





Abstract
Bacteriophage flux can cause the majority of genetic diversity in free-living bacteria. This tenet of bacterial genome evolution generally does not extend to obligate intracellular bacteria owing to their reduced contact with other microbes and a predominance of gene deletion over gene transfer. However, recent studies suggest intracellular coinfections in the same host can facilitate exchange of mobile elements between obligate intracellular bacteria—a means by which these bacteria can partially mitigate the reductive forces of the intracellular lifestyle. To test whether bacteriophages transfer as single genes or larger regions between coinfections, we sequenced the genome of the obligate intracellular Wolbachia strain wVitB from the parasitic wasp Nasonia vitripennis and compared it against the prophage sequences of the divergent wVitA coinfection. We applied, for the first time, a targeted sequence capture array to specifically trap the symbiont's DNA from a heterogeneous mixture of eukaryotic, bacterial, and viral DNA. The tiled array successfully captured the genome with 98.3% efficiency. Examination of the genome sequence revealed the largest transfer of bacteriophage and flanking genes (52.2 kb) to date between two obligate intracellular coinfections. The mobile element transfer occurred in the recent evolutionary past based on the 99.9% average nucleotide identity of the phage sequences between the two strains. In addition to discovering an evolutionary recent and large-scale horizontal phage transfer between coinfecting obligate intracellular bacteria, we demonstrate that “targeted genome capture” can enrich target DNA to alleviate the problem of isolating symbiotic microbes that are difficult to culture or purify from the conglomerate of organisms inside eukaryotes.
Introduction
Horizontal gene transfer (HGT) permits the acquisition of novel DNA sequences and adaptations through “the non-genealogical transmission of genetic material from one organism to another” (Goldenfeld and Woese 2007). In free-living bacteria, the rate of occurrence of HGT is very high and nearly universal. In contrast, obligate intracellular bacteria have few opportunities to acquire new genes due to being reproductively confined to a eukaryotic host cell. As a result of their restrained lifestyle, genome evolution of obligate intracellular bacteria is dominated by gene loss rather than gene insertion (Ochman et al. 2000; Mira et al. 2001, 2002; Andersson et al. 2002; Bordenstein and Reznikoff 2005). For instance, the pervasive amount of HGT in free-living bacteria contrasts with the extreme stability and downsizing observed for the small endosymbiont genomes of aphids (Shigenobu et al. 2000; Tamas et al. 2002; van Ham et al. 2003; Degnan et al. 2005), ants (Gil et al. 2003; Degnan et al. 2005), and flies (Akman et al. 2002), in which no rearrangements or inflow of genetic material have occurred over millions of years.
HGT can be driven by a genetic vehicle that transfers genes from one cell to another, typically in the form of bacteriophages, plasmids, or transposons (Frost et al. 2005). In contrast to the highly reduced genomes of host-restricted species that tend to manifest striking degrees of stability and gene synteny, obligate intracellular bacteria that host–switch often harbor mobile elements and the genetic toolkit to acquire new DNA (Bordenstein and Reznikoff 2005; Newton and Bordenstein 2010). Examples of extrachromosomal mobile elements in ancient, horizontally transmitted obligate intracellular bacteria include Rickettsial plasmids (Baldridge et al. 2010) and an integrative conjugative element (ICE; Blanc et al. 2007), Phytoplasma plasmids (Oshima et al. 2001) and Chlamydiaceae phages (Hsia et al. 2000; Read et al. 2000).
In free-living bacteria, bacteriophages are significant to the structure of bacterial ecosystems and adaptive evolution. Phage-mediated HGT (Ochman et al. 2000; Bushman 2002; Canchaya et al. 2003) accounts for the majority of intraspecific genome diversification among environmental bacteria and human pathogens (Ohnishi et al. 2001; Banks et al. 2002; Van Sluys et al. 2003). In contrast, the transfer of a full bacteriophage genome between the chromosomes of obligate intracellular bacteria in the same host has not been demonstrated. Of the obligate intracellular bacteria, Wolbachia is a likely target for large-scale HGT of mobile elements because up to 21% of the genome can be dedicated to mobile DNA, specifically transposons and the Wolbachia bacteriophage family, known as WO (Wu et al. 2004; Klasson et al. 2008; Klasson et al. 2009; Kent and Bordenstein 2010). Wolbachia are a genus of cytoplasmically transmitted α-proteobacteria and are among the most common bacterial infections in arthropods (Werren and Windsor 2000; Hilgenboecker et al. 2008). Recent interest in these bacteria has emerged because of their ability to 1) induce the major inflammation responses associated with filarial diseases (Bandi et al. 2001; Nutman 2001; Taylor et al. 2001), 2) reduce vector capacity of mosquitoes and decrease populations of vectorial insects (Zabalou et al. 2004; Evans et al. 2009; McMeniman et al. 2009; Turley et al. 2009), 3) transfer genes to the genomes of their host animals (Kondo et al. 2002; Hotopp et al. 2007; Nikoh et al. 2008), 4) modify host sex ratios and sex determination mechanisms for their own advantage (Werren et al. 2008), and 5) influence arthropod speciation (Bordenstein et al. 2001; Bordenstein 2003; Jaenike et al. 2006; Koukou et al. 2006; Miller et al. 2010).
Although mobile elements have been identified in Wolbachia and other obligate intracellular bacteria, there is sparse evidence of recent, large-scale HGTs outside of insertion sequences (Cordaux et al. 2008). In Wolbachia, minor capsid gene sequences from bacteriophage WO are identical between coinfections in five different insect species (Masui et al. 2001; Bordenstein and Wernegreen 2004; Chafee et al. 2010), suggesting that transfer between Wolbachia coinfections in the same host is very common. However, evidence of phage transfer beyond the ∼350 bp of this single gene has not previously been shown and the question remains if the whole phage or just single genes can transfer between Wolbachia in the same host. The implication of large mobile element transfers between obligate intracellular coinfections is significant to bacterial genome evolution. For some intracellular infections, especially those that are not confined to a single host or restricted to bacteriocytes (Bordenstein and Reznikoff 2005; Kent and Bordenstein 2010), the exposure and ability to take up novel DNA sequences creates an opportunity for these bacteria to partially mitigate the reductive nature of an intracellular lifestyle.
To determine if a complete bacteriophage WO transfer occurred between coinfections, we sequenced the genome of the B-Wolbachia infection of the parasitic wasp Nasonia vitripennis (wVitB) and compared it with the prophage sequences of the A-Wolbachia (wVitA) that coinfects the same host. These two ancient infections diverged ∼60 million years ago (Werren et al. 1995). In this study, the wVitA and wVitB infections were previously segregated into separate N. vitripennis strains to avoid DNA contamination. The wVitB and wVitA coinfections are likely candidates for phage transfer because 1) wVitB and wVitA share an identical minor capsid gene sequence (Bordenstein and Wernegreen 2004); 2) active phage has been visualized inside wVitA bacterial cells by EM (Bordenstein et al. 2006); 3) these phage-containing Wolbachia showed structural defects including a collapsed inner membrane, degraded DNA, and lysis coupled with phage release (Bordenstein et al. 2006); and 4) the WOVitA1 phage is the only phage in the wVitA genome that encodes a full set of structural genes, including head, tail, and baseplate genes (fig. 1), indicating it is able to form active phage particles. If transfer of the complete phage occurs between wVitA and wVitB, then it is likely that the several other examples of phage gene transfer between Wolbachia coinfections extend to the entire phage genome. Below, we provide evidence of a full bacteriophage transfer between these two coinfecting Wolbachia.
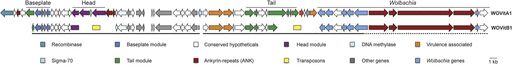
Gene presence and synteny between prophages WOVitB and WOVitA1. WO genes are syntenous and homologous between prophages WOVitB and WOVitA1. WOVitB genes contained within a single contig are denoted with an underline. WOVitB reads mapped to repetitive flanking regions of WOVitA1 are denoted with a dashed line. The other genes category includes genes annotated as a holliday junction resolvasome, a leucine-rich repeat protein, DNA polymerase III, an ATPase, and a methyl accepting chemotaxis protein.
Studying the genomes of fastidious bacteria that cannot be cultured outside of their eukaryotic host cells often presents practical problems owing to a difficulty in sequencing a genome that might be a small minority of the total DNA in a host. In addition, the amplification methods that have been used to overcome small DNA yields can introduce chimeras and mutations in the resulting sequence. Below we describe the first use of a high-density tiled oligonucleotide array for efficient enrichment and sequencing of this endosymbiont's DNA. Based on the results, we propose that targeted genome capture can be widely used to isolate the small genomes of symbiotic members of eukaryotic hosts.
Materials and Methods
Sequence Capture Array
The targeted sequence capture array of 385,000 tiled probes was designed by Roche NimbleGen from whole genome sequences and shotgun sequences from the Wolbachia infections of Culex pipiens Pel (NC_010981), Drosophila melanogaster yw (NC_002978), Drosophila simulans Riverside (NC_012416), Brugia malayi (NC_006833), Drosophila ananassae (NZ_AAGB00000000), Muscidifurax uniraptor (NZ_ACFP00000000), and Drosophila willistoni (NZ_AAQP00000000) and prophage sequences from the Wolbachia infections of Cadra cautella (AB161975.2, AB478515.1, AB478516.1) and Ephestia kuehniella (AB036666.1). Sequences also present in the insect genomes of Aedes aegypti (AAGE02000000), Apis mellifera (NC_007070–NC_007085), D. melanogaster (NC_004353, NC_004354, NS_000188, NT_033777, NT_033778, NT_03379, NT_037436), Drosophila sechellia (AAKO010000000), Tribolium castaneum (NC_007416–NC_007425), N. vitripennis (AAZX01000000), Nasonia giraulti (ADAO00000000), and Nasonia longicornis (ADAP00000000) were excluded from the array.
Genomic DNA Extraction and Sequencing
Genomic DNA was extracted from ∼200 abdomens of the B-Wolbachia infected N. vitripennis 4.9 using the QIAGEN DNeasy Kit insect tissue protocol. DNA was treated with 100 mg/ml RNaseA and incubated at room temperature for 2 min. The resulting DNA was quantified on a Nanodrop (ThermoScientific). Twenty micrograms of DNA were provided to the Genomic Services Lab of the HudsonAlpha Institute for Biotechnology for array hybridization, library construction, and sequencing using the Illumina GenomeAnalyzer IIx via 36 nt paired-end sequencing by synthesis reactions. DNA eluted from the array was size selected to isolate approximately 400-bp fragments. Two paired-end sequencing lanes yielded 5,046,411 reads.
Assembly
Contigs were assembled using the Velvet assembly package (Zerbino and Birney 2008). Various kmer lengths (21, 23, 25, and 27) and coverage cutoff values (15 or auto) were used to determine the best assembly parameters. Constant parameters included an insert length of 400 and minimum contig length of 100. To maximize contig assembly and genome coverage, contigs generated by Velvet were further assembled using the Geneious Custom Assembly algorithm with the parameters: word length 30, index word length 12, gap size 2, maximum gaps per read 20, maximum mismatches 3, and maximum ambiguity 16 (Drummond et al. 2009). The Geneious assembly of multiple Velvet contigs that were constructed under different parameters would identify any Velvet contigs that were improperly assembled. The initial assembly yielded 698 contigs with a maximum length of 22,329 bp, a mean of 2,267.6 bp, a G + C content of 34%, and a total length of 1,454,337 bp. All contigs were manually reviewed by analyzing overlap regions to ensure that no misassembly had occurred. Contigs assembled using Velvet can have errors on the ends that correspond to low coverage regions at the terminals (Gibbons et al. 2009). Due to these errors, some Velvet-generated contigs containing identical genomic regions were not assembled by Geneious to form a single consensus sequence. These duplicated genome regions were removed from the assembly and contigs that overlapped within a wPip reading frame were merged. Prior to manual scaffolding, each contig was searched using MegaBlast (Altschul et al. 1990) and BlastN against the NCBI nr database to determine the origin of the sequence. Contigs that contained insect or non-Wolbachia bacterial DNA were removed. The final assembly consisted of 426 contigs, with a maximum length of 38,705 bp, a mean of 2,645.5 bp, and a G + C content of 34%. The contigs were concatenated to determine the length of the genome sequence, which was 1,107,643 bp. Open reading frames (ORFs) were identified using the Geneious ORF finder algorithm for bacterial genomes. The wVitB assembly encodes 1,122 ORFs greater than 150 bp in length. 5,046,411 paired-end sequence reads, each 36 bp long, and differing in up to 3 positions from the reference sequence were simulated from the genome of the Wolbachia endosymbiont of Culex quinquefasciatus Pel (Klasson et al. 2008). The average library sequence fragment length was set to 400 bp. Simulation analysis was performed using the paired-end simulation script from the rmap package, version 2.0.5 (Smith et al. 2009). This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AERW00000000. The version described in this paper is the first version, AERW01000000. Sequences for phages WOVitA and WOVitB have been deposited under accession numbers HQ906662–HQ906666.
Comparison of the wVitB and wVitA Phage Regions
Coding sequences for phage WOVitA1, WOVitA2, and WOVitA4 and the 5′- and 3′-flanking regions from the genome project for wVitA (M.C., I.L.G.N., S. Richards, B.N.K., S.R.B, and J. Werren) were identified using Blast. WOVitA1 ORFs (supplementary table S1Supplementary Material online) used in this analysis were assigned using the Geneious ORF finder algorithm as described for wVitB. These coding sequences were searched against all wVitB contigs using MegaBlast to identify phage regions and determine percent homology between homologs. Raw reads were also assembled to WOVitA1 and its flanking regions to identify contiguous segments of WOVitB sequence. Oligonucleotide primers were designed so that polymerase chain reaction (PCR) could be used to 1) assemble the phage contigs into a full-length prophage where possible, 2) determine if phage genes that were not full-length when compared with their homologs in WOVitA1 were truly missing these sequences in the wVitB genome, and 3) determine if phage genes found in WOVitA1 but not in WOVitB were absent in the wVitB sequence (supplementary tables S1 and Supplementary Data, Supplementary Material online). PCR was performed using the Phusion High-Fidelity enzyme kit (New England Biolabs) with 25 ng of genomic DNA from N. vitripennis 4.9 (WOVitB), N. giraulti IntG[12.1]F9(13) (WOVitA1), or no DNA (data not shown). Maximum likelihood phylogenetic analysis was performed using the PhyML (Guindon and Gascuel 2003) plugin in Geneious (GTR substitution model, 100 bootstrap data sets, 4 substitution rate categories, estimated transition/transversion ratios, proportion of invariable sites, and gamma distribution parameters, and optimized tree topology, branch length, and substitution rate).
Comparison of wVitB to Wolbachia
To further explore the efficiency of capture by determining the presence or absence of genes pulled down and sequenced by the array, Blast was performed to compare each gene from the four previously sequenced whole Wolbachia genomes (B-Wolbachia wPip of C. pipiens; Klasson et al. 2008, A-Wolbachia infections wMel of D. melanogaster; Wu et al. 2004, wRi from D. simulans Riverside; Klasson et al. 2009, and D-Wolbachia wBm from Brugia maylai; Foster et al. 2005) with wVitB. Two parameters were used: 2) an E value less than 0.00001 and 2) a minimum of 30% of homologous sequence present between the query and the hit.
The core genome was identified by an expanded form of the Reciprocal Best Hit (RBH) algorithm (Tatusov et al. 1997; Salichos L and Rokas A, unpublished data) between the genomes of wPip, wMel, wRi, and wBm. Genes that occurred in all four sequences were considered core genes. To identify the core genes absent from wVitB, the list of absent genes from each Wolbachia genome described above was compared with the list of core genes. Average nucleotide identity between wPip and wVitB was calculated as the average of the total shared genome when wVitB ORFs were aligned to wPip. WO phage regions in wPip were defined as WPa_0239–0272, WPa_0297–0319, WPa_0320–0342, WPa_0411–0455, and WPa_1294–1340.
Results
Phage Genome Transfer
We array captured and sequenced the genome of the Wolbachia wVitB, which was previously segregated from its natural wVitA coinfection in N. vitripennis (Perrot-Minnot et al. 1996). Wolbachia wVitB is a low titer infection at 0.01 Wolbachia gene copies/1 Nasonia gene copy (Bordenstein et al. 2006), which extrapolates to approximately 25,000 Nasonia base pairs per one Wolbachia base pair.
Array capture and subsequent Illumina sequencing indicated a single bacteriophage WO haplotype, WOVitB, in wVitB. This single haplotype was compared with the three WOVitA prophages in the A-Wolbachia to determine if a complete phage WO transfer occurred between wVitA and wVitB (fig. 1). Seven contigs from the wVitB assembly were originally syntenous to sequences from WOVitA1. Subsequent mapping of raw Illumina reads to the WOVitA1 genome, PCR, and Sanger sequencing were employed to complete the WOVitB phage sequence in two contigs. Sixty-nine percent (35/51) of WOVitA1 genes were present in WOVitB (supplementary table S1, Supplementary Material online). Of these 35 genes, five WOVitB genes are not full coding length. The remaining genes in phage WOVitA1 have been deleted from WOVitB. In two cases, a 5 kb deletion in the head region and a 4.5 kb deletion in the tail region, genes have been replaced by transposon insertions of the ISSod13 family (fig. 1, supplementary table S1, Supplementary Material online). Inability to PCR-amplify genes that were absent in WOVitB, but present in WOVitA1, confirmed their loss from the wVitB genome.
The 35 prophage coding sequences and partial genes of the WOVitB genome are, on average, 99.9% similar to genes in phage WOVitA1 (range 98.7–100%) (fig. 2). This nucleotide identity is significantly higher than the 83.3% average nucleotide identity of the remaining phage genes in the wVitB genome (range 68.4–89.8%) (supplementary table S2, Supplementary Material online) that are not related to the transferred WOVitA1 (fig. 2, Mann–Whitney U, two-tailed, P < 0.0001). These genes are homologous to ankyrin-repeat, conserved hypothetical, or site-specific recombinases in other WO haplotypes. Also, the complete genomes of WOVitB and WOVitA1 phages have significantly higher nucleotide identity in comparison to the average 89% identity (range 85.1–93.4%) between eight previously sequenced Wolbachia protein-coding genes from wVitA and wVitB (fig. 2, MWU, two-tailed, P < 0.0001). The close relationship between WOVitA1 and WOVitB was further confirmed by a phylogenetic analysis of phage genes showing divergent phage gene topologies from the Wolbachia protein-coding genes (supplementary fig. S1, Supplementary Material online). Finally, the evolutionarily recent transfer of the phage between these coinfecting Wolbachia receives support if phage genetic divergence is lower than that of its two bacterial hosts despite the more rapid rate of nucleotide evolution in phage genes (Chafee et al. 2010). A Fisher's exact test indicates that the nucleotide identity in genes across the phage genome between WOVitB and WOVitA1 (VrlC, tail sheath, DNA methylase, Baseplate J) is significantly lower than the “expected” divergence based on the 11.8% nucleotide difference between the wVitA and the wVitB MLST genes (VrlC—0/1,197 bp vs. 141/1,197, tail sheath—0/1,176 vs. 139/1,176, DNA methylase—0/1,215 vs. 143/1,215, Baseplate J—0/786 vs. 93/786; P < 0.0001 for all genes).
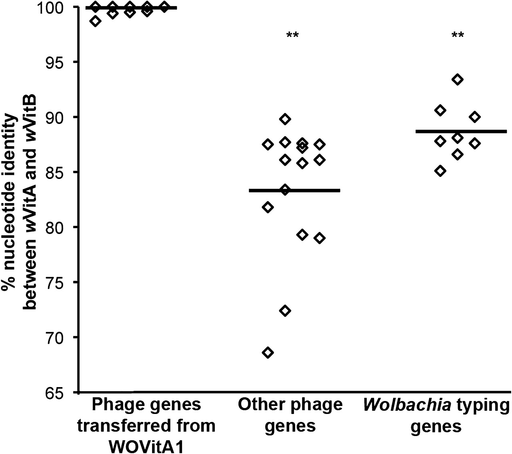
Percent nucleotide identity between prophage genes encoded on WOVitB, WOVitA1, and other phages. Nucleotide identity between transferred prophage genes is significantly elevated between the genomes of the coinfections. Percent nucleotide identity is compared between phage genes transferred between wVitA with wVitB, other phage genes present in wVitB that are not homologous to genes in WOVitA1, and wVitA and wVitB previously sequenced protein-coding genes. A list of phage gene homologs present in wVitB that are not found in WOVitA1 is provided in supplementary table S2 (Supplementary Material online).
The truncated tail region of WOVitB is adjacent to the same location as the tail region of WOVitA1, which contains helix-turn-helix transcriptional regulators, the gene encoding the DNA repair protein RadC, the gene mutL, a gene encoding a heat shock protein, conserved hypothetical genes, and ankyrin-repeat proteins (fig. 1). Many of these genes are involved in DNA binding or protein–protein interactions that could regulate the lytic or lysogenic state of the phage (Kolkhof et al. 1992; Tourasse and Kolsto 2008). Like the defined phage region, the homology between wVitA and wVitB is also nearly identical in these genes, indicating that they are part of the transferred phage. Interestingly, WO phages from strains wPip (Klasson et al. 2008), wMel (Wu et al. 2004), wCauB (Tanaka et al. 2009), and wRi (Klasson et al. 2009) are also integrated near some of these same genes, signifying that integration of some WO types may be preferential for this region or this region is part of the WO phage. Taken together, these findings indicate a recent whole genome transfer event followed by some erosion due to transposon insertions and gene loss.
Efficiency of Targeted Genome Capture
Illumina sequencing of the captured DNA yielded 5.04 million paired-end reads that were assembled and manually edited. The final assembly was comprised of 426 contigs (≥100 bp) totaling 1,107,643 bp. Only 1.7% of the assembled nucleotides before manual editing had non-Wolbachia sequence matches (fig. 3). Fifty-four contigs were from the host N. vitripennis (supplementary table S3, Supplementary Material online) and 28 contigs were from other bacteria such as Proteus, Arsenophonus, Providencia, and Xenorhabdus (supplementary table S4, Supplementary Material online), which are known infections of Nasonia (Werren et al. 2010). Forty-four contigs spanning 0.6% of the assembly had no significant match in GenBank, yet their average GC content was 36%, similar to the typical GC content in Wolbachia genomes. These contigs likely represent novel sequences in the wVitB genome that were array captured by either adjacent regions to hybridized DNA fragments or sequential hybridization of captured DNA acting as template for flanking DNA not printed on the array.
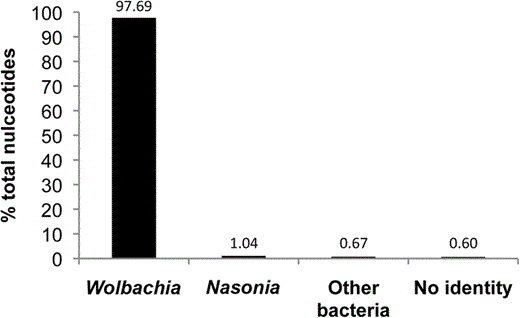
Enrichment efficiency of the Wolbachia capture array. Bar graph denotes % of the nucleotides out of the total assembled contigs (≥100 bp) that match to the four taxonomic categories. The no identity category (0.6%) that had no significant homology to any sequence in the NCBI nr database was left in the final assembly because it had a low GC content that is diagnostic of the Wolbachia genome. The Nasonia and other bacteria categories represent 1.04% and 0.67% of the total assembled DNA, respectively.
The above analyses indicate that the array capture was highly specific to the bacterial endosymbiont. Next we tested the completeness of the captured genome. We note five observations:
1) First, the assembly has 34 tRNAs, one 16S, 23S, and 5S rRNA gene, and a 34% GC content that typify all Wolbachia genomes thus far (Wu et al. 2004; Klasson et al. 2008; Klasson et al. 2009).
2) Second, of the 756 core genes present in four fully sequenced Wolbachia genomes, only 13 were absent in the captured wVitB, including a drug resistance transporter, genes of a phage HK97 family remnant not related to phage WO, and hypothetical proteins (supplementary table S5, Supplementary Material online). Absent genes from the core genome were compared with the array sequence to ensure that these sequences were available on the array for pull-down, and all 13 sequences were present.
3) Third, 81.6% of genes from the B-Wolbachia wPip of Culex quinquefasciatus Pel, the closest sequenced relative to wVitB, were present in wVitB (supplementary fig. S2, Supplementary Material online). The remaining wPip genes that did not have homologs in the wVitB sequence (supplementary fig. S3, Supplementary Material online), included 55 phage genes from the five phage types present in wPip, 56 transposases, and 30 ankyrin-repeat proteins, all of which have been shown to be highly variable between Wolbachia sequences (Wu et al. 2004; Iturbe-Ormaetxe et al. 2005; Klasson et al. 2008). A high number (76) of the additional absent genes are of unknown function. A full list of the wPip genes lacking orthologs in the pulled-down wVitB sequence is provided in supplementary table S6 (Supplementary Material online).
4) Fourth, eight previously sequenced protein-coding genes (Baldo et al. 2006; Paraskevopoulos et al. 2006) from wVitB are identical to the same genes in the captured wVitB sequence. In addition, the average nucleotide identity between the wVitB genes captured and wPip is 95.7% and the lowest nucleotide similarity between the wVitB sequence and the array sequence is 78.5% (316 bp of a transposase sequence), indicating the array methodology is sufficient to pull-down sequences of closely related strains, species, or even genera.
5) Finally, the distributions of the length and coverage of the assembled contigs are consistent with the distributions obtained by assembling the same number and type of paired-end Illumina reads simulated from the genome of the Wolbachia endosymbiont of Culex quinquefasciatus Pel (Klasson et al. 2008) (fig. 4). However, a sharp peak was observed in the simulated data at 15× coverage, whereas the array coverage data was more evenly distributed, likely due to the efficiency of the array capture. These data suggest that the capture is comprehensive, and that any genome fragments missing are due to either their true absence from the genomes or a failure to assemble them, rather than failure to capture them. The 1.1 Mb genome size is mostly consistent with previously sequenced Wolbachia genomes, including the Wolbachia wMel strain that is 1.27 Mb (Wu et al. 2004; Klasson et al. 2008; Klasson et al. 2009; Scott and Ghedin 2009).
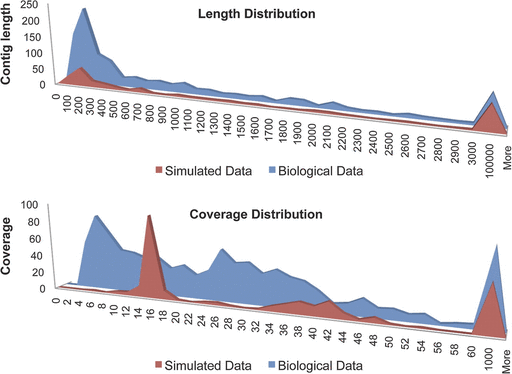
Analysis of genome assembly and coverage with real and simulated data. Top graph: Histogram distributions of assembled contig length in real and simulated sequence reads. The x and y axes correspond to contig length (in bins) and number of occurrences, respectively. Bottom graph: Histogram distributions of assembled contig coverage in real and simulated sequence reads. The x and y axes correspond to contig coverage (in bins) and number of occurrences, respectively.
Discussion
The implication of frequent mobile element transfers between obligate intracellular coinfections is significant to genome evolution in host-associated microbes. If eukaryotic hosts can be arenas for HGT between intracellular infections, especially those bacteria that frequently move between different hosts (Bordenstein and Reznikoff 2005; Kent and Bordenstein 2010), then these bacteria can partially ease the reductive processes of an intracellular lifestyle. Indeed, minor capsid gene sequences are identical between Wolbachia coinfections in every examined species thus far (Masui et al. 2001; Bordenstein and Wernegreen 2004; Chafee et al. 2010), implying that bacteriophage transfer between Wolbachia coinfections in the same host is likely the rule rather than the exception. However, evidence of phage transfer outside of this single gene had not previously been shown.
Horizontal Transfer of WO
Here, we demonstrated for the first time that large-scale HGT (52.2 kb) of phage and flanking genes occurred between the wVitA and the wVitB coinfections of N. vitripennis. The transferred haplotypes, WOVitB and WOVitA1, are distinct from the WO haplotypes found in other Wolbachia in both synteny and nucleotide identity (data not shown), demonstrating that the exchange happened within the wasp host. Complete and frequent transfer of temperate bacteriophage WO likely supports its spread in the Wolbachia-arthropod symbiosis (Bordenstein and Wernegreen 2004; Gavotte et al. 2007) and explains why the integrated prophage is associated with the most rapidly evolving portions of the Wolbachia genome (Ishmael et al. 2009).
By inference, this finding suggests that other phage-containing Wolbachia coinfections may also gain novel genes or functions through the complete transfer of WO. For instance, bacteriophage WO can increase the genetic repertoire of Wolbachia through vectoring nonstructural proteins such as ankyrin-repeat proteins, putative virulence factors, and transposons. The presence of these proteins can vary between phage types (Wu et al. 2004; Klasson et al. 2008; Klasson et al. 2009; Tanaka et al. 2009; Kent and Bordenstein 2010); thus diversity can increase with each phage transfer and could shape the interaction between the Wolbachia and the insect host. Indeed, ankyrin-repeat and virulence proteins that are encoded on the phage are candidates that could interact with the insect host (Iturbe-Ormaetxe et al. 2005; Sinkins et al. 2005; Sanogo and Dobson 2006; Tanaka et al. 2009), although no direct effect of these proteins on host reproductive alterations has been yet shown (Yamada et al. 2010). This evidence builds upon studies in N. vitripennis, where the phage's principle influence is associated with lysing Wolbachia and reducing the expressivity of Cytoplasmic Incompatibility (Bordenstein et al. 2006). These findings, however, do not preclude the role of these proteins in other eukaryotic cellular activities.
The transferred phage genes were 99.9% identical, suggesting a very recent transfer in an otherwise rapidly evolving segment of the genome. Despite the fixation of this newly acquired phage in the genome, approximately one-third of the genome was degraded by gene loss and insertion sequences. The rapid accumulation of insertion sequences in a recently acquired phage can be explained by the high rates of transposition both within and between Wolbachia genomes (Cordaux et al. 2008). Degradation of prophage WO genomes in other Wolbachia is common, and it remains to be determined why some Wolbachia genomes maintain intact phages, whereas others exhibit remnants of the phage genome.
Other examples of mobile element transfers larger than insertion sequences are restricted to just two cases—both in other widespread obligate intracellular bacteria. In Rickettsia, genome-sequencing projects have identified the transfer of a 54.6-kb ICE between a relative of Rickettsia bellii and Rickettsia massiliae (Blanc et al. 2007). In addition, clusters of Rickettsial plasmid conjugal transfer genes were found in the genome, which could indicate horizontal transfer of the plasmid followed by genome integration. Within Chlamydia pneumoniae, in vitro cocultivation experiments with different strains that were positive and negative for a small lytic bacteriophage family (4,524 bp) showed that bacteriophages can be transmissible between cells in the laboratory (Rupp et al. 2007).
Targeted Genome Capture of Microbial Symbionts
By array capturing the genome of an organism that lives within cells of a host carrying other bacterial species and viruses, we demonstrated that the “targeted genome capture” method is highly efficient for enriching and sequencing specific microorganisms from a heterogeneous mixture of DNA. Since this method relies on sorting DNA and not cells or tissues, it could be applied to samples that have been fixed or frozen. We propose that this method can be widely applied to other bacterial and viral symbiont genomes, including human pathogens that exist in low titers. However, some caution is required. First, our results show capture arrays unexpectedly have the capacity to enrich for divergent sequences, with as much as 78.5% nucleotide divergence. The risk is that multiple strains or species within the same host could be captured together; whereas the benefit is that the capture is not restricted to closely related strains. If multiple haplotypes are captured on the same array, binning heterologous DNA sequences bioinformatically could circumvent problems associated with assembling various haplotypes. Second, the extent of captured DNA can be constrained by the range of probes printed on the array, such that novel genomic sequences within the target genome may be problematic to capture. However, our results show that 0.6% of the nucleotides in the final genome are potentially new Wolbachia sequences based on their low GC content typical of the endosymbiont. We hypothesize that captured genes could act as single-stranded probes for the subsequent capture of genes not printed on the array.
Genomic analyses of fastidious symbionts that are difficult to culture or purify from their hosts often rely on isolations from sensitive host tissue collections, whole genome amplifications that subject the template DNA to artificial chimeras, or are not possible. Targeted genome capture has the potential to obviate these pitfalls for rapid enrichment and sequencing of organisms with relatively small genomes.
In summary, we have found that transfer of bacteriophage between coinfections of obligate intracellular bacteria in the same host can span entire phage genomes with many genes and that targeted sequence capture can trap genomes for de novo sequencing of new microbial or viral symbionts from eukaryotic hosts. Although the array capture method intrinsically depends on preexisting sequence information, we have shown that the captured sequences can be divergent from the arrayed DNA. The method offers significant improvements over labor-intensive alternatives for sequencing symbionts that are difficult to purify or culture.
We thank M. Cipriano for advice and assistance with computational analyses, S. Bordenstein for performing quantitative PCR analysis, P. Abbot and R. Brucker for providing feedback on the manuscript, and J. Werren and S. Richards for consent to publish the unpublished wVitA prophage in advance of finishing the full genome sequence. We thank S. Richards and colleagues for handling the sequencing of the wVitA genome. This work was supported by the National Science Foundation (grant number IOS-0852344 to S.R.B. and grant number DEB 0821936 to J. Werren) and the National Institutes of Health (grant number R01 GM085163-01 to S.R.B., grant number F31 AI091343 to J.G.G., and grant number R24 GM084917 to J. Werren).
References
Author notes
Associate editor: Richard Cordaux

{kind=link}
{kind=link}
{kind=link}
{kind=link}