (see Materials and Methods). The 110-bp promoters with/without the Cbf1 binding site were amplified from four K. lactis Module 5 genes: KLLA0D05082g (KlCOX4), KLLA0C00825g (KlQCR7), KLLA0F13838g (KlHAP4), and KLLA0E23639g (KlATP4). As observed in EMSA blots, the Cbf1 protein and all four wild-type promoters (with the predicted Cbf1 binding site) clearly formed one complex (lane 6 in fig. 4A–D). In contrast, none of the mutant promoter probes form a complex with Cbf1p (lanes 7, fig. 4A–D). As a control, the equivalent amounts of nonspecific protein (BSA) do not form a complex with any of the wild-type and mutant promoter probes (lanes 8 and 9, fig. 4A–D). To exclude the possibility that the promoter structure might be affected by motif deletion, we also replaced the 6-bp Cbf1 core binding sites with a randomized sequence. We successfully obtained three promoter probes with scrambled Cbf1 binding sites (KlCOX4, KlATP4, and KlHAP4). As shown in supplementary figure S4, Supplementary Material online, there was no binding complex between Cbf1p and any of the three promoter probes with scrambled sites. Furthermore, we also conducted EMSAs to determine whether Cbf1p could bind to the promoters of S. cerevisiae QCR7 and ATP4 that lack a Cbf1 motif according to our prediction and previous ChIP-chip assays. None of these promoter probes form a complex with Cbf1p (supplementary fig. S4, Supplementary Material online). These results strongly support the binding of Cbf1p to the predicted Cbf1 binding sites in the promoters of K. lactis Module 5 genes.
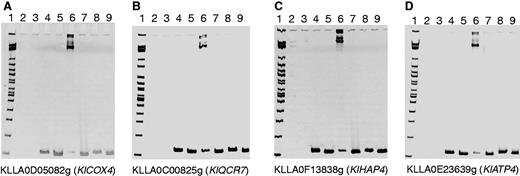
The binding of the Cbf1 protein to the predicted Cbf1 binding sites in Kluyveromyces lactis was validated by EMSA. (A) KLLA0D05082g (KlCOX4), (B) KLLA0C00825g (KlQCR7), (C) KLLA0F13838g (KlHAP4), and (D) KLLA0E23639g (KlATP4). Loading samples (from left to right) are lane 1: 2-log DNA marker; lane 2: BSA protein; lane 3: Cbf1 protein; lane 4: wild-type promoter; lane 5: Cbf1 binding site deleted promoter; lane 6: wild-type promoter + Cbf1 protein; lane 7: Cbf1 binding site deleted promoter + Cbf1 protein; lane 8: wild-type promoter + BSA protein; and lane 9: Cbf1 binding site deleted promoter + BSA protein. Binding complex bands were only formed between the Cbf1 protein and wild-type promoters with Cbf1 binding sites (lanes 6).
Deletion of the Cbf1 Binding Motif
The CBF1 gene is not essential to S. cerevisiae, but its deletion is lethal in K. lactis (Mulder et al. 1994). Therefore, the transcriptional control of Module 5 genes by Cbf1p in K. lactis cannot be examined by inactivation of KlCBF1. A feasible way is to delete the predicted Cbf1 binding sites from the promoter of a Module 5 gene and to evaluate the effects on its expression levels. We have successfully generated mutant strains with deletion of predicted Cbf1 binding sites in the promoters of three Module 5 genes: KLLA0D05082g (KlCOX4), KLLA0C00825g (KlQCR7), and KLLA0F13838g (KlHAP4). These mutants with deletion of 6 bp of Cbf1 core motif are named as Klcox4pr-Δ6, Klacr7pr-Δ6, and Klhap4pr-Δ6, respectively. Two of the three mutants, Klcox4pr-Δ6 and Klhap4pr-Δ6, showed a significant expression reduction. The expression levels in these two deletion strains are only 73% and 78% of that of the wild-type strain by quantitative real-time PCR (fig. 5). Intriguingly, a previous study by Mulder, Scholten, van Roon, et al. (1994) has demonstrated that the deletion of the Cbf1 binding site in the KlQCR7 promoter severely lowered the mRNA expression during growth on both glucose and ethanol/glycerol. However, no significant difference in the expression level of KlQCR7 was observed between mutant and wild-type in our study. In the study by Mulder et al. (1994) a 35-bp region surrounding the Cbf1 motif was deleted, but we only deleted the 6-bp core region, which might partly explained the discrepancy. This difference notwithstanding, the results suggest that the Cbf1 binding motifs in the promoter regions of respiration-related genes are important for the activation of expression, supporting the view of an activator role of Cbf1p for respiration-related genes.
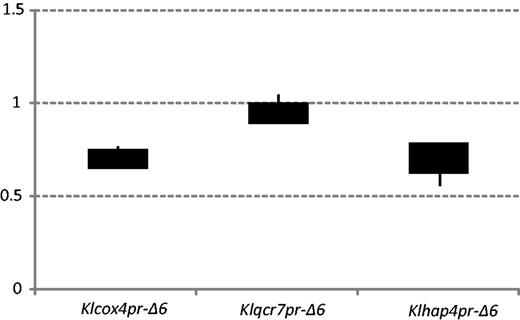
Deletion of Cbf1 binding motifs reduced the expression levels of Module 5 genes. Two of the three mutants, KlCox4pr-Δ6 and Klhap4pr-Δ6, showed a significant expression level reduction (only 73% and 78% relative to the wild-type strain), according to the quantitative real-time PCR data.
Discussion
Rewiring transcriptional circuitry is now generally recognized as an important mechanism for evolution of organismal complexity (Tsong et al. 2003, 2006; Carroll et al. 2005; Wray 2007; Tuch et al. 2008; Perez and Groisman 2009). The prevalence of this strategy is becoming evident as more cases are examined. In this study, we showed that the genes of Module 5, which are mainly involved in the mitochondrial respiration process, have experienced most significant reduction in expression level in the fermentative yeasts during the evolution of aerobic fermentation. The prevalence of Cbf1 motifs in the promoters of Module 5 genes in respiratory species suggested that Cbf1p is likely a general regulator for these genes. The binding of Cbf1p to the predicted binding sites and the activator role of Cbf1p were confirmed by our experimental assays. We also observed massive loss of Cbf1 binding sites in the promoters of Module 5 genes in fermentative yeasts, which could explain why these genes have experienced significant downregulation. These results provide a new insight into the evolution of yeast aerobic fermentation.
In addition, our study explained the change of essentiality of the CBF1 gene during the evolution of fermentative yeasts. In S. cerevisiae, the CBF1 gene is involved in both chromosome segregation and transcription activation (Mellor et al. 1990). Disruption of CBF1 in S. cerevisiae causes some minor effects including slow growth, partial chromosome loss, and methionine auxotrophy but is not lethal (Mellor et al. 1990). By transferring KlCBF1 into S. cerevisiae CBF1 deletion strain, it rescued the mutant strain (Mulder et al. 1994). Reversely, S. cerevisiae CBF1 rescued the K lactis CBF1 deletion strain (Mulder et al. 1994). Therefore, the CBF1 orthologs are functionally interchangeable between S. cerevisiae and K. lactis, suggesting that, as a transcription factor, the function of Cbf1p and its binding sites has been well conserved between respiratory and fermentative yeasts. However, despite the functional conservation of CBF1, a K. lactis strain with inactivation of the CBF1 gene is not viable, indicating that CBF1 is essential for K. lactis (Mulder et al. 1994). Therefore, the essentiality of CBF1 in cell survival has been changed during the evolution of aerobic fermentation, but it was not clear about the cause of lethality. Unlike fermentative species S. cerevisiae, the respiratory yeasts, including K. lactis, predominantly rely on a respiratory metabolism of glucose (Bianchi et al. 1996). In addition, loss of mitochondrial function is lethal for K. lactis because K. lactis cells are not able to tolerate the absence of electron-proton transport pumping and ATP synthesis components of oxidative phosphorylation (Clark-Walker and Chen 2001). In this study, we showed that Cbf1p plays a general activator role for respiration-related genes in respiratory yeasts. In fact, the activator role of Cbf1p has been noticed on some individual respiration-related genes in several previous studies. For example, the K. lactis QCR7 promoter contains a Cbf1 consensus binding site, which is absent from S. cerevisiae QCR7. Deletion of this site severely lowers the mRNA expression during growth on both glucose and ethanol/glycerol (Mulder, Scholten, van Roon, et al. 1994). In addition, K. lactis QCR8 contains the binding site for Cbf1p in their promoter regions. Mutation of Cbf1 binding sites slightly lowers the expression of QCR8, demonstrating that Cbf1p plays a role in transcriptional activation of the QCR8 gene in K. lactis (Mulder et al. 1995b). In this study, through genome-wide gene expression and promoter sequence analyses, we found that Cbf1p, as a general transcriptional regulator, plays a much more important role in respiratory yeasts than previously recognized. The inactivation of CBF1 in respiratory yeasts could lead to dysfunction of mitochondria, explaining why CBF1 is essential for respiratory species.
One might ask how Cbf1p activates the expression of respiration-related genes in respiratory yeasts. In S. cerevisiae, Cbf1p forms a homodimer, which may function as an activator recruiter and a chromatin remodeler of some MET genes, which are involved in methionine biosynthesis (Cai and Davis 1990; Mellor et al. 1990). The function of CBF1 in respiratory yeasts has not been well characterized. In view of the functional conservation of CBF1 between fermentative and respiratory yeasts (Mulder et al. 1994), the functional characterizations of CBF1 in S. cerevisiae might provide insights into its role in respiratory yeasts. The regulatory role of Cbf1p in methionine biosynthesis appears to be conserved between S. cerevisiae and C. albicans according to ChIP-chip assays (Lavoie et al. 2010). In addition to these amino acid synthesis genes, C. albicans Cbf1p also binds to the upstream region of other targets such as RP genes and glycolytic genes (Hogues et al. 2008; Lavoie et al. 2010). It has been shown that Cbf1p is involved directly or indirectly in establishing a nucleosome-free gap within some promoter regions which increases accessibility (Kent et al. 2004). Field et al. (2009) found that the respiration-related genes in respiratory yeasts have nucleosome-depleted type promoters, but they are nucleosome-occupied in fermentative species, so they proposed that changes in promoter chromatin organization change linked to evolution of aerobic fermentation. The connection between Cbf1 binding motif and promoter chromatin organization was confirmed by a recent genome-wide nucleosome survey in multiple species (Tsankov et al. 2010). It was found that Cbf1p acts as a general regulatory factor (GRF) that contributes to the establishment of a nucleosome-free region in respiratory yeasts, because promoters with the Cbf1 binding motif are strongly nucleosome depleted (Tsankov et al. 2010). They also found that the GRF role of Cbf1p was taken over by Reb1p after the WGD event with the presence of Cbf1 motifs. The authors compared the nucleosome occupancy level over Cbf1 binding sites between non-WGD and post-WGD species, and they found that the Cbf1 binding sequence is nucleosome depleted in vivo in most non-WGD species but not in most post-WGD species (Tsankov et al. 2010). An alternative hypothesis is that the reduction of expression level of Module 5 genes is due to changes in the intrinsic nucleosome occupancy pattern in their promoters. To test this hypothesis, we also analyzed the intrinsic nucleosome occupancy data, which were calculated using the promoter sequences for the 12 yeast species (Field et al. 2008). We calculated the occupancy over the promoter nucleosome-depleted region (PNDR, which was defined as average nucleosome occupancy of most depleted 100-bp region, within 200 bp upstream of the translation start site) for each Module 5 gene in the 12 species. We then did the K-S test of the PNDR between the two types of yeasts, and we obtained D = 0.0979 (P value = 0.1086), which is much smaller than the D value of gene expression. Therefore, it is not likely that the expression difference of Module 5 genes between the two types of yeasts is due to their intrinsic nucleosome occupancy difference. In addition, to evaluate whether the gene expression evolution could be attributed to the combined effect of Cbf1 motif loss and changes in intrinsic nucleosome occupancy, we compared the average PNDRs of the genes with Cbf1 motifs and genes without Cbf1 motifs in each of the 12 species. As shown in supplementary table S10, Supplementary Material online, none of the 12 species shows more depleted intrinsic nucleosome occupancy in genes with Cbf1 motif than in genes without Cbf1 motif, suggesting that the presence/absence of Cbf1 motif makes no difference in the level of intrinsic nucleosome occupancy in any of the species examined.
Therefore, our study suggests that the changes in the promoter chromatin organization of respiration-related genes was more likely due to loss of the Cbf1 motif, rather than changes of the in vivo nucleosome occupancy of these motifs. Based on our studies and previous knowledge about CBF1, it is reasonable to propose that: 1) the presence of the Cbf1 motif in the promoters of respiration-related genes in fermentative species may create nucleosome-deplete regions, facilitating the expression of these genes; 2) Cbf1 binding sites have been lost in the promoters of these genes in fermentative species and their promoters switch to nucleosome-occupied that could interfere or prevent the binding of transcriptional activators to their binding sites; and 3) the expression levels of respiration-related genes in fermentative species are significant downregulated, so that the assimilation of glucose is switched to the fermentation pathway.
Furthermore, our study also suggested that the roles of HAP complex have been changed during evolution of aerobic fermentation. In S. cerevisiae, the HAP complex is known to regulate transcriptional expression of genes involved in respiratory metabolism in response to carbon. For example, deletion of the consensus HAP complex binding sequence could lead to significantly reduced expression of S. cerevisiae QCR8 gene and caused severely impaired growth on respiration-only medium (de Winde and Grivell 1992). The HAP complex members are functionally conserved between the two types of yeasts (Mulder, Scholten, de Boer, et al. 1994; Bourgarel et al. 1999). Although our data showed that the HAP complex binding motifs are also enriched in the Module 5 genes in respiratory yeasts (fig. 2B), it appears that HAP complex does not have significant impacts on expression of Module 5 genes in respiratory yeasts for the following reasons: 1) unlike in S. cerevisiae, disruption of the functional homologs of HAP2 or HAP3 in K. lactis had no significant effect on the growth on respiratory substrates (Mulder, Scholten, de Boer, et al. 1994); 2) elimination of putative HAP binding sites in the CYC1 promoter revealed that they are not associated with functional glucose repression/glycerol derepression (Freire-Picos et al. 1995). These observations indicated that despite the presence of HAP complex binding sites in promoter regions in respiratory yeasts, the HAP complex is not likely a major regulator for these respiration-related genes. Therefore, the general regulator of respiration-related genes has been switched from Cbf1p in respiratory yeasts to HAP complex in fermentative yeasts. If the HAP complex does not play an important role of respiration-related genes in respiratory yeasts, we would expect gradual loss of the HAP complex motif from these genes due to accumulation of mutations. It is thus intriguing that the HAP motifs are still retained in the promoter of these genes. The function of the HAP complex in respiratory yeasts has not been thoroughly examined under various conditions. However, in S. cerevisiae, the HAP complex activates respiration-related genes only under the condition that glucose is getting depleted (the diauxic shift). We therefore speculate that the HAP complex might have a function in the regulation of respiration-related gene under stress conditions in respiratory yeasts.
It should be mentioned that the evolution of aerobic fermentation was a complicated process, involving many genetic changes (Ihmels et al. 2005; Thomson et al. 2005; Jiang et al. 2008; Field et al. 2009; Lin and Li 2011b) and that these changes would not have occurred in a short time period. Thus, some extant yeasts show very strong aerobic fermentation, whereas some others show only very weak aerobic fermentation. For example, S. kluyveri can produce some ethanol after a period of aerobic growth (Moller et al. 2002) and was thought to be a fermentative yeast (Moller et al. 2002). However, considering that the ability of converting glucose into ethanol under aerobic condition is much lower in S. kluyveri than in S. cerevisiae (Merico et al. 2007) and that the codon usage pattern of nuclear-encoded mitochondrial genes in S. kluyveri is similar to other respiratory yeasts, Jiang et al. (2008) classified S. kluyveri as a respiratory yeast. In this study, we showed that the expression levels of respiration-related genes are much higher in S. kluyveri than in all fermentative species (fig. 1C) and that the Cbf1 motif is more prevalent in S. kluyveri than in the fermentative species (fig. 2D). Therefore, it is reasonable to include S. kluyveri in the group of respiratory yeasts.
With all the above observations, it is reasonable to postulate that loss of the Cbf1 binding motifs in the fermentative species was responsible for the gene expression divergence of respiration-related genes. Our study and two previous studies (Ihmels et al. 2005; Field et al. 2009) all suggest that reprogramming of genes involved in the respiration pathway was associated with the evolution of aerobic fermentation. Another question is whether the transcriptional reprogramming of respiration-related genes was sufficient for the emergence of aerobic fermentation. As shown here, deletion of the Cbf1 motif reduced the expression level of respiration-related genes (fig. 5). However, increase in ethanol production was not observed, suggesting that reduced expression of a single respiration-related gene is not sufficient to force K. lactis to switch to the fermentation pathway. Because Cbf1 motifs have massively lost in respiration-related genes in fermentative species, the switch from respiration to fermentation in K. lactis might rely on deletions of Cbf1 motifs in many of these genes. On the other hand, activation of the fermentation pathway under the aerobic condition may also require changes either in the expression level or in the enzymatic activity of genes involved in converting pyruvate into ethanol. Because only two biochemical reactions are needed to convert pyruvate into ethanol, mainly two enzymes, PDC1 and ADH1, are involved in this process in S. cerevisiae (Pronk et al. 1996). It will be of great interest to study what changes in these fermentation-relate genes were associated with the evolution of aerobic fermentation.
Acknowledgments
The authors thank Dr Zhenglong Gu for providing the yeast strain, K. lactis KB101. They thank Dr Jure Piskur for valuable comments and I-Li Wang and Ya-Wun Cai for experimental assistance. This study was supported by James Watson Professorship, University of Chicago, and by Academia Sinica, Taiwan.
Literature Cited
Author notes
Associate editor: Yoshihito Niimura

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}