




Abstract
We have established that the frequency of LRRK2 mutations in a series of 118 cases of familial Parkinson's disease is 5.1%. In the largest family with autosomal dominant, late-onset Parkinson's disease where affected subjects share a Y1699C missense mutation we provide a detailed clinical, pathological and imaging report. The phenotype in this large British kindred included asymmetrical, levodopa-responsive parkinsonism where unilateral leg tremor at onset and foot dystonia were prominent features. There was no significant abnormality of cognition but there was prominent behavioural disorder. We observed a lower age of onset in successive generations. Histopathology in one patient showed substantia nigra cell loss and Lewy body formation, with small numbers of cortical Lewy bodies. 18F-dopa positron emission tomography (PET) in another patient showed a pattern of nigrostriatal dysfunction typical of idiopathic Parkinson's disease. 18F-dopa-PET scans in unaffected family members prior to identifying the disease locus did not detect subclinical nigrostriatal dysfunction. Olfaction was assessed in affected subjects and Lewy bodies were identified in the olfactory bulb as well as cortex and brainstem of one deceased patient. In order to assess the role of mutations in this gene in other familial cases we undertook a mutation screen of all 51 exons of LRRK2 in 117 other smaller British kindreds with familial Parkinson's disease. The commonest mutation was G2019S and we also identified two novel mutations, R1941H and T2356I, in the coding sequence. These data suggest that parkinsonism caused by mutations in LRRK2 is likely to represent the commonest locus for autosomal dominant Parkinson's disease with a phenotype, pathology and in vivo imaging similar to idiopathic, late-onset Parkinson's disease.
Introduction
We previously reported the largest British kindred (the Lincolnshire kindred) with autosomal dominant parkinsonism; at that time, no post-mortem data were available and genetic studies had excluded linkage to other known loci (Nicholl et al., 2002). Recently, disease-causing mutations were identified in a novel gene LRRK2 (leucine-rich, repeat kinase 2) both in this kindred and the Basque kindreds (Paisan-Ruiz et al., 2004). Here we present a detailed study of the LRRK2 phenotype, neuropathological data of brain including the olfactory bulb, functional imaging using 18F-dopa positron emission tomography (PET) and olfactory testing on the largest British kindred known to have late-onset, familial Parkinson's disease with disease attributable to a mutation in the LRRK2 gene.
We also report a mutation screen of all 51 exons in LRRK2 in 117 unrelated British patients with familial Parkinson's disease identifying both common and novel mutations.
Patients and methods
Patients
All subjects gave informed consent. The study was approved by the Ethics Committees of the National Hospital for Neurology and Neurosurgery (NHNN).
The Lincolnshire kindred. A clinical study that included a standardized clinical proforma was performed. Cognition was assessed using the Folstein Mini-Mental State Examination (Folstein et al., 1975). Neuropsychometry was performed on subject III.1 (Fig. 1). Genealogical data were collected through civil and church records of birth, marriages and deaths, medical records and accounts from family members. The University of Pennsylvania Smell Identification Test (UPSIT) was used to test olfaction (Doty et al., 1984). Scores range from 0 to 40; higher scores denote better olfaction.
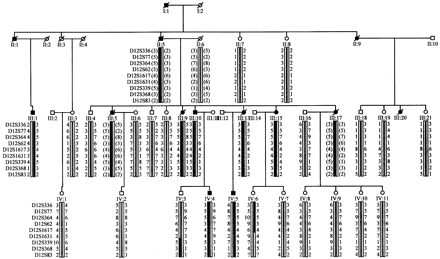
A simplified pedigree and haplotype analysis of the region on chromosome 12 linked to disease in this kindred. A total of 6 affected and 19 unaffected, at risk members were used in a genome-wide search using 382 microsatellite markers; a haplotype segregated in all clinically affected subjects between ABI LMS markers D12S99 to D12S83; D12S364 gave the highest LOD score of 3.55 (𝛉 = 0.00). Haplotypes of family members were constructed based on the genotyping of 9 microsatellite markers. Black symbols denote individuals affected by Parkinson's disease. Deceased members are marked by diagonal bars. The black bar denotes the haplotype segregating with disease and all other haplotypes are represented by a white bar. Inferred haplotypes are represented in parentheses.
Unrelated familial Parkinson's disease patients. A total of 117 cases with one or more affected first-degree relatives were identified fulfilling the UK Brain Bank criteria for the diagnosis of Parkinson's disease (Gibb et al., 1988) except for the presence of a family history. One proband was also included with prominent postural tremor with a family history of parkinsonism (II.2, Family 4). An autosomal dominant mode of inheritance (one or more affected first-degree relatives across two generations) was present in 60 patients, the remaining 57 subjects were sibling-pairs.
Molecular analysis. Subject III.1 (Fig. 1) from the Lincolnshire kindred was screened for mutations in HD, DRPLA, SCA3 and for alpha-synuclein rearrangements using methods previously reported (Singleton et al., 2003). All other subjects in this study were screened for LRRK2 mutations by methods previously reported (Paisan-Ruiz et al., 2004; Zimprich et al., 2004a).
Functional imaging. 18F-dopa PET scans were performed on subjects from the Lincolnshire kindred prior to the linkage study and analysed using methods previously reported (Khan et al., 2002). Subject III.1 (Fig. 2) has previously been reported (Nicholl et al., 2002). Five unaffected members [III.3, III.4, III.8, III.21, IV.8, (Fig. 1)] were also scanned prior to the genome screen. Ethics Committees of the NHNN and the Hammersmith Hospitals Trust, London, UK approved the project. Permission to administer radiation was licensed by the Administration of Radioactive Substances Advisory Committee (ARSAC) UK.
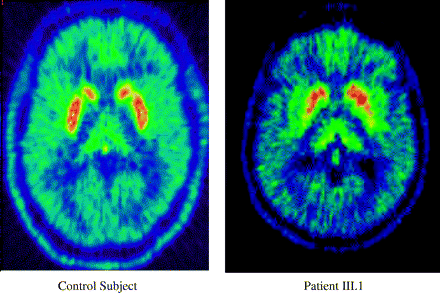
18F-dopa PET scans showing nigrostriatal dysfunction in Patient III.1 (Fig. 1) and a healthy control.
Neuropathology
Consent for post-mortem examination of the brain of Patient III.13 (Lincolnshire kindred, Fig. 1) was obtained from the next of kin according to the standard procedures of the Queen Square Brain Bank. The anterior frontal, anterior cingulate, temporal and parietal neocortices in addition to olfactory bulb, basal ganglia, mid-brain, pons, medulla and cerebellum were sampled and stained using haematoxylin and eosin or Bielschowsky's silver impregnation according to standard protocols. For immunohistochemical staining endogenous peroxidase activity was blocked by treating sections with methanol/H2O2 solution, and pre-treating by pressure cooking and/or 99% formic acid to unmask antigenic activity. Non-specific antigen binding was blocked by normal horse serum (Vectastain Elite ABC kit). Sections were incubated with primary antibodies, washed and incubated in the secondary antibody (Vectastain Elite ABC kit) and following washes the ABC reagent was applied. Immunolabelling was visualized using diaminobenzidine with Mayer's haematoxylin counterstaining. The primary antibodies used were as follows: tau (rabbit polyclonal, 1:200, no pre-treatment), DAKO, UK; tau (AT8, mouse monoclonal recognizing phosphorylated Ser202/Thr205, 1:600, pressure cooking pre-treatment), Autogen Bioclear, UK; Aβ (mouse monoclonal, 1:50, formic acid and pressure cooking pre-treatment), Novocastra, UK; neurofilaments (mouse monoclonal, 1:20, no pre-treatment), ICN Pharmaceuticals Inc., USA.
Results
The Lincolnshire kindred
Clinical data. Ancestors of I.1 (Fig. 1) can be traced to a family of farmers living in rural Lincolnshire, England in the 16th century. Accounts from living and deceased family members and death certificates identify affected members back to the early 1800s.
Affected subjects. A total of 25 affected members have been identified over four generations; only part of the pedigree is shown in Fig. 1. At the time of the previous description (Nicholl et al., 2002), there were five living subjects with Parkinson's disease (III.1, III.9, III.10, III.13, III.15) and a sixth (IV.5) had just developed disease. At follow-up, subject IV.4 had developed Parkinson's disease and Patients III.9 and III.13 had died; post-mortem examination was available on III.13.
Clinical features are emphasized here and in Table 1 with detailed descriptions in the Appendix
Clinical characteristics of affected subjects with LRRK2 mutation
| Subject | Sex/age | Age at onset | Symptoms at onset | DD | Response to L-dopa | Other features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The Lincolnshire kindred | ||||||||||||
| I.1 | M/67 | 65 | Tremor | 2 | Not taken | Born late 1800s, mild disease, died aged 67 years | ||||||
| II.1 | M/66 | 61 | Unknown | 5 | Not taken | Born 1890s, shuffling gait, severe parkinsonism, died aged 66 years | ||||||
| II.5 | M/70 | 50 | Unknown | 20 | Not taken | Born 1890s late disease, severe parkinsonism, died aged 70 years | ||||||
| II.9 | F/85 | 75 | Unknown | 10 | Unknown | Born 1890s; mild disease, died aged 85 years | ||||||
| III.1 | M/70 | 56 | Rest tremor big toe | 14 | Excellent | Anxiety at onset, foot dystonia and dykinesias after 4 years treatment, MMSE 30 at age 70 years | ||||||
| III.5 | M/54 | 50 | Unknown | 4 | Good | 10 years of anxiety and neurosis prior to the onset parkinsonism, died aged 70 years | ||||||
| III.9 | F/77 | 57 | Unknown | 20 | Significant | After 10 years: severe offs, bulbar symptoms, akinetic and mute, died aged 77 years | ||||||
| III.10 | M/70 | 45 | Unilateral leg tremor | 25 | Significant | Suicide attempt at 66, depression for 10 years, continued L-dopa response, no dyskinesias. MMSE 30 | ||||||
| III.13 | F/70 | 50 | Unilateral leg stiffness and tremor | 20 | Moderate | After 10 years: depression, foot dystonia (rest/exercise induced), peak-dose dyskinesias; died aged 70 years (stroke) | ||||||
| III.15 | F/68 | 44 | Unilateral leg tremor and stiffness | 24 | Significant | Claustrophobia, anxiety and depression after 14 years, MMSE 28 at dyskinesia after 8 years | ||||||
| III.17 | F/57 | 45 | Unilateral leg tremor and stiffness | 12 | Significant | Paranoia, anxiety and depression after 10 years, foot dystonia, little dyskinesias on treatment, died aged 57 years | ||||||
| III.20 | F/57 | 48 | Unknown | 9 | Unknown | Hemi-parkinsonism, died from bronchopneumonia. No PM | ||||||
| IV.4 | M/47 | 44 | Unilateral leg tremor | 3 | Unmedicated | Most recent subject who has developed disease | ||||||
| IV.5 | M/49 | 40 | Unilateral leg tremor | 9 | Significant | Anxiety 2 years prior to disease onset, foot dystonia before L-dopa MMSE 30 | ||||||
| Other unrelated LRRK2 subjects | ||||||||||||
| Family 1 | ||||||||||||
| III.1 | M/61 | 59 | Bradykinesia, micrographia | 2 | Excellent | Sustained response to L-dopa | ||||||
| Family 2 | ||||||||||||
| III.2 | M/56 | 42 | Dragging left leg | 14 | Very good | Depression, hypomania, alcohol-dependency L-dopa addiction, FH depression | ||||||
| Family 3 | ||||||||||||
| I.2 | F/73 | 70 | Unknown | 3 | Excellent | Depression preceding motor symptoms | ||||||
| Family 4 | ||||||||||||
| II.2 | F/unknown | Unknown | Tremor | Unknown | Unknown | FH healthy carrier of T23561 and phenocopy | ||||||
| III.1 | F/unknown | Unknown | Tremor | Unknown | Unknown | |||||||
| Family 5 | ||||||||||||
| II.2 | F/81 | 67 | Tremor, bradykinesia micrographia | 14 | Excellent | FH healthy carrier of G2019S and phenocopy | ||||||
| Subject | Sex/age | Age at onset | Symptoms at onset | DD | Response to L-dopa | Other features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The Lincolnshire kindred | ||||||||||||
| I.1 | M/67 | 65 | Tremor | 2 | Not taken | Born late 1800s, mild disease, died aged 67 years | ||||||
| II.1 | M/66 | 61 | Unknown | 5 | Not taken | Born 1890s, shuffling gait, severe parkinsonism, died aged 66 years | ||||||
| II.5 | M/70 | 50 | Unknown | 20 | Not taken | Born 1890s late disease, severe parkinsonism, died aged 70 years | ||||||
| II.9 | F/85 | 75 | Unknown | 10 | Unknown | Born 1890s; mild disease, died aged 85 years | ||||||
| III.1 | M/70 | 56 | Rest tremor big toe | 14 | Excellent | Anxiety at onset, foot dystonia and dykinesias after 4 years treatment, MMSE 30 at age 70 years | ||||||
| III.5 | M/54 | 50 | Unknown | 4 | Good | 10 years of anxiety and neurosis prior to the onset parkinsonism, died aged 70 years | ||||||
| III.9 | F/77 | 57 | Unknown | 20 | Significant | After 10 years: severe offs, bulbar symptoms, akinetic and mute, died aged 77 years | ||||||
| III.10 | M/70 | 45 | Unilateral leg tremor | 25 | Significant | Suicide attempt at 66, depression for 10 years, continued L-dopa response, no dyskinesias. MMSE 30 | ||||||
| III.13 | F/70 | 50 | Unilateral leg stiffness and tremor | 20 | Moderate | After 10 years: depression, foot dystonia (rest/exercise induced), peak-dose dyskinesias; died aged 70 years (stroke) | ||||||
| III.15 | F/68 | 44 | Unilateral leg tremor and stiffness | 24 | Significant | Claustrophobia, anxiety and depression after 14 years, MMSE 28 at dyskinesia after 8 years | ||||||
| III.17 | F/57 | 45 | Unilateral leg tremor and stiffness | 12 | Significant | Paranoia, anxiety and depression after 10 years, foot dystonia, little dyskinesias on treatment, died aged 57 years | ||||||
| III.20 | F/57 | 48 | Unknown | 9 | Unknown | Hemi-parkinsonism, died from bronchopneumonia. No PM | ||||||
| IV.4 | M/47 | 44 | Unilateral leg tremor | 3 | Unmedicated | Most recent subject who has developed disease | ||||||
| IV.5 | M/49 | 40 | Unilateral leg tremor | 9 | Significant | Anxiety 2 years prior to disease onset, foot dystonia before L-dopa MMSE 30 | ||||||
| Other unrelated LRRK2 subjects | ||||||||||||
| Family 1 | ||||||||||||
| III.1 | M/61 | 59 | Bradykinesia, micrographia | 2 | Excellent | Sustained response to L-dopa | ||||||
| Family 2 | ||||||||||||
| III.2 | M/56 | 42 | Dragging left leg | 14 | Very good | Depression, hypomania, alcohol-dependency L-dopa addiction, FH depression | ||||||
| Family 3 | ||||||||||||
| I.2 | F/73 | 70 | Unknown | 3 | Excellent | Depression preceding motor symptoms | ||||||
| Family 4 | ||||||||||||
| II.2 | F/unknown | Unknown | Tremor | Unknown | Unknown | FH healthy carrier of T23561 and phenocopy | ||||||
| III.1 | F/unknown | Unknown | Tremor | Unknown | Unknown | |||||||
| Family 5 | ||||||||||||
| II.2 | F/81 | 67 | Tremor, bradykinesia micrographia | 14 | Excellent | FH healthy carrier of G2019S and phenocopy | ||||||
DD = disease duration; M = male; F = female; FH = family history.
Clinical characteristics of affected subjects with LRRK2 mutation
| Subject | Sex/age | Age at onset | Symptoms at onset | DD | Response to L-dopa | Other features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The Lincolnshire kindred | ||||||||||||
| I.1 | M/67 | 65 | Tremor | 2 | Not taken | Born late 1800s, mild disease, died aged 67 years | ||||||
| II.1 | M/66 | 61 | Unknown | 5 | Not taken | Born 1890s, shuffling gait, severe parkinsonism, died aged 66 years | ||||||
| II.5 | M/70 | 50 | Unknown | 20 | Not taken | Born 1890s late disease, severe parkinsonism, died aged 70 years | ||||||
| II.9 | F/85 | 75 | Unknown | 10 | Unknown | Born 1890s; mild disease, died aged 85 years | ||||||
| III.1 | M/70 | 56 | Rest tremor big toe | 14 | Excellent | Anxiety at onset, foot dystonia and dykinesias after 4 years treatment, MMSE 30 at age 70 years | ||||||
| III.5 | M/54 | 50 | Unknown | 4 | Good | 10 years of anxiety and neurosis prior to the onset parkinsonism, died aged 70 years | ||||||
| III.9 | F/77 | 57 | Unknown | 20 | Significant | After 10 years: severe offs, bulbar symptoms, akinetic and mute, died aged 77 years | ||||||
| III.10 | M/70 | 45 | Unilateral leg tremor | 25 | Significant | Suicide attempt at 66, depression for 10 years, continued L-dopa response, no dyskinesias. MMSE 30 | ||||||
| III.13 | F/70 | 50 | Unilateral leg stiffness and tremor | 20 | Moderate | After 10 years: depression, foot dystonia (rest/exercise induced), peak-dose dyskinesias; died aged 70 years (stroke) | ||||||
| III.15 | F/68 | 44 | Unilateral leg tremor and stiffness | 24 | Significant | Claustrophobia, anxiety and depression after 14 years, MMSE 28 at dyskinesia after 8 years | ||||||
| III.17 | F/57 | 45 | Unilateral leg tremor and stiffness | 12 | Significant | Paranoia, anxiety and depression after 10 years, foot dystonia, little dyskinesias on treatment, died aged 57 years | ||||||
| III.20 | F/57 | 48 | Unknown | 9 | Unknown | Hemi-parkinsonism, died from bronchopneumonia. No PM | ||||||
| IV.4 | M/47 | 44 | Unilateral leg tremor | 3 | Unmedicated | Most recent subject who has developed disease | ||||||
| IV.5 | M/49 | 40 | Unilateral leg tremor | 9 | Significant | Anxiety 2 years prior to disease onset, foot dystonia before L-dopa MMSE 30 | ||||||
| Other unrelated LRRK2 subjects | ||||||||||||
| Family 1 | ||||||||||||
| III.1 | M/61 | 59 | Bradykinesia, micrographia | 2 | Excellent | Sustained response to L-dopa | ||||||
| Family 2 | ||||||||||||
| III.2 | M/56 | 42 | Dragging left leg | 14 | Very good | Depression, hypomania, alcohol-dependency L-dopa addiction, FH depression | ||||||
| Family 3 | ||||||||||||
| I.2 | F/73 | 70 | Unknown | 3 | Excellent | Depression preceding motor symptoms | ||||||
| Family 4 | ||||||||||||
| II.2 | F/unknown | Unknown | Tremor | Unknown | Unknown | FH healthy carrier of T23561 and phenocopy | ||||||
| III.1 | F/unknown | Unknown | Tremor | Unknown | Unknown | |||||||
| Family 5 | ||||||||||||
| II.2 | F/81 | 67 | Tremor, bradykinesia micrographia | 14 | Excellent | FH healthy carrier of G2019S and phenocopy | ||||||
| Subject | Sex/age | Age at onset | Symptoms at onset | DD | Response to L-dopa | Other features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The Lincolnshire kindred | ||||||||||||
| I.1 | M/67 | 65 | Tremor | 2 | Not taken | Born late 1800s, mild disease, died aged 67 years | ||||||
| II.1 | M/66 | 61 | Unknown | 5 | Not taken | Born 1890s, shuffling gait, severe parkinsonism, died aged 66 years | ||||||
| II.5 | M/70 | 50 | Unknown | 20 | Not taken | Born 1890s late disease, severe parkinsonism, died aged 70 years | ||||||
| II.9 | F/85 | 75 | Unknown | 10 | Unknown | Born 1890s; mild disease, died aged 85 years | ||||||
| III.1 | M/70 | 56 | Rest tremor big toe | 14 | Excellent | Anxiety at onset, foot dystonia and dykinesias after 4 years treatment, MMSE 30 at age 70 years | ||||||
| III.5 | M/54 | 50 | Unknown | 4 | Good | 10 years of anxiety and neurosis prior to the onset parkinsonism, died aged 70 years | ||||||
| III.9 | F/77 | 57 | Unknown | 20 | Significant | After 10 years: severe offs, bulbar symptoms, akinetic and mute, died aged 77 years | ||||||
| III.10 | M/70 | 45 | Unilateral leg tremor | 25 | Significant | Suicide attempt at 66, depression for 10 years, continued L-dopa response, no dyskinesias. MMSE 30 | ||||||
| III.13 | F/70 | 50 | Unilateral leg stiffness and tremor | 20 | Moderate | After 10 years: depression, foot dystonia (rest/exercise induced), peak-dose dyskinesias; died aged 70 years (stroke) | ||||||
| III.15 | F/68 | 44 | Unilateral leg tremor and stiffness | 24 | Significant | Claustrophobia, anxiety and depression after 14 years, MMSE 28 at dyskinesia after 8 years | ||||||
| III.17 | F/57 | 45 | Unilateral leg tremor and stiffness | 12 | Significant | Paranoia, anxiety and depression after 10 years, foot dystonia, little dyskinesias on treatment, died aged 57 years | ||||||
| III.20 | F/57 | 48 | Unknown | 9 | Unknown | Hemi-parkinsonism, died from bronchopneumonia. No PM | ||||||
| IV.4 | M/47 | 44 | Unilateral leg tremor | 3 | Unmedicated | Most recent subject who has developed disease | ||||||
| IV.5 | M/49 | 40 | Unilateral leg tremor | 9 | Significant | Anxiety 2 years prior to disease onset, foot dystonia before L-dopa MMSE 30 | ||||||
| Other unrelated LRRK2 subjects | ||||||||||||
| Family 1 | ||||||||||||
| III.1 | M/61 | 59 | Bradykinesia, micrographia | 2 | Excellent | Sustained response to L-dopa | ||||||
| Family 2 | ||||||||||||
| III.2 | M/56 | 42 | Dragging left leg | 14 | Very good | Depression, hypomania, alcohol-dependency L-dopa addiction, FH depression | ||||||
| Family 3 | ||||||||||||
| I.2 | F/73 | 70 | Unknown | 3 | Excellent | Depression preceding motor symptoms | ||||||
| Family 4 | ||||||||||||
| II.2 | F/unknown | Unknown | Tremor | Unknown | Unknown | FH healthy carrier of T23561 and phenocopy | ||||||
| III.1 | F/unknown | Unknown | Tremor | Unknown | Unknown | |||||||
| Family 5 | ||||||||||||
| II.2 | F/81 | 67 | Tremor, bradykinesia micrographia | 14 | Excellent | FH healthy carrier of G2019S and phenocopy | ||||||
DD = disease duration; M = male; F = female; FH = family history.
Age of onset. Mean age of onset of living subjects was 57 years and decreased in successive generations; generation II: 62.0 years, III: 50.1 years and IV: 40.0 years.
Tremor. All reported unilateral leg symptoms at onset. Subject III.5, aged 50 years, reported rest tremor of the big toe of the right foot progressing to both feet and legs. Subject III.10 reported an initial symptom of unilateral tremor in one leg that was prominent only when seated. IV.5 first noticed tremor and heaviness in the right leg that initially occurred only while driving. Subject III.17, aged 45 years, first noticed tripping over her left foot and intermittent dragging of her left foot and leg; she subsequently developed rest tremor in left arm and left-sided foot dystonia.
Dystonia. Foot dystonia (rest and exercise-induced) was reported prior to and during L-dopa treatment; the latter developed early or later on in disease.
Other motor features. All subjects reported hemi-parkinsonian symptoms at onset with symptoms of slowly progressive parkinsonism. Freezing episodes and falls occurred late. Only one subject (III.9) reported significant bulbar symptoms late in disease.
Autonomic symptoms. Subject (III.13) reported symptoms of orthostatic hypotension after 15 years of disease.
Cognitive function. Cognition in subjects examined in detail (III.1, III.10, III.15 and IV.5) was normal (MMSE >28 years). In particular subject III.15 was normal despite disease duration of up to 26 years. Formal neuropsychometric testing in III.1 showed mild word finding difficulties, mild executive dysfunction with moderate speed and attention difficulties suggesting a mild degree of cognitive impairment predominantly affecting subcortical and anterior cortical regions despite 14 years of disease.
Response to treatment. Disease was characterized by significant response to l-dopa; subject III.10 reported sustained improvement after 25 years of disease and no dyskinesias despite 18 years of L-dopa treatment. Others reported little dyskinesia. Foot dystonia was more prominent at peak dose of l-dopa medication. Subject IV.5 had significant benefit from ropinirole and pramipexole as first line treatment. Subject III.15 reported significant improvement on benzhexol for 10 years and continued to report sustained improvement after substitution with l-dopa treatment.
Behavioural disorder. Seven subjects reported behavioural disorder (anxiety attacks, depression, paranoia and suicide): six reported symptoms after the onset of parkinsonism and subject IV.5 described symptoms 2 years prior to disease onset.
Clinical features are summarized in Table 1.
Olfaction testing. Patients III.1, III.10, III.15 and IV.5 had scores of 34, 24, 25 and 36 out of 40, respectively on UPSIT testing.
Unaffected subjects. Subjects IV.3 (aged 42 years) and IV.6 (aged 48 years) declined a neurological examination or a PET scan. They were asymptomatic and are not reported to have signs of parkinsonism. Subjects III.3, III.4, III.7, III.21 and IV.7 (aged 77, 83, 78, 82 and 42 years, respectively) had normal examinations. Subject II.7 was aged 103 years, and is not reported to have developed parkinsonism.
Molecular data. Patient III.1 did not have HD, DRPLA, SCA3 expansions or alpha-synuclein rearrangements. A Y1699C missense mutation in LRRK2 was identified in Patients III.1, III.9, III.10, III.13, III.15, IV.4, IV.5 (Fig. 1). The mutation was not present in 1300 control chromosomes (Paisan-Ruiz et al., 2004) or unaffected subjects III.3, III.4, III.18, III.21, IV.2, IV.3, IV.6 and IV.7.
PET data. 18F-dopa PET of an affected Patient III.1 has previously been reported (Nichol et al., 2002). This showed a pattern of nigrostriatal dysfunction (presynaptic reduction of putamenal 18F-dopa uptake with relative sparing of the caudate) typical of idiopathic Parkinson's disease (Fig. 2). A further five subjects, III.3 (aged 77), III.4 (aged 83,), III.7 (aged 78), III.21 (aged 82) and IV.7 (aged 42), in whom the clinical examination was normal were also scanned; their 18F-dopa uptake was in the normal range (data not shown).
Neuropathology
The brain of Patient III.13 weighed 1075 g and showed no evidence of atrophy. Coronal slices of the right cerebral hemisphere showed reduction in size of the lateral ventricle due to brain swelling secondary to hypoxia. The cortical ribbon and underlying white matter were unremarkable except for a region of haemorrhagic infarction in the medial occipital vascular watershed region. The caudate, putamen, globus pallidus and thalamus appeared normal. There was extreme pallor of the substantia nigra with some preservation of pigmentation in the medial aspect. The locus coeruleus was indiscernible. The medulla and cerebellum appeared normal.
Histological examination showed severe loss of pigmented neurons in the dorsal and ventral tiers of the substantia nigra with marked gliosis, although the medial part of the nucleus showed some preservation. Small numbers of Lewy bodies were seen in the H&E preparation (Fig. 3A). α-Synuclein immunohistochemistry confirmed this finding and also demonstrated Lewy neurites (Fig. 3B and C). Lewy bodies and Lewy neurites were present in the locus coeruleus. In the olfactory bulb scattered α-synuclein-positive neurites and scanty Lewy bodies were present (Fig. 3D). Cortical Lewy bodies were observed with a frequency corresponding to brainstem-predominant Lewy body disease (McKeith et al., 1996) (Fig. 3E). Tau immunohistochemistry showed neurofibrillary tangles (NFTs) in the hippocampus, subiculum, entorhinal and transentorhinal cortices corresponding to Braak and Braak Stage II. The olfactory bulb was similarly affected. No NFTs were found in the neocortex, basal ganglia, brainstem or dentate nucleus and there was no glial tau pathology. Occasional diffuse Aβ deposits were found in the neocortex; neuritic plaques were absent using Bielschowsky's silver impregnation. In the cerebellum there was mild Purkinje cell loss with empty baskets and occasional axonal torpedoes on neurofilament immunohistochemistry. There was widespread hypoxic neuronal damage and oedema in the neocortex and hippocampus.
![Histopathology of Patient III.13 (Lincolnshire kindred, Fig. 1). In the substantia nigra there was very severe loss of pigmented neurons with small numbers of Lewy bodies [arrow and insert (A)]. Lewy bodies (B) and Lewy neurites (C) were identified in the substantia nigra using α-synuclein immunohistochemistry. Similarly there were small numbers of Lewy neurites (D) and occasional Lewy bodies [inset in (D)] in the olfactory bulb. Lewy bodies were scanty in the neocortex (E). Bar represents 28 µm in (A–E) and 18 (μm in the insert in (A). (A): H&E; (B–E): α-synuclein immunohistochemistry.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/128/12/10.1093/brain/awh667/2/m_awh667f3.gif?Expires=1716340767&Signature=4uamHN7qqvWORRRGkM0cgRUIfgPsGUHN7k3g3ui2U1Cf0WP1U8sh-zgRVhlur0jZ3QCAaLakJp7C6kI3txkhjayQb4Z3ruYt3S1TXwEgWH9v88-2Yd7nVupHdkxU-ApBgRMwvprCHlVVzXLzLdR3pDUMQh~szigl2iE7xGMNLi~eSZa8iUsZ~aaDhfgQDwH9YC5r5hFDPp82~s-r0YY8nTXAelxE5YbbkUJmwGVO5Oog-RYAlCJaoEUDzxX7GutZCx4RXpGM~Und3U7IbBtNOY37oOaZZ7tuLX2oiknB6g2zKEa3Gldi3g6qyz7crE2Hz6P59rEGo6a1x1vUwA3jTQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Histopathology of Patient III.13 (Lincolnshire kindred, Fig. 1). In the substantia nigra there was very severe loss of pigmented neurons with small numbers of Lewy bodies [arrow and insert (A)]. Lewy bodies (B) and Lewy neurites (C) were identified in the substantia nigra using α-synuclein immunohistochemistry. Similarly there were small numbers of Lewy neurites (D) and occasional Lewy bodies [inset in (D)] in the olfactory bulb. Lewy bodies were scanty in the neocortex (E). Bar represents 28 µm in (A–E) and 18 (μm in the insert in (A). (A): H&E; (B–E): α-synuclein immunohistochemistry.
Other LRRK2 patients
Molecular analysis. A series of 117 patients with a family history of Parkinson's disease, 5 (4 autosomal dominant, 1 sib-pair) had LRRK2 substitutions; G2019S was identified in three patients (Family 1, 2 and 5; Table 1 and Fig. 4), R1941H in one subject (Family 3; Fig. 4) and T2356I mutation in another (Family 4; Table 1 and Fig. 4). Overall, mutations were found in 5.1% of the 118 families screened. In addition healthy subjects with LRRK2 mutations were also identified: III.3, aged 55 years (Family 4; Fig. 4) and II.5, age unknown (Family 5; Fig. 4). These mutations were not found in a total of 1438 control chromosomes.
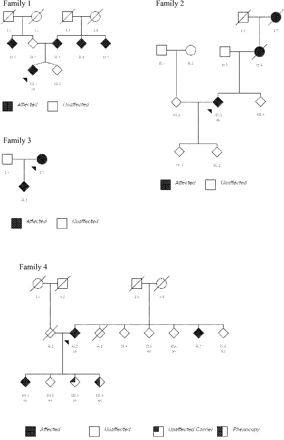
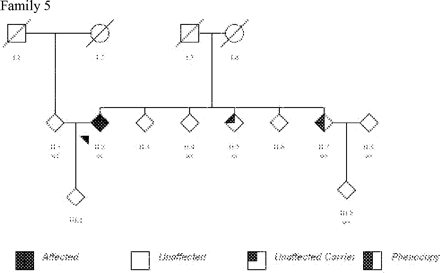
Pedigrees of other British families with LRRK2 mutations.
Clinical features. Clinical details are summarized in Table 1 and outlined in detail in the Appendix. However, in brief, the phenotype was of an asymmetrical parkinsonism with rest tremor, bradykinesia and rigidity, except for Family 4 who displayed a prominent postural tremor. The age of disease-onset ranged from 42 to 70 years with a mean age of onset of 59.5 years. Levodopa responsiveness was observed in all five families. Behavioural disorder was noted in Families 2 and 3. In addition, two phenocopies (clinical signs without a LRRK2 mutation) were identified: III.4 (Family 4) and II.7 (Family 5).
Discussion
Here we provide the first detailed clinical reports and detailed pathological description of Parkinson's disease associated with mutations in a novel gene, LRRK2, encoding the protein dardarin. The frequency of LRRK2 mutations in this series of 118 patients with a family history of Parkinson's disease is 5.1%.
The phenotype in all these familial cases carrying a mutation included a mean age of onset in index patients of 58.8 years (range 40–75) with unilateral signs at onset, l-dopa responsiveness, disease progression and duration that was similar to idiopathic Parkinson's disease. A more detailed study of the Lincolnshire kindred noted a sustained response to l-dopa despite years of disease and treatment; subject III.10 continued to benefit without dyskinesias after 25 years of disease and 18 years of l-dopa. Subject III.15 reported a significant improvement on benzhexol for 10 years. Overall, patients did not report troublesome dyskinesias complicating treatment. Foot dystonia (at rest and exercise induced) prior to and during drug treatment was observed analogous to that reported in parkin disease (Khan et al., 2003).
Unilateral symptom onset of tremor in the foot or leg was reported by most subjects. Prominent symptoms of tremor have also been observed in the Basque kindreds and it is for this reason that the protein has been named ‘dardarin’ derived from the Basque word ‘dardara’ meaning ‘tremor’ (Paisan-Ruiz et al., 2004).
Cognition in this kindred was not significantly abnormal despite lengthy disease duration in some subjects suggesting that neurodegeneration involving aberrant LRRK2 protein does not significantly impair structures critical to cognitive function.
We observed behavioural disorder in seven patients in this kindred; the significance of this is unclear. However, behavioural disorder was notable in patients with a younger age of onset (mostly <50 years) analogous to that in parkin disease (Khan et al., 2003) and DJ-1 (Dekker et al., 2003).
Olfactory dysfunction is found in 70–100% of Parkinson's disease patients rendering it as common a clinical sign as pill rolling tremor (Doty et al., 1988; Katzenschlager and Lees, 2004) and is associated with neuronal loss and Lewy body deposition in the olfactory pathway (Daniel and Hawkes, 1992). The identification of Lewy bodies in the olfactory bulb in a LRRK2 patient implies that altered kinase activation by mutant LRRK2-encoded protein extends to the olfactory neural networks. Olfaction in four other affected subjects, however, did not uniformly detect anosmia despite lengthy disease duration in these subjects. Although patient numbers were too small for meaningful statistical analysis, their mean UPSIT score was 29.7 similar to normal, age-matched British controls (mean of 27.6) contrasting a cohort of unrelated British patients with idiopathic Parkinson's disease (mean 17.1) (Katzenschlager et al., 2004). Discordance between pathological findings and UPSIT score in those affected may be explained by the fact that up to 30% of Parkinson's disease patients have normal smell despite Lewy body disease (Katzenschlager and Lees, 2004) or, that the Lewy body load in the olfactory pathway is not as deleterious in LRRK2-associated Parkinson's disease. Moreover, we were unable to perform a direct correlation of pathology with UPSIT score; the latter was not available on subject III.13.
Prior to commencing the genome screen, five unaffected family members underwent 18F-dopa PET. The purpose was to identify pre-symptomatic nigrostriatal dysfunction and extend the ‘affected’ phenotype to increase the power/logarithm of odds (LOD) score in linkage analysis. Imaging was normal and subsequent mutation analysis confirmed that LRRK2 in this kindred is fully penetrant unlike the original Japanese report in which penetrance was estimated at 70% (Funayama et al., 2002) and in other European kindreds where penetrance appears to be age-dependent (17% at 50 years and 85% at 70 years) (Kachergus et al., 2005).
This kindred has typical Lewy body pathology with marked loss of pigmented neurons and gliosis of the substantia nigra and small numbers of cortical and brainstem Lewy bodies (Fig. 3). This complements in vivo functional imaging performed on another affected subject, Patient III.1 (Fig. 1), in whom 18F-dopa PET findings showed a pattern of nigrostriatal dysfunction typically seen in Parkinson's disease; a preferential degeneration of the posterior dorsal putamen with relative sparing of the anterior dorsal putamen and head of the caudate (Bernheimer et al., 1973). More recently, 18Fdopa PET studies in patients with LRRK2 mutations have also confirmed this (Adams et al., 2005, Hernandez et al., 2005). A similar pattern was observed in patients from the Greek American kindred with dominant Parkinson's disease with a G209A mutation in alpha synuclein (Samii et al., 1999) and contrasts other forms of familial parkinsonism including some parkin and PINK1 patients, where there is more uniform loss of caudate and putamen (Khan et al., 2002).
Pathology that is typical of Parkinson's disease has also been reported in other LRRK2 kindreds described elsewhere (Wszolek et al., 2004; Zimprich et al., 2004b). However, in the original description of the Japanese kindred nigral degeneration without Lewy bodies was reported (Funayama et al., 2002). Two other LRRK2 kindreds are reported to have diverse pathologies; histopathology from patients in Family D, is more consistent with diffuse Lewy body, nigral degeneration and progressive supranuclear palsy-like pathology (Zimprich et al., 2004). A second family, Family A, has clinical and pathological features consistent with anterior horn cell degeneration. Interestingly, this family shares the same LRRK2 mutation as the Lincolnshire kindred whose phenotype, pathology and functional imaging is strikingly similar to idiopathic Parkinson's disease. The discordance between the British kindred and Family A may be attributed to the presence of two separate diseases and, therefore, two distinct types of pathology in the latter. Alternatively heterogeneous pathology, which includes Lewy bodies, tau, and amyloid, raises the possibility that dardarin may interact directly or indirectly with other pathways that lead to neurodegeneration.
LRRK2 encodes a protein of 2527 amino acids with five domains: (i) a tyrosine kinase-like MAPKKK-related (mitogen-activated protein kinase kinase kinase) domain, (ii) WD40 repeats, (iii) Ras-like small GTPase family (Roc) domain, (iv) C-terminal of Roc (COR) domain and (v) an LRRK. Until recently mutations have been reported in all but the WD40 domain (Mata et al., 2005); we have also identified a substitution (T2356I) in this domain. WD40 repeat proteins have diverse functions including signal transduction, cytoskeletal assembly, and vesicle formation and trafficking. The repeats have no intrinsic enzymatic activity but act to facilitate protein–protein interaction (Smith et al., 1999).
A screen of all 51 exons in 118 patients detected mutations in 5.1%. The G2019S mutation was the most frequent with an overall frequency of 2.5%. This mutation has been reported previously in 1.6% of isolated cases (Gilks et al., 2005), up to 6.6% in familial Caucasian patients (Di Fonzo et al., 2005; Nichols et al., 2005) and up to 41% of familial patients of North African descent (Lesage et al., 2005).
We identified healthy subjects in the cohort of familial Parkinson's disease cases with mutant LRRK2. Incomplete penetrance has been reported in the original PARK8 kindred (Funayama et al., 2002) and in other European kindreds where findings appear to be age-dependent, being 17% at 50 years and 85% at 70 years (Kachergus et al., 2005). Moreover, a recent case has been described of an octogenarian with a G2019S mutation who has an entirely normal examination (Kay et al., 2005) suggesting other genetic/environmental protective modifiers co-exist. We also report the presence of phenocopies; an observation that has been described previously in other LRRK2 families (Nichols et al., 2005). Explanations for this include: idiopathic disease is common and that this is an independent disease process that has coincidentally occurred, or, that the phenocopies share other susceptibility alleles or environmental risk factors in common with patients who have mutations in LRRK2, which predisposes to disease.
The clinical features of LRRK2-associated Parkinson's disease appear remarkably similar to idiopathic Parkinson's disease. Dardarin is a multidomain protein of uncertain function and there is now evidence of mutations in all five domains that may cause Parkinson's disease. A deeper understanding of the molecular pathways involved in LRRK2-mediated Parkinson's disease will be invaluable.
Appendix
The Lincolnshire kindred.
Patient I.1
This subject, born in the late 1800s, is reported to have developed tremor in his 60s. Several grandchildren have commented that this was relatively mild compared with subsequent generations. He died aged 67 years.
Patient II.1 and offspring III.1
II.1 developed parkinsonism at 61 years. Other family members report the subsequent development of a shuffling gait, falls and severe parkinsonism within 5 years. He died aged 66 years from bronchopneumonia in 1969 and did not have a post-mortem examination.
His only child, subject III.1, aged 70 years, first reported symptoms at the age of 56 years. He described anxiety, rest tremor of the big toe on the right foot which progressed to rest tremor of both legs. One year after the onset of symptoms he was noted to have a paucity of facial expression and poor arm swing and commenced l-dopa reporting a 90% improvement. Four years after treatment, he developed peak dose dyskinesias and right-sided foot dystonia both at rest and with exercise. He reported one fall per month for the past 6 years and freezing episodes for the past 4 years. He does not report any symptoms of autonomic dysfunction. There is no history of sleep benefit, motor fluctuations prior to starting treatment or any form of behavioural disorder prior to the onset of, or with disease. MMSE score was 30 aged 70 years. This patient underwent an 18f-dopa PET scan (Nichol et al., 2003).
Patient II.5 and affected offspring (III.5, III.9, III.10, III.13, III.15, III.17)
II.5 developed parkinsonism aged 50. Family members report that late disease associated with severe disability, marked facial hypomimia, severe rest tremor, akinesia and rigidity. He died aged 70 years in 1966.
Patient III.5. This patient had a longstanding history of anxiety and neurosis before the onset l-dopa responsive parkinsonism aged 50 years. He died aged 54 years.
Patient III.9 developed hemi-parkinsonism at the age of 57; there was a definitive response to l-dopa but this was complicated by end of dose akinesia. After 10 years disease duration, aged 67, there was severe on–off phenomena and difficulty with speech and swallowing when off. At that time, when `off', she was mute, with left sided hemi-parkinsonism and some restriction of upgaze only. The disease became marked disabling requiring care in a nursing home for 9 years until the patient died aged 77. A post-mortem examination was not possible.
Patient III.10. This 70 year old man noticed tremor in the left leg, especially prominent when seated aged 45 years. At age of 50 years he developed stiffness of the left arm and leg, foor dystonia and poor swing on the left. Two years later he commenced l-dopa and reported a significant improvement. After 25 years of disease he continues to report improvement with l-dopa, has no dyskinesias, reports falls over the past 18 months and freezing over the past 12 months. He attempted suicide aged 66 years and has been treated for depression since, MMSE score was 30.
Patient III.13 developed parkinsonism aged 50 years, with dragging of the left leg and subsequent tremor of the left arm and leg. l-dopa commenced 2 years after symptom onset with some improvement of symptoms. Peak dose dyskinesias, foot dystonia (rest and exercise-induced) and depression occurred 10 years later. Freezing, falls and postural-related symptoms occurred in the last 5 years of disease. The patient died, aged 70 years, in 2000 after a stroke. The post-mortem examination and histopathology is reported in the main text of the article.
Patient III.15. This 68 year old, reported both rest tremor and dragging of the left leg and foot at the age of 44 years. At that time, she commenced benzhexol and reported a significant improvement of symptoms. Within 2 years she developed a prominent tremor of the left hand, flexed posturing, poor arm swing and dystonia of the left foot at rest and with exercise. l-dopa was started 10 years after disease onset, on which she reported 80% improvement of symptoms. Dyskinesias started after 8 years of l-dopa therapy. Falls and freezing have complicated disease in the past year. There are no urinary or postural-related symptoms. She developed severe claustrophobia, panic attacks and depression 14 years after disease onset. MMSE was 28 aged 68 years.
III.17 developed parkinsonism aged 45 years. At disease onset she would often trip over and began to drag the left foot and leg. She subsequently developed rest tremor in the left arm and significant left-sided foot dystonia. l-dopa therapy started 1 year after disease onset with significant improvement. Significant depression, paranoia and panic attacks occurred 10 years after disease onset. At age 53, a hemi-colectomy was performed after developing complications of chronic constipation. This subject had little dyskinesia and was predominantly akinetic and died after disease duration of 12 years.
Patient II.9 and offspring III.20
II.1 is reported by family members to have had mild signs of parkinsonism in her early seventies and died in 1976, aged 85 years. One of her children, III.20 developed parkinsonism at the age of 48 years but died aged 57 from bronchopneumonia. A post-mortem was not performed on the brain.
Patient IV.4.
This 47 year old first started to drag the left leg aged 44 years. At 45 years he developed poor arm swing on left and stiffness of left arm and leg. He has not taken any medication.
Patient IV.5.
This 49 year old patient (offspring of patient III.13) first noticed tremulousness of the right leg whilst driving aged 40 years. There was also heaviness of the right leg and intermittent slurring of speech. Three years later, following a fall, he developed intermittent, right-sided foot dystonia at rest. Initial treatment commenced aged 44 years with ropinirole and then pramipexole on which he reported 75% improvement of symptoms. Three years after treatment he commenced l-dopa and described a significant improvement of symptoms. He reported no dyskinesias, falls or freezing episodes. He developed prominent panic attacks two years prior to the onset of disease.
Other unrelated British LRRK2 patients
Family 1
The index case (III.1, Family 1, Figure 4) was examined aged 61 years following an 18-month history of micrographia, hypophonia and slowed walking. There was bradykinesia and right arm rigidity but no tremor or postural instability. He was treated with 100 mg of slow-release levodopa twice daily, with immediate and sustained effect.
Five years after onset he has remained tremor-free but has required the addition of pramipexole for bradykinesia and rigidity. His father developed PD in his 70s. Two paternal and one maternal uncle also have PD.
Family 2
The index case of this family (III.2) presented age 42 years complaining of flat-footedness and dragging of the left leg. Examination at that time revealed facial hypomimia and left sided signs including poor arm swing, bradykinesia and intermittent left-sided leg tremor. After 2 years of disease there was bilateral upper limb rest tremor, bradykinesia and cogwheel rigidity, which was more marked on the left side. Levodopa replacement was commenced with good response. He also suffered low mood, obsessive and hypomanic behaviour and drank excessive amounts of alcohol. After 3 years of treatment this man developed dopa-related complications including early “wearing-off”, motor fluctuations and mild peak-dose dyskinesia. Currently, 14 years after disease onset he has severe symmetrical rigidity and severe drug-induced dyskinesia requiring a continuous apomorphine infusion pump. He also suffered a levodopa-replacement addiction, and at one point, in addition to his prescribed medications, he was consuming 100 tablets of madopar (250mg) tablets every week.
In the family history, this patient's mother, who is now deceased, developed Parkinson's disease aged 54 years. Her phenotype was characteristic of idiopathic Parkinson's disease and responded to levodopa. She also suffered from anxiety-depression, treated with thioridazine but not thought to have contributed to her parkinsonism. A maternal grandmother also had the disease, but very little is known about this individual.
Family 3
The index case of this family, I.2 developed PD aged 70. She has a background of mild depression. Currently, 3 years after the onset of the first symptom of PD, she has rest tremor in the right arm with mild, asymmetrical right-sided rigidity and bradykinesia. She continues to benefit from half-Sinemet CR four times daily and amantidine 100 mg BD. She also has lower limb peak-dose dyskinesia and has reported occasional visual hallucinations. This patient has one daughter with PD, on whom there is no data.
Family 4
Two family members (III.1 and the II.2), carry a T2356I mutation and at the last examination eight years ago, had postural tremor (without rest tremor) and subtle extrapyramidal features.
III.4 who did not carry the LRRK2 mutation developed symptoms of a left sided rest tremor of the hand aged 44 years. She was initially treated with trihexyphenidyl hydrochloride, primidone and propranolol without benefit. Within 7 years, the tremor worsened and she also developed rest tremor of her right hand, jaw, tongue and left leg. Currently, this tremor has rest, postural and action components, and both postural and action tremor improve with alcohol consumption. She also has left sided rigidity and bradykinesia. All showed a good response to levodopa. Seven years after disease onset the dose of levodopa was 300 mg.
III.3 carries the mutation but was reported to be healthy at 55 years.
Family 5
The index case in this family, II.2 presented aged 67 years with asymmetric bradykinesia, rest tremor and micrographia. She later developed rigidity and had a very good response to levodopa. At 81 years of age, she was taking 300 mg of levodopa, and selegiline and suffered mild peak-dose dyskinesias.
A sibling, II.7 who did not carry the LRRK2 mutation, developed asymmetric micrographia and bradykinesia aged 54 years and represents a phenocopy. He later developed unilateral rest tremor and had an excellent response to levodopa. When last examined, 12 years following the onset of PD, he required 1200 mg of levodopa. He has also developed dyskinesias. The remaining four siblings in the kindred did not have signs of PD including II.5 who carried a mutation, and another who had a wild-type genotype.
We wish to thank colleagues in the Department of Molecular Neuroscience: Asra Siddiqui, Victoria Stinton, Tunde Akinbode, Elizabeth Graham (deceased), Peter Dixon and Catherine Woodward for extremely helpful discussions throughout this work. We also wish to thank Hope McDevitt, Stella Ahier and Andrew Blyth for their expert help with PET scanning. We thank all family members who have agreed to participate in research over the past 10 years. N.L.K. is funded by the Parkinson's Disease Society, the Brain Research Trust and a Doris Hillyer Award from the British Medical Association. J.M.L. is supported by a research contract with UCB Pharma. N.W.W. acknowledges support from the Brain Research Trust, the Parkinson's Disease Society and the Medical Research Council.
References
Adams JR, van Netten H, Schulzer M, Mak E, McKenzie J, Strongosky A, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation.
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations.
Daniel S, Hawkes C. Preliminary diagnosis of Parkinsons' disease using olfactory bulb pathology.
Dekker MCJ, Bonifati V, van Duijn CM. Parkinson's disease: piecing together a genetic jigsaw.
Di Fonzo A, Rohé CF, Ferreira J, Chien HF, Vacca L, Stocchi F, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease.
Doty R, Deems D, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurological signs, disease stage or disease duration.
Doty R, Shaman P, Dann M. Development of the University of Pennsylvania Smell Identification Test: a standardised microencapsulated test of olfactory function.
Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician.
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1.
Gibb W, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease.
Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, et al. A common LRRK2 mutation in idiopathic Parkinson's disease.
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A, Jain S, Meyer-Lindenberg A, Evans EW et al. Clinical and positron emission tomography of Parkinson's disease caused by LRRK2.
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations.
Katzenschlager R, Lees AJ. Olfaction and Parkinson's syndromes: its role in differential diagnosis.
Katzenschlager R, Zijlmans J, Evans A, Watt H, Lees AJ. Olfaction distinguishes vascular Parkinsonism from Parkinson's disease.
Kay DM, Kramer P, Higgins D, Zabetian CP, Payami H. Escaping Parkinson's disease: A neurologically healthy octogenarian with the LRRK2 G2019S mutation.
Khan NL, Valente E, Bentivoglio A, Wood NW, Albanese A, Brooks DJ, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-Dopa PET study.
Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series.
Lesage S, Ibanez P, Lohmann E, Agrid Y, Durr A, Brice A. The G2019S LRRK2 mutation in autosomal dominant European and North African Parkinson's disease is frequent and its penetrance is age-dependent.
Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, et al. LRRK2 pathogenic substitutions in Parkinson's disease.
McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop.
Nicholl D, Vaughan J, Khan NL, Ho SL, Aldous DE, Lincoln S, et al. Two large British kindreds with familial Parkinson's disease: a clinico-pathological and genetic study.
Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, Van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease.
Samii A, Markopoulou K, Wszolek ZK, Sossi V, Dobko T, Mak E, et al. PET studies of parkinsonism associated with mutation in the alpha-synuclein gene.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson's disease.
Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions,
Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant Parkinsonism with pleomorphic pathology.
Author notes
1Department of Molecular Neuroscience, 2Sobell Department of Motor Neuroscience and Movement Disorders, 3Queen Square Brain Bank, Institute of Neurology, 4Reta Lila Weston Unit of Neurological Studies, Royal Free Hospital and University College Medical School, 5MRC Clinical Sciences Centre and Division of Neuroscience, Faculty of Medicine, Imperial College, Hammersmith Hospital, London, 6Exeter University, Exeter, 7Department of Neurology, Addenbrooke's Hospital, Hills Road, Cambridge, 8Department of Neurology, Queen Elizabeth Hospital, Birmingham, UK and 9Laboratory of Neurogenetics, National Institute of Aging, National Institutes of Health, Bethesda, MD, USA


{kind=link}
{kind=link}
{kind=link}
{kind=link}