

Abstract
New materials for electrochemical energy storage and conversion are the key to the electrification and sustainable development of our modern societies. Molecular modelling based on the principles of quantum mechanics and statistical mechanics as well as empowered by machine learning techniques can help us to understand, control and design electrochemical energy materials at atomistic precision. Therefore, this roadmap, which is a collection of authoritative opinions, serves as a gateway for both the experts and the beginners to have a quick overview of the current status and corresponding challenges in molecular modelling of electrochemical energy materials for batteries, supercapacitors, CO2 reduction reaction, and fuel cell applications.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction—where are we heading in computational electrochemistry?
Chao Zhang1,2,4 and Jun Cheng3,4
1 Department of Chemistry—Ångström Laboratory, Uppsala University, Box 538, 75121 Uppsala, Sweden
2 Wallenberg Initiative Materials Science for Sustainability, Uppsala University, 75121 Uppsala, Sweden
3 State Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, People's Republic of China
4 Guest editors of the Roadmap.
E-mail: chao.zhang@kemi.uu.se and chengjun@xmu.edu.cn
On the very first pages of his Lecture (notes) on Physics, Richard Feynman already said that the single most important scientific knowledge is 'all things are made of atoms' [1]. Indeed, materials are the foundation of our modern societies. In particular, new materials related to electrochemical energy storage and conversion are the key to electrification and sustainable development of our world. That is why different initiatives have been taken place across the globe, e.g. Energy Frontier Research Centres in the US, the Collaborative Innovation Centre of Chemistry for Energy Materials in China, and the Wallenberg Initiative Materials Science for Sustainability in Sweden.
These new and large initiatives come in sharp contrast with the fact that electrochemistry on its own is a rather old discipline, even just counting from the era of Michael Faraday (1791–1867) instead of tracking back to its origin in physiology. This contrast highlights the motivation and the ambition behind these collaborative efforts, which is to understand, control and design electrochemical energy materials at atomistic precision. In fact, one of the grand challenges is to establish the quantitative relationship between the macroscopic observables such as current and voltage measured in electrochemical experiments, and the structural, dynamical, and compositional evolutions of corresponding bulk materials and interfaces (interphases) at the microscopic scales. In this regard, molecular modelling based on the principles of quantum mechanics and statistical mechanics as well as empowered by machine learning techniques can provide both new physical insights and predictive solutions to this outstanding problem.
When the field first started about 40 years ago, Woods Halley as one of the pioneers wrote that 'Electrochemistry is not widely regarded as a forefront area for the condensed matter theorist. Here we will try to show with two examples that the charged solid-liquid interface at which all the basic phenomena of electrochemistry take place is a promising area for research for condensed matter theory' [2]. Today, we no longer need to convince young theoreticians that molecular modelling of electrochemical energy materials is an exciting field to work on. Nevertheless, the key phrase 'charged solid-liquid interface' does reveal the focus and the uniqueness of our research area.
Electrochemistry involves the interface between electrode and electrolyte, in which the transition between ionic current and electronic current happens [3]. This transition involves the fluctuation of electronic levels and the solvent (electrolyte) molecules. Therefore, it is crucial to have a molecular and dynamical understanding of both electrode and electrolyte materials. In this roadmap, Chan and her team provide a succinct overview of integrating materials modelling, characterization techniques, and machine learning approaches for the development of transition metal oxide-based cathode materials. This is followed by an up-to-date report from Cai on her account of the current status and challenges of modelling metal anodes for rechargeable batteries. To explore the low-cost and sustainable electrode materials, Araujo and his collaborators present a snapshot of the molecular modelling-assisted design of organic electrode materials using both the density functional theory (DFT) calculations and the surrogate models based on machine learning (ML). Similarly, organic materials can also be used as electrode materials for supercapacitors and the recent advances in molecular dynamics (MD) simulations of metal-organic frameworks with the constant potential method are summarized by Feng's group. On the electrolyte side, we have two contributions focusing on liquid electrolytes, in which the ion solvation and its dynamics are crucial for solving the conundrum. The PHENIX team focuses on ionic liquids and carbon-based supercapacitors with an emphasis on the polarizable force fields used in the MD simulations. The joint piece from the Swedish and Japanese teams highlights the difference in the development and the modelling of liquid electrolytes for multivalent batteries as compared to more matured lithium-ion batteries.
As distinguished from energy storage systems, the focus of electrochemical interface modelling in energy conversions, such as CO2 reduction reaction (CO2RR) and fuel cell reactions, lies on local reactivity. This is particularly true for single-atom electrocatalysis, as laid out and discussed by Xiao's group, where the strong static correlation plays an important role and high-level wavefunction methods are often necessary for quantitative predictions. On a similar note, Eikerling provides a holistic view of the role of theory and computation in describing the local reaction environment within the context of the oxygen reduction reaction in polymer electrolyte fuel cells. This comes together nicely with a multi-scale perspective on fuel cell modelling from Jinnouchi, in which ML-based simulation techniques are anticipated to bridge the first-principles method and the coarse-grained MD simulations.
In addition to the central position of solid–liquid interfaces in computational electrochemistry regardless of whether it is for energy storage or conversion applications, the second aspect of the key phase mentioned above is 'charged' or electrified (interfaces). This is the core difference in molecular modelling of electrochemical energy materials, as compared to other branches of computational (theoretical) chemistry or materials modelling. The boundary condition matters in computational electrochemistry as much as it does in potentiostatic, galvanostatic, or coulostatic measurements in electrochemical experiments. In this regard, we have two contributions to the method developments of grand canonical DFT and DFTMD from Melander with Kastlunger and Bouzid with Pasquarello respectively. This comes hand-in-hand with perspectives on how to reduce the computational cost of describing the solvent (electrolyte) degrees of freedom from Kim's group with a quantum mechanics/molecular mechanics approach and from Schwarz and Sundararaman on implicit solvation models.
Overall, this roadmap originating from 20 groups in 11 countries serves as a gateway for both the experts and the beginners to have a quick overview of the current status and corresponding challenges in molecular modelling of electrochemical energy materials for batteries, supercapacitors, CO2RR, and fuel cell applications. It is not intended to be a comprehensive review, rather they are opinions of leading experts in their respective domains. Therefore, topics such as solid electrolytes (both ceramic and polymer), descriptor engineering of electrocatalysts and their ML discovery, are not included in this collection and can be found elsewhere [4–8].
Looking into the future, we are optimistic about the acceleration brought by ML techniques to molecular modelling. Being hopeful about ML techniques, it is also clear that the physics-based approaches will still play a central role in tackling unconventional systems (e.g. non-pristine electrode surfaces in the double layer modelling) [9] and generating new inspirations (e.g. the connections between different types of constant potential simulation techniques used in molecular simulation and electronic structure calculation communities) [10]. Keeping these in mind, our field will contribute more significantly to the R&D of new materials for electrochemical applications; the common physical chemistry principles behind a plethora of seemingly different experimental phenomena can be revealed and understood; and the gap between theory and experiment will be further narrowed down with collaborative efforts at both national and international levels.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Unions Horizon 2020 research and innovation programme (Grant Agreement No. 949012). This work was partially supported by the Wallenberg Initiative Materials Science for Sustainability (WISE) funded by the Knut and Alice Wallenberg Foundation (KAW). J C is grateful for the funding support from the National Natural Science Foundation of China (Grant Nos. 21861132015, 21991151, 21991150 and 22021001).
2. Modelling and characterization of transition metal oxide electrodes
Yiming Chen and Maria K Y Chan
Center for Nanoscale Materials, Argonne National Laboratory, 9700 Cass Ave, Lemont, IL 60439, United States of America
E-mail: mchan@anl.gov
Status
Transition metal oxides (TMOs) have been playing crucial roles in the family of Li-ion battery cathodes since the 1980s when layered LiCoO2 was developed [11]. As its name indicates, TMO is a group of materials that adopt a general formula of Lix My Oz where M indicates single or multiple TM elements. There are three major structural prototypes for TMO cathode materials: layered, spinel, and disordered rocksalt (DRX). Layered TMOs usually possess alternating layers of TM-O2 octahedra and Li along the c-axis. Spinel materials, such as well-investigated LiMn2O4, mostly have a cubic lattice symmetry where oxygen anions form a face-centred cubic lattice. DRX which has Li and TM mixing in its cation site has attracted significant attention in recent years.
In addition to electrochemical experiments, modelling and characterization are vital to explain the material properties of TMO electrodes (as shown in figure 1). Modelling methodologies, including first principles density functional theory (DFT) calculations, molecular dynamics or Monte Carlo simulations, and continuum approaches, have been proven to be reliable and versatile tools to understand these properties. For example, DFT calculations are widely applied to predict the voltage profiles, structure stability, and diffusion barriers based on thermodynamics and kinetics [12]. Molecular dynamics and Monte Carlo simulations are used to understand lithium diffusion dynamics and thermodynamic ground states. Characterization techniques, including microscopy and spectroscopy, have become indispensable to identifying materials properties and explaining mechanisms. In-situ and operando measurements allow researchers to identify dynamic evolution during cycling and synthesis. In recent years, simulated characterization techniques have emerged as a valuable complement to experimental ones to explain data and identify mechanisms [13–15].
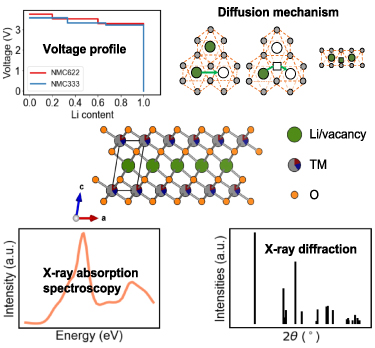
Figure 1. Schematic diagram for modelling and characterization of TMOs. The top panel represents a few typical properties that can be solved with modelling tools. The middle panel showcases the crystal structure of a typical TMO, NMC material. The bottom panel exemplifies several characterization techniques that are widely used for TMO studies.
Download figure:
Standard image High-resolution imageWith those tools, the investigation of TMO electrodes has gained substantial progress toward commercialization. For instance, LiNix Mny Co1−x−y O2 (NMC) was first discovered in 2001 [16, 17] and is now one of the prevailing cathode materials in high-energy applications such as electric vehicles. Yet, there still exists considerable room for improved understanding and performance with the help of modelling and characterization techniques.
Current and future challenges
An ideal cathode for lithium-ion batteries needs to satisfy the following requirements: high gravimetric and/or volumetric energy density for the appropriate storage application, high rate capability to allow fast charging, low cost of production for large-scale commercialization, and stability for safety and long-term cyclability. Unfortunately, there exists no single material that can satisfy all requirements. In the family of TMOs, spinel materials have excellent rate performance because of the 3D percolating diffusion channels. However, its low energy density makes it less appealing to high-energy applications. DRX has the highest energy density among all TMOs [18] but it also suffers from materials degradation. On the other hand, layered TMOs have high energy density and good stability, explaining its popularity in the industry. The common problem for layered TMOs lies in their usage of Co, which is becoming increasingly expensive and raises ethical issues during Co mining processes.
One of the biggest obstacles for TMO cathode improvement is understanding their degradation mechanisms. In NMC in particular, with an increasing effort to replace costly Co with cheaper and more abundant Ni, NMC gains structural instability that leads to defects and performance decrease. Although defects in NMC are well-observed in experiments, its formation mechanism and how it impacts battery performance remain inconclusive. In addition to experimental efforts, computational material scientists have been solving this problem using first principles calculations [19, 20]. Both studies unveiled the relevance of the lithiation state to the degradation of NMC cathodes during electrochemical cycling. One study explicitly explored its impact on mechanical properties, such as elastic modulus and hardness, while the other one investigated how the formation of defects can be related to delithiation. First principles calculations revealed that Jahn–Teller distortion, depletion of electrostatic interactions of Li–O, and weak ionic TM–O bonding contribute to the change in mechanical properties as Li is extracted from NMC materials. Simultaneously, the reduction in Li content also diminishes the formation energy of defects, including oxygen vacancy, thereby increasing the likelihood of defect formation. In particular, the role of oxygen instability was found by DFT to be particularly enhanced at grain boundaries [21].
Another challenge lies in the oxidation state change during electrochemical cycling. With advanced characterization techniques such as x-ray absorption spectroscopy, researchers can qualitatively determine the oxidation states of TM in different states of charge [22, 23]. However, such changes are not always straightforward to interpret from spectral data. Scientists are interested in the complex interplay between TM redox and oxygen redox [24], details of the atomistic processes involving degradation and oxygen instability, the role of defects, and the effect of stoichiometry on the above.
Advances in science and technology to meet challenges
Advances in modelling, in concert with advances in advanced characterization, are needed to tackle the above challenges. In modelling, the need to model larger simulation cells due to cation order and disorder as well as grain boundaries, and to dynamic simulations covering ion diffusion and degradation time scales, requires the use of efficient interatomic potentials such as those trained using machine learning methods. The use of high throughput simulations together with ML will also allow the discovery of new compositions with desired properties. On the characterization side, the development of spatially and temporally resolved diffraction and spectroscopy (x-ray and electron) will allow the simultaneous, multi-modal characterization of TMO cathode materials. For instance, Quilty et al demonstrated the necessity to use multiple characterization techniques to unveil the full picture of the capacity fade of NMC in real time [25].
Moreover, with an increasing trend to embrace open-source data and code, data sharing will significantly enrich both the data quality and quantity available to the public, spurring the usage of machine learning methods in the electrochemistry field. Researchers can not only train more reliable machine learning models but also can develop novel machine learning architectures that are limited by the data size beforehand. For instance, a recent work by Chen et al employed random forest models to discern the fundamental driving forces of battery aging modes, achieving an accuracy of 86% in classifying aging modes. This investigation revealed that a combination of active materials loss and a reduction in Li inventory predominately governs the aging behaviours observed across diverse NMC compositions [26]. Such machine learning applications will pave the way to understanding materials aging and diminishing performance degradation of NMC materials.
The most exciting future developments involve the intersection of modelling, characterization, and ML. Using computational spectroscopy and microscopy, ML models and iterative learning approaches will allow real time inference on experimental characterization data [27], allowing understanding of redox reaction and degradation mechanisms, and even spatial inhomogeneity of these mechanisms with mapping techniques.
Concluding remarks
The computational materials science field has come a long way towards predictive modelling of TMO cathode materials. The semi-quantitative prediction of voltages and stability of cathode materials from DFT calculations is now a matter of routine. The frontier has moved towards the discovery, understanding, and improvement of existing and yet-to-be-discovered classes of TMO, aided by advanced characterization, computational microscopy and spectroscopy, as well as machine learning approaches.
Acknowledgments
This work is supported by the U.S. Department of Energy (DOE) Office of Science Scientific User Facilities project titled 'Integrated Platform for Multimodal Data Capture, Exploration and Discovery Driven by AI Tools'. M C acknowledges the support from the BES SUFD Early Career award. Work performed at the Center for Nanoscale Materials, a U.S. Department of Energy Office of Science User Facility, was supported by the U.S. DOE, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
3. Metal anodes for rechargeable next-generation batteries
Qiong Cai
School of Chemistry and Chemical Engineering, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford GU2 7XH, United Kingdom
The Faraday Institution, Quad One, Harwell Campus, Didcot, OX11 0RA, United Kingdom
E-mail: q.cai@surrey.ac.uk
Status
Utilizing metals as anodes is an ultimate solution towards achieving high energy density rechargeable batteries, due to their high storage capacities and low electrochemical potentials. Among the studied metals (Li, Na, K, Zn, Mg, Ca, Al), Li metal anode (LMA) is considered as the most promising due to the extremely high theoretical capacity and low reduction potential (see figure 2), for next-generation batteries such as Li-metal batteries, Li–oxygen batteries, and Li–sulphur batteries. LMAs started in 1970s but were mainly used for primary Li metal batteries. The roadblocks for the application of LMAs in researchable Li batteries come from the formation and growth of Li dendrite, the loss of active Li, and the huge volume change during the repeated charge and discharge process, causing breakage of solid electrolyte interface (SEI), significant capacity fade, piercing of separator, short circuit, and fatal failure of batteries [28]. Therefore, Li-ion batteries utilizing stable graphite as anodes have been dominant in practical applications.
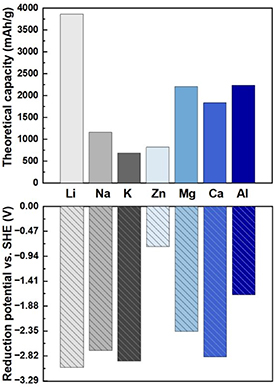
Figure 2. The theoretical capacity and reduction potential of different metal anodes.
Download figure:
Standard image High-resolution imageNevertheless, the promises and challenges of LMAs have attracted significant research interest and especially extensive research efforts over the past decade, driven by the desire to improve the energy density of Li batteries for longer driving range of electric vehicles. Through intensive research, four strategies have been proposed to enable longer cycle life for LMAs: (1) novel electrolyte additives to regulate the Li mobility and suppress the Li dendrite growth [29]; (2) novel separators/solid electrolyte with high mechanical modulus and flexibility to suppress Li dendrite and accommodate the big volume change [30, 31]; (3) interface engineering to design protective layers at the metal surface [31, 32] hosting structures to stabilize LMAs [32]. These strategies have prolonged the cycle life to above 100 cycles; longer cycle life of 500–1000 cycles might be achievable with continuous research and breakthrough.
Recently, Na metal anodes (NMAs) [33] and K metal anodes (KMAs) [34] have gained increasing interest due to their abundance, low cost, and sustainability. NMAs and KMAs are still in their infancy, showing even more severe capacity fade with more un-controlled dendrite growth and different dendrite morphology/properties. More research efforts are needed for developing NMAs and KMAs, with the anticipation that the strategies for LMAs would also be effective but different materials solutions might be required for each metal anode. Although multi-valent metals (i.e. Zn, Al) have achieved much longer cycle life than alkali metals [35, 36], it lacks a good understanding on their exact working mechanisms during the charge/discharge process and why they behave differently from alkali metals.
Current and future challenges
To design high-performance metal anodes, it is pivotal to understand the fundamental mechanisms of dendrite formation and growth. However, the high reactiveness of metals (especially alkali metals) and the interfacial process at the anode–electrolyte interface present great challenges for experimental techniques [37]. Microscopy techniques (e.g. optical microscopy, cryogenic electron microscopy) can directly observe the meso-scale dendrite growth process during battery operation, whilst electrochemical measurement (e.g. electrochemical impedance spectroscopy, galvanostatic intermittent titration technique) can be used to interpret the likely processes at metal anodes [38]. The atomic-scale mechanisms, which are difficult to probe using experimental techniques, have been poorly understood.
Atomistic modelling using density functional theory (DFT), molecular dynamics (MD), and ab initio molecular dynamics (AIMD) simulations can provide insights into atomic-scale mechanisms (see figure 3). So far, atomistic modelling activities are centred around the following areas: (1) classical DFT calculations to determine bulk and surface properties of metal anodes, surface energies and thermodynamic stability of crystal faces, adsorption energies on the crystal surfaces [39], diffusion pathways and diffusion barriers of metal ions on crystal faces, Young's moduli and shear moduli of crystalline metals, the impact of Cu current collectors on the surface diffusion barriers of Li, the formation of coordinated clusters as nucleation seeds on Li and Cu surfaces, the nucleation barriers and nucleation overpotentials [40]; (2) potential-dependent grand canonical DFT simulations to investigate the thermodynamic origin of different dendrite growth regimes (e.g. epitaxial, mossy, fractal) and SEI formation, linking with intrinsic metal surface properties [41]; (3) MD simulations to study the grain boundary (GB) structures of the polycrystalline metal anodes, Li diffusion through the GB structures, atomic-scale structure mismatch between Li metal and Cu current collectors, generation of vacancies [42]; (4) large-scale MD simulations to study the defects and pore formation within Li metals at the Li metal-solid state electrolyte [43]; (5) AIMD simulations to study the reactions of metal anodes with electrolyte and the formation of SEIs [44, 45].

Figure 3. Schematic illustrating the complex processes happening at metal anodes.
Download figure:
Standard image High-resolution imageMost of the atomistic modelling work has been on LMA systems, with very limited work on Na and K, and multivalent metals. A systematic approach is needed to compare different metals which will provide insights in how the chemical and physical nature of different metals dictates different behaviours of metal anodes. To better understand dendrite formation and growth process, bigger nucleation clusters beyond tens of atoms need to be simulated to identify the critical nucleation size and track the initial dendrite morphology change. Furthermore, the model systems need to explicitly include the influence of the realistic battery environment (e.g. presence of electrolytes, potential SEI layers, potential defects, charge/discharge processes). However, such systematic and thorough studies are computational demanding and require huge computational power and time.
Advances in science and technology to meet challenges
Significant development and improvement of computational power and atomistic modelling methods (e.g. involvement of smart machines or robotic techniques to automate the simulation processes) are required, to allow: (1) systematic investigations of different metal anodes to shed lights on fundamental mechanisms of the different behaviours of metal anodes; (2) implementation of model systems that include the fluences of the realistic battery environment with the presence of electrolyte, SEI and defects, battery operation temperatures, charge/discharge process; (3) capture of long-range or time-dependent phenomena such as small to larger nucleation clusters and continuous growth of dendrite during battery operation. With AIMD simulations, it is now possible to study the time-dependent breakdown of electrolyte and formation of SEI at the metal–electrolyte interfaces. The ability to link the SEI with metal dendrite formation and growth will accelerate systematic materials design and simulations of realistic phenomenon.
Correlating atomistic modelling with advanced operando measurements is important to verify the model systems and enhance our understanding of the metal anodes at atomic scale. This calls for development of operando techniques that can probe atomic or nano-scale chemical and morphological information and processes in 3D space as a function of time. At the other hand, atomistic modelling could also be used to help design materials for high performance metal anodes. For example, atomistic modelling could be used to design highly effective 3D host materials for metal anodes to stabilize the plating/striping process during battery operation, and provide fundamental insights into the underlying mechanisms.
The conventional atomistic modelling studies using DFT, MD, and AIMD are computationally intensive and time consuming as they normally involve the examination of a large number of structures and calculations. Machine learning methods could help speed up the atomistic modelling by reducing and optimizing the number of structures and simulations needed, thereby boosting the predictive capabilities and expanding the boundaries of atomistic simulations. Machine learning methods could also help develop force fields based on DFT calculations and experimental inputs, to enable simulations of larger metal anode systems (e.g. metals with grain boundaries, systems containing both metal anodes and SEI layers) [46]. For example, neural network potentials and inverse generative network have been developed for Li-ion batteries and could be adapted for batteries based on metal anodes [44, 46].
Concluding remarks
Atomistic computational research based on DFT, MD, and AIMD simulations provides atomic-scale fundamental understanding of metal anodes and critical processes such as dendrite formation and growth, which cannot be obtained with meso- and macro-scale approaches. The atomic-scale understanding of metal anodes is still limited and patchy, with current studies focusing on small and partial metal anode systems. The full potential of atomistic computational research is yet to be unleashed, for systematic and thorough studies of metal anodes to understand the underlying mechanisms of different metal behaviours. Development of machine learning methods are required to speed up such atomistic simulations. Machine learning methods will be useful in assisting DFT calculations by reducing and optimizing the number of structures and calculations, enabling fast screening. Machine learning methods will also be helpful in developing force fields for large-scale MD simulations of metal anode systems considering more complex and realistic battery environment surrounding metal anodes. Atomistic modelling is foreseen to play a crucial role in helping design high-performance metal anodes systems and providing insights in fundamental mechanisms.
Acknowledgments
Qiong Cai would like to acknowledge financial support by the Faraday Institution through the LiSTAR programme (Grants FIRG014, FIRG058) and Horizon Europe through the OPERA consortium (Grants Number 101103834).
4. Organic electrode materials
Rodrigo P Carvalho1,2, Cleber F N Marchiori3, Daniel Brandell2 and C Moyses Araujo1,3
1 Materials Theory Division, Department of Physics and Astronomy, Uppsala University, Box 516, 75120 Uppsala, Sweden
2 Department of Chemistry—Ångström Laboratory, Uppsala University, Box 538, 75121 Uppsala, Sweden
3 Department of Engineering and Physics, Karlstad University, 65188 Karlstad, Sweden
E-mail: rodrigo.carvalho@physics.uu.se, Cleber.Marchiori@kau.se, daniel.brandell@kemi.uu.se and Moyses.Araujo@kau.se
Status
Organic electrode materials (OEMs) are arising as promising alternatives to enable next generation battery technologies thanks to a unique combination of key sustainability aspects (e.g. green chemistry synthetic routes, renewable resources and easier end-of-life treatments) and molecular versatility (a large possible variation of chemical compositions and structures) [47–50]. While such materials have been explored for a long time [51], the success of the inorganic counterparts has to some degree hampered the investigation and further development of OEMs. Now, with the enormous increase in the demand for sustainable batteries, the research on OEMs has been revitalized, and is emerging as a subfield within battery research and technology. Due to limitations in volumetric energy density, these are primarily targeting cells for large-scale energy storage, while using printed electronics and other cost-efficient techniques.
OEMs are usually grouped as p-, n- or bipolar-type materials depending on their charge state in the redox reactions [48]. For instance, during battery cycling the n-type redox units will change reversibly between the negatively charged (O− ) and neutral (O) states while the p-type ones change between neutral (O) and positively charged (O+ ) states. The direction of such reactions will determine whether they become anode or cathode active materials. These definitions and the most common chemistries are displayed in figures 4(a) and (b), respectively. OEMs are generally not very ion-specific, so that progress achieved for Li-ion battery materials would generally also impact other battery chemistries, e.g. Na-ion batteries [48].
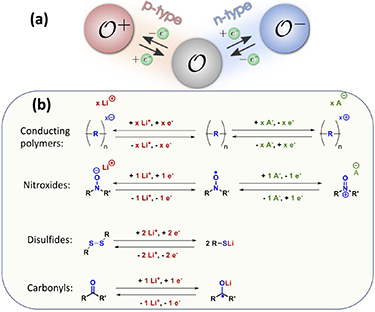
Figure 4. (a) Definition of p- and n-type materials ([48] John Wiley & Sons. © 2018 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) and (b) most common redox chemistry in organic materials (reprinted (adapted) with permission from [52]. Copyright 2016 American Chemical Society).
Download figure:
Standard image High-resolution imageThe most straight-forward OEMs are based on low-Mw crystalline compounds with different redox-active groups incorporated. Since this gives rise to molecular packing and a high concentration of electrochemically active sites, the capacity is comparatively high. In fact, exceptionally high levels of reversible ion insertion have been reported for unsaturated compounds for a phenomenon called 'superlithiation' [52]. Furthermore, they display fairly stable electrochemical potentials spanning between 0 and 4 V vs. Li+/Li depending on the used electroactive moiety, and thus rendering it possible to construct high-voltage full cells.
The major issue plaguing the development of low-Mw OEMs are their easy dissolution in most common electrolyte systems, rendering significant capacity fade of the corresponding cells. One apparent compromise is thereby to copolymerize organic redox-active unit by attaching it to a polymer backbone [53, 54] or turn the conjugated backbone itself redox-active [55–58]. The polymer-based electrodes bring also other advantages concerning mechanical flexibility, easy casting and film formation and eventually electrochemical stability. However, this renders a significant compromise with the energy density.
The fundamental understanding of the underlying electrochemistry taking place during the charge and discharging of OEMs is still lacking. Advanced atomic-scale modelling approaches are here playing a prominent role. However, these are still limited considering the relevance of the research field. Earlier works have employed molecular modelling within density functional theory (DFT) to investigate the thermodynamics of redox process, and has provided some relevant insights [59–63]. However, the need for a proper modelling of the crystalline environment has prompted a combination of DFT with crystal structure prediction approaches (e.g. evolutionary algorithms) in a number of studies [64–67]. Moreover, recent studies have been exploring artificial intelligence methodologies to accelerate the discovery of novel OEMs [68–70].
Current and future challenges
There are still fundamental issues related to energy density, rate capability and cycling stability that need to be resolved for OEMs to become a competitive technology. Such drawbacks are intrinsically connected to the low concentration of redox-active units, low electronic and ionic conductivity and high reactivity during battery cycling.
Rational design methodologies need to be developed to explore the huge chemical space provided by the organic realm [71] in order to accelerate the discovery of suitable OEMs. This could only be achieved through a fundamental understanding of the electrochemical reactions at molecular level, which remains as a challenge. The nature of the organic compounds makes it difficult to employ the standard experimental techniques used for inorganic battery electrodes, e.g. operando-XRD or XPS to explore the bulk structure and solid electrolyte interphase, respectively. The different elements (dominated by C, N, O) can be difficult to distinguish from each other with electron scattering techniques, the crystallinity is often incomplete, and the elements are similar to those used in the electrolytes and electrode additives. Moreover, synthesis and characterization of the huge amounts of organic materials theoretically available, to fully explore all possibilities, would certainly be unfeasible.
There are several problems to resolve for the critical understanding of the bulk properties of OEMs during battery operation. This is at present a bottle-neck for materials development, since the interplay between chemical composition, OEM structure and electrode morphology and the resulting electrochemical properties are far less developed than for inorganic counterparts. Key issues regard reaction kinetics and energetics of the OEMs, mass transport and structural chemistry. The operating voltage is controlled both through the electroactive moiety in the compound and its chemical surrounding, while the rate performance is controlled by ionic and electronic transport. Their interrelations are far from being properly elucidated.
Yet one property that requires considerably more attention are the interfacial properties of OEMs in battery cells. These will control the possible decomposition reactions of both electrolyte (as for solid electrolyte interface (SEI) layer formation for inorganic counterparts) and the electrode materials themselves, while they will also strongly affect the dissolution of active material into the electrolyte. Such interfacial chemistry of OEMs has only started to be mapped, and only for a few selected systems. The field is decades behind that of inorganic counterparts. There is thus a great opportunity here for the development of modelling approaches at different scales in space and time.
An important development was the implementation of evolutionary algorithms interplayed with DFT calculations to resolve the crystal structure (without need of experimental inputs), which allowed a more fundamental understanding of the solid-state electrochemistry of OEMs [64–67]. However, this constitutes a computational demanding strategy that hinders its use for more extended high-throughput screening purposes. In this sense, there is a need for alternative methodologies to explore the vast number of possible candidates. Surrogate models to fast and efficient prediction of the materials properties, based on artificial neural networks, have been developed [68–71]; but it is still a challenge to achieve accurate results with small training datasets. Another great challenge in this field is the development of multiscale models to assess the electrochemical properties of the amorphous and crystalline polymeric electrodes. This is in fact much needed as it is known that organic materials can display polymorphism [72], be completely amorphous as is the case of polymeric materials, and even lose crystallinity upon cycling.
Advances in science and technology to meet challenges
The interplay between first-principles modelling (based on DFT) and artificial intelligence techniques (e.g. machine learning and evolutionary algorithms) is opening new horizons in materials science by accelerating the discovery of novel compounds. These data-driven methodologies are already having an impact on the design of novel OEMs and they will continue playing an important role in the consolidation of these technologies. Figure 5 displays a workflow of such computational materials design approach that we have recently proposed [68]. This AI-driven design was extended to post-lithium chemistries and also to provide an initial assessment of the materials redox stability [69].

Figure 5. Workflow for the AI-driven in-silicon design of novel OEMS (reproduced from [68]. CC BY 4.0). Two datasets are developed (crystals and molecules) to train the AI-kernel including linear and neural models, which in turn facilitate the high-throughput screening of millions of molecules.
Download figure:
Standard image High-resolution imageIn a first assessment, the gas-phase molecular modelling has given important insights about the structure-properties relationships. For instance, it successfully described the thermodynamics trend for superlithiation process in dilithium benzene-dipropiolate (Li2BDP) [52]. In a broader context it has also been employed to assess lithiation limits of high-capacity organic battery anodes in combination with so-called atomic charge derivative analysis [73]. The molecular modelling will certainly continue being developed to acquire knowledge on the electrochemical trends using cheaper computational methods.
The evolutionary algorithms in connection with DFT calculations will be further explored as a powerful tool to determine the crystal structure when having only the chemical composition as input. Here, by starting with an initial set of randomly generated crystal structures, the geometry optimization is performed using DFT calculation and the free energy of each structure is computed. The energetically most favourable structures are selected to form the first population of individuals. With those individuals, a series of evolutionary inspired operations are performed to form the next generation of individuals. This is followed by another round of DFT based geometry optimization and energy evaluations. This procedure is repeated until the halt criteria, e.g. variation of free energy, is reached. Such methodology must be connected with less costly computational methods like tight-binding DFT to explore more complex systems and larger materials libraries. Novel solid-state approaches allowing the investigation of metastable structures may also be further advanced [74].
Molecular dynamics (MD) simulations will also be critical to understand both the interplay between atomic-scale structure and dynamics in these complex systems, as well as elucidating transport processes in electrodes and in the active materials itself, which is largely unexplored today. There is plenty of force field development made for organic compounds from simulations of biological systems which are adequate to use for OEMs, while AI-developed force fields could also provide useful solutions in the near future. The simulation of interfacial chemistry could be especially rewarding, where a rapid methodology improvement has been seen for inorganic counterparts in recent years, but should be transferred also to OEM-based systems.
Concluding remarks
The great demand for the development of sustainable battery technologies has revitalized the research on organic electrode materials. It is in turn prompting the development of novel molecular modelling approaches not only to achieve the fundamental understanding of their electrochemistry but also to accelerate the discovery of new materials. For instance, DFT based calculations have been interplayed with evolutionary algorithms to resolve the crystal structures at different Li-ion insertion stages while surrogate models, based on artificial neural networks, have been developed to predict materials properties at much lower computational costs. The latter has made it feasible to explore a vast materials library and it has been integrated in a novel computational materials design platform for OEMs. Molecular modelling is foreseen to continue playing an important role in the consolidation of these technologies. Multiscale approaches should be further developed to investigate the morphology effects establishing the structure-properties relationships. Furthermore, the interfacial modelling of OEMs in battery cells is also very important to understand the possible decomposition reactions of both electrolyte and the electrode materials themselves, which may lead to formation of SEI. Finally, MD simulations, using AI-developed force fields, will be critical to understand ion transport mechanism in these complex systems.
Acknowledgments
The authors acknowledge support from the Swedish Research Council (Grant Numbers 2018-04506 and 2020-05223), the Swedish Energy Agency (Grant Number. 45420-1) and STandUP for Energy.
5. MOF-based supercapacitors
Ming Chen, Xiangyu Ji and Guang Feng
State Key Laboratory of Coal Combustion, School of Energy and Power Engineering, Huazhong University of Science and Technology, Wuhan 430074, People's Republic of China
E-mail: gfeng@hust.edu.cn
Status
Electrical double layer capacitors (EDLCs), also called supercapacitors, have played an increasing role in the energy storage community, because they bridge the gap between batteries and conventional capacitors [75]. With the significant progress over the last decades, commercial EDLCs can provide a high power density of approximately 30 kW kg−1 but a moderate energy density (∼10 Wh kg−1), which is noticeably lower than that of lithium-ion batteries (∼300 Wh kg−1) [76, 77]. Therefore, the primary focus of current research and development in the field of supercapacitors is to improve their energy density while maintaining high power density.
Metal-organic frameworks (MOFs), characterized as a class of crystalline sponge-like materials with a periodic network structure formed by metal nodes and organic ligands, have been identified as a type of promising electrode candidates for supercapacitors, on account of their exceptional specific surface area, controllable pore size, and tuneable chemical functional groups [78–80]. As shown in figure 6, owing to the successful synthesis of exceptionally stable and highly porous MOFs in the 1990s [81, 82], tens of thousands of MOFs have been reported and developed [78–80]. However, the majority of these MOFs were characterized as electrical insulators with low charge mobility. Consequently, MOFs have traditionally been employed as sacrificial templates or precursors to yield MOF derivatives as electrode materials [80]. Despite certain advantages (e.g. high conductivity or high specific capacity) obtained from such modification, it can also cause an inadvertent collapse of some unstrengthened MOF-derived electrodes during charging and discharging, deteriorating their electrochemical performance [83, 84].
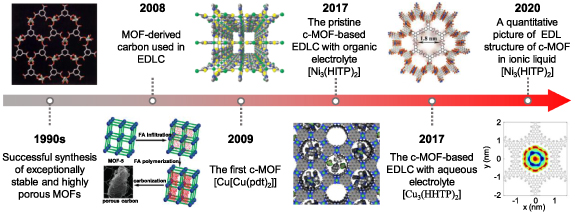
Figure 6. The timeline of MOFs for supercapacitors [81, 84, 85, 88, 89, 91]. Adapted from [81], with permission from Springer Nature. Reprinted (adapted) with permission from [84]. Copyright 2008 American Chemical Society. Reprinted (adapted) with permission from [85]. Copyright 2009 American Chemical Society. Adapted from [88], with permission from Springer Nature. [89] John Wiley & Sons. © 2017 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced with permission from [91]. Copyright © 2020, The Author(s), under exclusive licence to Springer Nature Limited.
Download figure:
Standard image High-resolution imageThe first conductive MOF (c-MOF), named Cu[Cu(pdt)2] (pdt = 2,3-pyrazinedithiolate), was reported in 2009, delivering an electrical conductivity of 6 × 10−4 S cm−1 [85]. Since then, many c-MOFs with distinctive electronic structures have begun to sprout [86]. In particular, a 2D c-MOF, Ni3(HITP)2 (HITP = 2,3,6,7,10,11-hexaiminotriphenylene) was developed to show high conductivity with bulk and film values up to 2 and 40 S cm−1, respectively [87]. When this 2D pristine c-MOF was used as a sole electrode without any binders and additivities, it demonstrated excellent capacitive performance with a specific capacitance of up to 111 F g−1 in organic electrolytes, indicating that pristine c-MOFs can be employed as supercapacitor electrodes without any modifications [88]. This landmark work has opened up a new opportunity for using the pristine MOF as supercapacitor electrodes. Subsequently, c-MOFs with organic ligands formed by triphenylene or hexaaminobenzene have been successfully employed as electrodes with extraordinary capacitive performance in aqueous electrolytes [89, 90]. Meanwhile, the structure and energy storage performance of c-MOF immersed in ionic liquid electrolyte were described quantitatively for the first time through a joint work of molecular dynamic (MD) simulation and experiment [91]. Future advances in this field could potentially see the realization of appropriate c-MOF electrodes in EDLC applications.
Current and future challenges
Despite recent significant advances in research to enhance the capacitive performance of supercapacitors with pristine c-MOF electrodes, their practical realization with high energy storage performance is still in its early stages [92, 93]. The major challenge of c-MOF-based EDLCs lies in the lack of fundamental understanding of the EDL structure formed by the MOF electrode and the electrolyte at the nanoscale and the behaviour of ions confined in complex nanopores, making it intolerable to sift out pleasing c-MOF electrodes through the standard trial and error methods.
Specifically, various combinations of metal nodes and organic ligands would generate dramatically diverse c-MOFs topologies, presenting different pore sizes and pore shapes. The non-ideal pore shape can have a significant effect on the distribution of electrolyte ions inside the pore. Meanwhile, the classical theory based on the ideal-solution model (e.g. Poisson–Nernst–Planck equation) fails to describe the charge storage and ion transport inside nanopores [75, 94]. MD simulations are useful to elucidate the mechanism of the capacitance performance of energy storage properties of the c-MOFs by monitoring the fluxes and electrosorption of the ion in nanopores during the charging and discharging process [80, 94].
In the past decade, the majority of EDLC modelling has been done with the constant-charge method, which can provide a qualitative comprehension of the EDL structure on the ideal surface [95]. However, this approach is limited in its ability to properly reproduce the behaviour of ions within the polarized c-MOF systems, as the partial charges of the c-MOF atoms remain static during the simulation in an unphysical way [91]. The MD simulations with the constant potential method (CPM), where the electrons of the metallic surface respond to the motion of nearby ions to maintain a constant potential at the surface by dynamically adjusting an electrostatic field and in turn affects the behaviour of ions, are required to gain a deeper understanding of the energy storage mechanism in c-MOF-based EDLCs. However, molecular simulations on c-MOF-based EDLCs are still scarce [91]. Moreover, current CPM studies have been limited to the potentiostatic mode, which does not accurately reflect the galvanostatic charge–discharge (GCD) mode widely used in experimental settings. CPM simulations on c-MOF EDLCs with more realistic charging conditions are required for future advances [96, 97].
In addition, the electrochemical stability of c-MOF, as another crucial factor in determining the capacitive performance, has been investigated based on the electronic structure of pure c-MOF via density functional theory (DFT) [86]. However, the effect of electrolytes on the physicochemical properties of c-MOF electrodes under polarization cannot be ignored. An accurate prediction on the electrochemical window of c-MOF-based EDLC remains a virgin area.
Despite the ability of CPM simulations to capture the interfacial structure and ion transport in nanopores of c-MOF electrodes clearly, there is still a gap between the microscopic details of ion transport and the macroscale patterns underlying the charging dynamics. In particular, the time constant of charging obtained from MD simulation deviates significantly from that observed in experiments. This highlights an urgent request to develop multiscale theory or modelling to link microdynamics to macroscopical charging-discharging performance quantitatively.
Advances in science and technology to meet challenges
Understanding capacitive behaviour in c-MOF hosts in favour of the design of innovative electrode materials and constructing sophisticated c-MOF-based EDLC systems. As shown in figure 7, the first atomic-level perception of the EDL structure and ion transport inside c-MOF electrodes has been unravelled via CPM simulation [91]. A quantitative agreement has been achieved between the MD-predicted capacitance and experimental measurements, establishing a link between the EDL structure and energy storage performance to some extent [91]. Besides, on the basis of the conventional CPM method, a GCD-aimed CPM was developed to physically control the electric current and maintain a constant potential across each electrode atom [96]. Further investigation with this advanced MD simulation method (figure 7) is required to further improve the understanding of ion transport behaviour in c-MOF under more realistic charging-discharging conditions, such as the hysteresis in ion adsorption–desorption dynamics during charging and discharging which has been observed in nanoporous electrode experimentally but not observed in c-MOF electrodes yet [96].
Figure 7. Advances in science and technology to meet challenges of electrochemical interface modelling of c-MOF-based supercapacitors. Reproduced with permission from [91]. Copyright © 2020, The Author(s), under exclusive licence to Springer Nature Limited. Reprinted (adapted) with permission from [102]. Copyright 2020 American Chemical Society.
Download figure:
Standard image High-resolution imageMoreover, the physicochemical properties of c-MOF have been extensively investigated via DFT calculation [86]. However, the influence of the electrolytes on the c-MOF electrode was not taken into account. To accurately describe the electrical structure of c-MOF electrodes under charging conditions, an interactive MD-DFT model within the EDL should be developed (figure 7). Specifically, advanced CPM simulations based on a fine-tuned force field are carried out to capture the structures and statistics in the EDL. Then, DFT calculations are employed to extract the physicochemical properties of c-MOF and the corresponding reduction potentials of the representative species in the EDL. In addition to the previously postulated models, an alternative approach involving the amalgamation of quantum mechanical/molecular mechanical (QM/MM) principles can be employed to achieve commensurate functionality [98, 99]. This model elucidates the interface by DFT, while solvent effects are captured through molecular mechanical frameworks [98, 99]. Currently, research on MOFs via QM/MM models predominantly fixates upon the intrinsic properties of MOF materials, such as reaction mechanisms [100, 101]. There is still a lack of research on MOF-based EDLCs via QM/MM models.
To make up for the gap between the charging and discharging characteristics at the nanoscale and power density at the macroscale, rationalizing the MD-obtained charging dynamics is required (figure 7) [94]. One approach to interpreting the charging process is multiscale modelling with an equivalent circuit model, which includes the transmission line model, where the circuit properties are assumed to be distributed continuously throughout the material [91]. Establishing a bridge from the micro to macro level via dimensionless is another feasible method [96].
Although the relationship between structure and performance can be established to a certain degree through the aforementioned MD simulations, DFT calculations, and multiscale analyses, it becomes inefficient to exhaust all possible combinations and to perform effective analysis to identify a few c-MOFs with glorious performance. As an emerging computer technology to train and analyse input data to disclose previously hidden trends, machine learning could offer a promising research direction to speed up the discovery and design processes for the c-MOF electrodes (figure) [80, 102]. It is worth highlighting that a fundamental grasp of the physics and chemistry of these systems is crucial, since decisions cannot be haphazardly left to artificial intelligence.
Concluding remarks
In the pursuit of high energy density and power density of EDLC technologies, c-MOFs with unique properties show emerging potential in supercapacitors as promising electrode candidates. The ordered nanostructure with well-controlled atomistic architectures serves as a platform to establish structure-function relationships and reliably predict their performance. However, modelling electrochemical interface of c-MOF-based supercapacitors is still a challenging task. Further developments in simulation methods, such as the constant potential method with more realistic charging conditions, and MD-DFT model on the electrochemical properties of MOFs, as well as the multiscale modelling are required to advance the understanding of the electrochemical behaviour of MOF-based supercapacitors. Additionally, machine learning is an auspicious tool to accelerate the discovery and design of c-MOF supercapacitors. Therefore, further in-depth and systematic research with the aforementioned methodologies is essential to bridge the gap between the c-MOF laboratory studies and commercial applications.
Acknowledgments
The authors acknowledge the funding support from the National Natural Science Foundation of China (52106090 and 52161135104) and the Program for HUST Academic Frontier Youth Team. M C also thanks to China Postdoctoral Science Foundation (No. 2022T150228).
6. Ionic liquids and carbon-based supercapacitors
Kateryna Goloviznina1, Alessandra Serva1 and Mathieu Salanne1,2
1 Sorbonne Université, CNRS, Physico-chimie des Électrolytes et Nanosystèmes Interfaciaux, PHENIX, F-75005 Paris, France
2 Institut Universitaire de France (IUF), 75231 Paris, France
E-mail: kateryna.goloviznina@sorbonne-universite.fr, alessandra.serva@sorbonne-universite.fr and mathieu.salanne@sorbonne-universite.fr
Status
Supercapacitors are energy storage devices characterized by fast charging/discharging times. This is enabled by a mechanism based on the adsorption of ions from a liquid electrolyte at the surface of high surface area electrodes. Unlike Li-ion batteries, the operation is not slowed down by the diffusion of ions inside the bulk of solid materials. In addition, supercapacitors display larger cycle life. In the absence of Faradaic reactions, and due to the small volume change during operations, the chemical and mechanical stresses are limited. Within a cycle, their performance depends on three characteristic properties, the capacitance, the applied voltage and the total resistance: the two formers have to be maximized while the latter should be kept as small as possible.
For the electrode, carbon-based materials are the best candidates due to their highly accessible surface area, low cost, and good electrical conductivity. The liquid electrolyte is traditionally made of organic ions (e.g. tetraethylammonium tetrafluoroborate) dissolved in acetonitrile, which displays a much larger electrochemical stability window (ESW) than water. When ionic liquids (ILs) were 'rediscovered', supercapacitors appeared quickly as one of the most promising applications. The main advantage of ILs is their wide ESW, which leads to a substantial increase of the applied voltage [103]. In addition, since ILs are made of ions only, they were expected to maximize the charge accumulated at the surface of the electrode, hence the capacitance.
However, the main weakness of ILs is their low fluidity. This is all the more important since it was discovered that carbon materials with narrow pores, of dimension similar to ionic species, yielded much larger capacitances due to the adsorption of desolvated ions [104]. Over the past two decades, most of the experimental work aimed at formulating ILs with optimized properties [105]. In parallel, the charging mechanisms inside carbon nanopores were studied by combining many techniques. In this context, molecular dynamics (MD) plays a prominent role together with in situ spectroscopic and diffraction techniques [106].
Current and future challenges
The theoretical study of bulk ILs has now become routine, yielding accurate structural and dynamic properties, but the study of electrochemical interfaces revealed much more challenging. Conventional double-layer theories were shown to lack many ingredients linked to the highly concentrated characters of these liquids [107]. Introducing them lead to a deeper understanding of the phenomenon at play, and in particular to the discovery of a 'superionic state' in which ions of the same sign pack together inside electrified nanopores [108].
In MD studies, electrodes were first treated as homogeneously charged surfaces and model nanoporous materials such as carbon nanotubes could be studied, yielding a qualitative view on the ion de-coordination mechanisms [109]. To reach quantitative agreements with the experimental capacitances, it was necessary to develop a method in which the electrodes are held at the constant potential to account for the charge polarization at the surface of the electrodes and to use realistic representations of the disordered nanoporous carbons as shown on figure 8(a) [110]. The charging mechanisms of nanopores were shown to be dominated by ion exchange with the bulk, the adsorbed ions being partly decoordinated due to confinement (figure 8(b)). The confirmation of the existence of a superionic state was further validated by in situ diffraction studies [111]. The impact of pore size, geometry, and roughness were also investigated [112, 113] as illustrated in figure 8(c). Concerning the dynamics, the confinement effects lead to a sluggish diffusion of the ions which is enhanced by crowding effects [114]. Efficient charging protocols were proposed to overcome this difficulty [115].
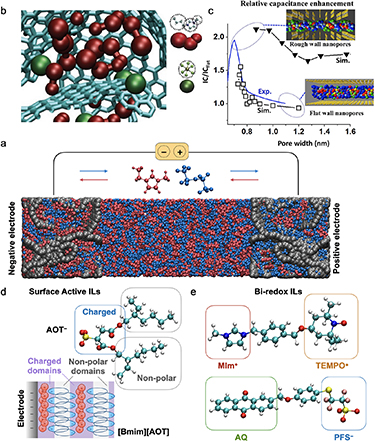
Figure 8. (a) Example of a supercapacitor consisting of the [Emim][TFSI] ionic liquid between two porous electrodes (CDC-1200) held at constant potential. (b) Typical structure of the [Bmim][PF6] ionic liquid inside electrified pores of the CDC-1200 material from coarse-grained molecular dynamics. Reproduced from [110], with permission from Springer Nature. (c) Integral capacitances for atomically flat and rough slit pores as a function of pore dimensions for [Emim][TFSI] ionic liquid. The corresponding simulation snapshots are also shown. Reprinted with permission from [112]. Copyright 2015 American Chemical Society. (d) Schematics of hypothesized ion arrangements for the [Bmim][AOT] surface active ionic liquid at a negatively charged interface held at = −2 V. Adapted from [116], with permission from Springer Nature. (e) Structural formula of the [MIm–TEMPO][AQ-PFS] bi-redox ionic liquid. The ionic liquid moieties are highlighted in red and blue, while the redox moieties in orange and green.
Download figure:
Standard image High-resolution imageIn complement to recent experiments, MD simulations have recently focused on systems made of functionalized ILs. For example, surface-active ILs with amphiphilic structures were proposed for increasing the charge accumulation at the interface (figure 8(d)) [116]. MD simulations showed that this effect was due to the exclusion of the non-polar alkyl tails from the electrode surface. In another work, redox-active ILs were used (figure 8(e)), leading to a two-fold increase of the capacitance [117]. In these systems, the charge is stored inside the ionic species through electron transfer reactions in addition to the charge accumulated on the carbon surface. Here also, simulations could provide insight into the peculiar structure of the ILs: due to the large concentration of ions, and thus of redox groups, the latter adopt a percolating structure throughout the whole liquid that could favour electron transfers beyond the first adsorbed layer [118].
Advances in science and technology to meet challenges
Despite the recent advances, several effects remain only partially addressed in current MD studies, preventing quantitative description of complex ILs at electrochemical interfaces. On the electrolyte side, the interaction potentials were often based on a coarse-grained representation at first, before the advances in computational power and in software efficiency allowed for the use of all-atom force fields. However, polarization effects are known to have a strong impact on the dynamics properties of these systems. In the case of interfaces, they can also change the adsorption properties of the ions, hence the capacitive properties of the devices. It is only recently that an approach allowing to account for the coupling of electrolyte and electrode polarization was introduced in the framework of the Ewald summation method, and implemented in a public software [119].
The next challenge consists in the development of adequate force fields for interfaces. A recipe for parametrizing polarizable potentials is now available, which is based on the most popular force field for ILs [120]. It is now necessary to extend it to the carbonaceous materials used for supercapacitor electrodes. This will require performing careful electronic structure calculations since it was shown using highly accurate reference quantum Monte-Carlo calculations that the various density functional approximations yield very different results for the interaction between a liquid and carbon nanostructures [121]. This is due to the importance of the van der Waals interactions in these systems. Accounting for the flexibility of the carbon materials would also be a significant step forward. Although supercapacitor electrodes do not undergo as significant structural changes as Li-ion batteries ones, the small changes may have consequences on the ion adsorption properties.
An interesting alternative to the polarizable interaction potentials is the use of machine-learning force fields. These have proven very useful for modelling systems, and the case of electrolytes was recently put forward [122]. However, the electrochemical interfaces are a particularly challenging case since electrostatic interactions play a key role, and due to the necessity to account for the electrode potential. Machine-learning force fields often have a short-ranged character, but physics inspired approaches combining them with conventional Coulomb potentials provide an interesting lead [123, 124]. It is very likely that many research efforts will be put in this field over the next years.
Concluding remarks
The modelling of IL-based supercapacitors has certainly played an important role on the improvements of the devices over the years. By providing a more and more precise picture of the structure and dynamics of the ions at electrified interfaces, simulations have provided experimentalists the necessary mechanistic insight to design electrode structures with better performances. The recent advances in the models, as well as the potentialities opened by machine learning, can bring the field even forward, in particular by allowing the simulation of complex functionalized ILs. In addition, the work performed on this topic has benefited other fields, such as the study of electrocatalytic surfaces or of battery interfaces, for which MD is emerging as a new tool to understand the impact of the electrolyte composition and structure on the overall performances.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 771294). It was supported by the French National Research Agency (Labex STORE-EX, Grant No. ANR-10-LABX-0076).
7. Liquid electrolytes for multivalent batteries
Toshihiko Mandai1, Tomooki Hosaka2, Mirna Alhanash3 and Patrik Johansson3,4
1 Center for Green Research on Energy and Environmental Materials, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
2 Department of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo, Japan
3 Department of Physics, Chalmers University of Technology, 412 96 Göteborg, Sweden
4 Alistore-ERI, CNRS FR 3104, 15 Rue Baudelocque, 80039 Amiens, France
E-mail: mandai.toshihiko@nims.go.jp, hosaka@rs.tus.ac.jp, mirna.alhanash@chalmers.se and patrik.johansson@chalmers.se
Status
Multivalent batteries come in different designs and with different promises; less dependent on scarce metals, enabling higher capacities and energy densities, lower price-tags, etc. Here we focus on magnesium (Mg), calcium (Ca), and aluminium (Al) battery electrolytes (figure 9) [125, 126]. Compared to mature battery technologies, i.e. lithium-ion batteries (LIBs), the role of molecular modelling concerning liquid electrolyte R&D is here more diverse. It is used to interpret experimental data, design the molecules to be used—both salts and solvents, address electrolytes for conceptual issues and promises with respect to the cell designs, and screen possible electrolyte materials prior to experimental efforts. While bulk electrolyte studies mostly use strategies and modelling protocols developed for LIBs, metal plating and stripping are fundamental electrochemical reactions of uttermost importance for multivalent batteries. Thus, much modelling effort should be devoted to understanding these mechanisms, especially for the often very special electrolytes employed.

Figure 9. A Venn diagram emphasizing the communalities between types of liquid electrolytes (targeted by molecular modelling) for different multivalent battery technologies: A: Organo-haloaluminates, B: Organic solvent based, and C: Ionic liquid based.
Download figure:
Standard image High-resolution imageTo date, experimental efforts have clearly paved the way for electrolyte development and therefore the interpretation assistance from molecular modelling to understand mechanisms at the electrolyte/electrode interfaces has been a major focus. For Mg batteries, the electrolytes are often still 'classical' organo-haloaluminate based, and the modelling focuses on the speciation in the bulk electrolyte [127, 128], with some notable and recent exceptions focusing on the role of Cl− anions at the electrolyte/electrode interface—i.e. for improved plating (and stripping) [129, 130]. More recently, borate and aluminate anions have been addressed [131], expanding electrolyte composition options for Ca batteries using the very same anions [132] together with the limited set of 'conventional' Ca-salts available [133–135]. For Al batteries, the situation is quite different as plating and stripping is fundamentally less of a problem in conventional AlCl4 ‒ based systems [136]. Despite the large need for Cl− free electrolytes to reduce corrosion issues [137], very few novel electrolytes have been proposed experimentally and computationally. Looking at properties targeted by the molecular modelling, most are physico-chemical, including local structure/coordination (figure 10) [138], solvation [128], and dynamics [127], rather than electrochemical, but there are some notable exceptions on e.g. electrochemical stability windows [130, 139] and electrode passivation [140].
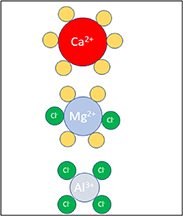
Figure 10. The most common cation 1st solvation shells modelled for Ca2+, Mg2+ and Al3+ electrolytes—showing the important role of Cl− anions for the two latter battery chemistries.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Going forward, it is important to move from basic physico-chemical, speciation and basic ion transport, etc, to more 'real' performance related modelling, including more realistic reactions, dynamics, and kinetics. To do this, we must be able to calculate ion transport under the influence of electric field, include dynamic ion correlation, look at the electrolyte decomposition and passivation on the Mg, Ca and Al electrode surfaces—which in turn demands detailed desolvation processes and activation energies and the kinetics of each of these processes. While we so far have not even mentioned the cathode side, there are also challenges as how to model the electrolyte (active) species both for organic and sulphur cathodes—popular choice is to match the high capacities of the metal anodes and to avoid the sluggish kinetics of hard cations in inorganic intercalation hosts. There are some recent approaches that seem promising in these aspects, such as the grand-canonical DFT calculations for electrochemical reactions [130] and MD simulations with ML (reactive) force-fields (FFs) [141]. Moving from methodology to scientific challenges, the modelling should be able to reveal how, and to some extent explain why, Cl− (Mg and Al) and B (Ca) seem to be ions/elements so crucial for efficient plating & stripping of Mg/Ca/Al at their respective metal anodes. This has already been touched upon by modelling efforts looking at how to prevent passivation [130, 139] and what role ion-pairs in the electrolyte have in the observed decreased overpotential [129, 130]. Another challenge is to use modelling to create electrolyte compositional maps and correlate with the generated electrochemically active species, including equilibria formed species such as AlCl4 −–Al2Cl7 − [136]. Finally, there are also many prospects for other liquid electrolyte designs than the 'traditional' salt-in-solvent. Concepts such as (localized) highly concentrated electrolytes ((L)HCEs) may both enhance functionality and safety, and have recently been modelled for multivalent cation conductors [142]. Another possibility is electrolytes based on deep eutectic solvents, to be further explored by modelling also for multivalent charge carriers [137].
Advances in science and technology to meet challenges
To enable proper and accurate treatment of both structure and dynamics, including reactions at interfaces under influence of electric field and phenomena such as Stern layer and inner and outer Helmholtz layers, better MD FFs are required, ideally also being able to handle and properly describe overpotentials. This is general to the battery field, but due to the inherently slower dynamics and stronger electrostatic interactions combined with the use of metal anodes, AIMD simulations are not feasible. This issue is even more pressing for multivalent battery technologies. Alongside this, there is then need for much more experimental data in order to train ML models and verify these classical FFs needed—all to enable faster simulations and eventually electrolyte design by ML. One route for this would be to do combined experimental and computational high-throughput screening—and ML approaches are indeed expected to both push the limits of how much molecular modelling, especially MD simulations, can contribute as well as bridge the gap to actual electrochemical experiments. However, relying on ML means locking in on concepts where training data is plentiful could prevent further realization of any revolutionary electrolyte designs—that are desperately needed for the Mg, Ca, and Al batteries alike. Hence, other routes allowing unbiased treatment of any kind of chemistry, such as today's popular AIMD and DFT protocols and strategies, will still have a place, and advances in pure computational power may still be a large contributing factor for success.
Concluding remarks
Molecular modelling efforts of (liquid) electrolytes for multivalent battery technologies follow closely both in scope, concepts, and method development that of LIBs. The main notable differences originate in the common use of special counter-anions and metal anodes, and the need to accurately predict and understand the plating & stripping phenomena. Recent advances in modelling methods and computational capabilities are steadily paving the way for modelling the complex electrochemical reactions that occur at the electrode–electrolyte interface. Such advanced molecular modelling has deepened the understanding of electrolyte-dependent electrochemical reactions and has facilitated electrolyte design. However, the paradigm change of AI and modelling truly guiding experimental efforts—the goal of BIG-MAP [143] and the Electrolyte Genome project [144]—is not (yet) a reality.
Acknowledgments
P J acknowledges the support from the Swedish Energy Agency (Grant P50638-1) to M A, the European Union's Horizon 2020 research and innovation programme under Grant Agreement 957189 (BIG-MAP), a part of Battery 2030+, his Swedish Research Council's Distinguished Professor Grant, and Sweden's Innovation Agency (VINNOVA) through Battery Alliance Sweden (BASE). T M acknowledges financial support from the NEXT Center of Innovation Program (COI-NEXT, Grant Number JPMJPF2016) of the Japan Science and Technology Agency and a Grant-in-Aid for Scientific Research (KAKENHI, Grant Number 21K05263) of the Japan Society for the Promotion of Science (JSPS). T H thanks the JSPS for the support through Grant-in-Aid for Scientific Research (KAKENHI, Grant Number 22K14772).
8. Theoretical understanding of single-atom electrocatalysis
Yun-Ze Qiu and Hai Xiao
Department of Chemistry, Tsinghua University, Beijing 100084, People's Republic of China
E-mail: haixiao@tsinghua.edu.cn
Status
Since the concept of single-atom catalyst (SAC) was first coined in 2011 [145, 146], there has been tremendous progress on developing and implementing SACs for a wide spectrum of topics in heterogeneous catalysis including electrocatalysis [147–149]. And the applications of SACs to electrocatalysis have demonstrated great potentials toward industrial deployment for a few vital electrochemical reactions including the hydrogen evolution reactions (HERs), oxygen evolution/reduction reactions (OER/ORR) and CO2 reduction reaction (CO2RR) [149], which compose promising technologies essential to energy and environmental engineering. The remarkable achievements of SAC are likely owing to its appealing merits including high stability, activity, selectivity and atomic efficiency [150], as well as its precise tunability that is a key feature for the rational pursuit of optimal catalysts. The precise tunability of SAC arises from its intrinsic nature by definition, i.e. its active centres are composed of singly dispersed single-atomic sites, so their structures are well-defined and uniform. This lays the very foundation for establishing explicit structure-performance relationships and thus formulating guidelines and theories for designing and optimizing the catalysts. Thus, the SAC presents an ideal design platform that can exploit the synergy between experimental and theoretical investigations to deliver high-performing electrocatalysis that meets the requirements for practical implementations.
In pursuit of the rational design of heterogeneous catalysts, theoretical investigations have been playing an indispensable role in elaborating the atomistic understanding of catalytic performances and formulating descriptor-based structure-performance relationships. This is perfectly illustrated by a dazzling line of research, which paved a road from the scaling relations and Brønsted–Evans–Polanyi relation to the volcano plot that quantifies the Sabatier principle to predict the optimal catalysts [151, 152]. In particular, density functional theory (DFT) calculations were universally employed, and the descriptors from the well-celebrated d-band theory [153] were widely adopted to rationalize the trends in catalytic performance and found great success in predicting highly efficient novel electrocatalysts for HER, OER, ORR, and CO2RR with volcano plots [152]. This research paradigm was seamlessly integrated into the studies of single-atom electrocatalysts (SAECs), in which the theoretical understanding based on DFT calculations has become a necessary component that often covers identification of active site structures, investigation of reaction mechanisms underlying catalytic performance, and preferably descriptor-based guidelines [154]. This routine, however, is confronted with challenges in every aspect from theoretical methods and catalytic mechanisms to formulations of understanding, which necessitate future endeavours (see figure 11).
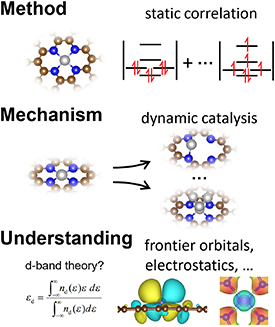
Figure 11. Challenges in theoretical studies of single-atom electrocatalysis including methods to tackle the static correlation, approaches to explore potential-dependent dynamic reaction pathways, and strategies to formulate quantitative understanding.
Download figure:
Standard image High-resolution imageCurrent and future challenges
The unparalleled popularity of DFT calculations in computational electrocatalysis is rooted in the balance between computational cost and accuracy offered by the commonly used exchange–correlation (XC) functionals mostly at the level of generalized gradient approximation (GGA). Besides, DFT calculations are versatile to include empirical dispersion corrections and implicit solvation effects, as well as the applied potential in electrocatalysis using the implicit computational hydrogen electrode (CHE) approach [155] or the explicit grand-canonical DFT method [156]. Explicit solvation and ions can be further included with DFT-based ab initio molecular dynamics (AIMD) simulations [157]. However, conventional XC functionals in DFT are plagued by the delocalization error and static correlation error [158]. The former may be alleviated with +U corrections or hybrid functionals, while the latter requires methods beyond the single-reference Kohn–Sham scheme. Unfortunately, the active sites of SAECs are often composed of transition metal atoms with strong static correlation, and this may greatly compromise the reliability of common DFT results. In quantum chemistry, there are standard high-level wavefunction theory (WFT) methods such as CASSCF and MRCI to tackle the static correlation error, but they are computationally too expensive to be practical in computational electrocatalysis. Therefore, there is a demand for new theoretical methods that are effective in describing static correlation while computationally viable.
In elucidating electrochemical reaction mechanisms, theoretical investigations are often simplified with the CHE approach that enables thermodynamics-only calculations to estimate the overpotentials and thus connect to the catalytic performance. Recently, there have been a growing number of studies that further included kinetics, i.e., transition states and associated barriers, and employed the microkinetic modelling to calculate the current densities that can be directly compared with experiments [154]. Complications in reaction mechanisms by participations of electrolyte species can also be examined with AIMD simulations. However, catalytic reactions at electrochemical interfaces are coupled with the applied potential, and a strong potential-dependence may alter the mechanism underlying catalysis in a dramatic manner [159–165]. A striking example is provided by operando characterizations that identify the reversible transformation of active sites in Cu SAEC from Cu single-atoms to metallic nanoparticles (clustering) under negative potentials in CO2RR conditions, and it is the nanoparticle that is responsible for electrocatalysis [159]. This shakes the common assumption in theoretical studies of reaction mechanisms on SAECs that the single-atomic sites are responsible for electrocatalysis, and calls particular attentions on the coupling between applied potentials and catalytic mechanisms starting from the dynamic transformation of active site structure. This clustering also presents a possible channel for the degradation of SAECs, and future efforts may be necessary to develop systematic strategies/algorithms to generate potential-dependent reaction networks that include the active site transformations, in order to understand both the catalytic activity and stability of SAECs.
In regard of the ultimate goal to design the optimal SAECs, theoretical understanding should provide quantitative guidelines in the form of mathematical functions of structure-performance relationship in terms of descriptor-based variables. This likely requires establishing approximate theories like the d-band theory. However, there are intricacies in formulating such understandings of single-atom electrocatalysis. A recent study showed that the symmetry of frontier orbitals in SACs can play a decisive role in catalysis and this results in the failure of d-band theory in characterizing the catalytic performance of SACs [166]. It was also demonstrated that the electrostatic interactions arising from local atomic heterogeneity around the active sites of SACs can play a significant role in delivering the catalysis [167]. Therefore, delicate considerations may be required in future endeavours to formulate design principles for SAECs.
Advances in science and technology to meet challenges
Quantum embedding methods present a promising approach to tackle the static correlation problem in a cost-efficient manner [168]. The basic idea underlying these methods is to decompose the whole system into subsystems that are treated with different levels of theories. This strategy may be a perfect match for the theoretical modelling of SAEC, where the local structure around the transition metal single-atomic site can be treated with high-level WFT methods, and the rest (support and environment) is described by cost-efficient methods such as DFT or even force fields. Very recently, there have emerged studies applying quantum embedding methods to computational electrocatalysis. A notable case study is from Carter's group [169], in which they applied the approach with embedded correlated wavefunction to study the electrochemical CO2RR on Cu(111) surface. Developments along this line for the studies of SAECs are much-anticipated and should strengthen the synergy between computation and experiments in understanding and designing high-performing SAECs.
AIMD simulations have seen rapidly increasing applications to investigate the dynamic catalysis on SAECs, and found great success in revealing the roles of electrolyte species in electrocatalysis. Nevertheless, exploration of reaction mechanisms by common AIMD simulations is limited to the chemical space predefined by the collective variables (CVs). Subtle complications like the dramatic transformation of active site structures in SAECs might be easily overlooked and missed in CVs, and would not emerge naturally in AIMD simulations as they are rare events. These can be further complicated by the potential-dependences of reaction mechanisms. Very recently, Bai et al [170] elucidated the dynamic evolution of active site structure in Cu SAECs from Cu single-atoms to clusters that is initiated by the adsorptions of H under negative potentials. Future developments toward automatic and comprehensive exploration of potential-dependent reaction pathways can greatly facilitate the pursuit of a true picture underlying single-atom electrocatalysis.
There is a rich toolbox accumulated so far for analysing and extracting intrinsic features in catalysis, as partly showcased in the previous study [167]. Yet it remains challenging to compose relevant descriptors and thus formulate quantitative structure-performance relationships because of intertwined intricacies present in single-atom electrocatalysis, and it relies highly on chemical intuitions. Nowadays, the surge of machine learning (ML) techniques has made a thrilling impact on computational catalysis, and great progress has been made on descriptor-based catalyst discovery with ML techniques [152]. With future advancements toward systematic extraction of chemically intuitive descriptors and automatic formulation of quantitative structure-performance relationships based on physical laws, ML techniques may spark a wave of truly breakthrough studies in the rational design of SAECs.
Concluding remarks
Theoretical understanding of single-atom electrocatalysis constitutes a core element for the design of high-performing SAECs that unleash their full potentials for industrial applications in energy and environment. Yet challenges are present in theoretical studies of single-atom electrocatalysis, from efficient methods to tackle the static correlation, to comprehensive approaches to explore potential-dependent reaction mechanisms, and to systematic strategies to formulate quantitative and physically sensible structure-performance relationships. Nevertheless, there are promising directions to face up these challenges, and hopefully the field will be advancing rapidly.
Acknowledgments
We are grateful to the financial support from National Natural Science Foundation of China (Nos. 22122304 and 92261111), Tsinghua University Dushi Program, National Key Research and Development Project (2022YFA1503000) and Tsinghua University Initiative Scientific Research Program (20221080 065).
9. Theory and computation of the local reaction environment in electrocatalytic media
Michael Eikerling
Forschungszentrum Jülich GmbH and RWTH Aachen University, Jülich, Germany
E-mail: m.eikerling@fz-juelich.de
Status
Hydrogen fuel cells and water electrolysers will be enablers of the epochal green energy transition [171, 172]. Among the materials and components that these technologies require to function, the electrocatalytically active layers or catalyst layers (CLs), a type of porous composite electrode, are of critical importance [173]. The most advanced CLs are those for the oxygen reduction reaction (ORR) in polymer electrolyte fuel cells (PEFCs). Their development history unveils important lessons, described in [171, 174], in order to speed up CL development for other technologies.
Theory and computation
Theory and computation provide multifarious approaches to understand, analyse, and predict relations among structure, properties and performance of CLs. Figure 12 comprehensively depicts the relevant phenomena, model systems, and approaches. Physical models can be devised to rationalize impacts on performance, durability and economic viability that are exerted by (1) the catalyst material's electronic structure, (2) the catalyst surface atomic structure and chemisorption state, (3) the local reaction environment (LRE) at the catalyst–electrolyte interface, (4) catalyst–support interactions, (5) a near-surface ionomer-skin layer [175], (6) pore size and contact angle distributions in the CL [175, 176], (7) percolation-controlled transport properties [177].
Figure 12. Full-cycle integration of methods in theory and computation to rationalize how catalyst layers form and function, fade and fail. The arrow that closes the loop from device modelling to ab initio simulations applied of atomistic structure and dynamics symbolizes the importance of knowing the real conditions that prevail at the surface of the catalysts, as a prerequisite of AI-enabled materials design and development.
Download figure:
Standard image High-resolution imageWhere to seek improvements?
Major efforts in the scientific community exploring energy materials for hydrogen technology have been focusing on the discovery and design of novel electrocatalysts, relying on descriptors (or feature vectors) that represent electronic structure effects, surface atom arrangement, and chemisorption properties [178, 179]. However, the staggering increase in the mass-specific activity of platinum in PEFCs—by about a factor 103 over the last 60 years has not been achieved primarily through modification of these properties but mostly through advanced engineering and optimization of the electrode medium that the catalyst is surrounded with. This assertion and the hierarchy of effects in figure 12 imply that more complex features, beyond those directly related to electronic structure and chemisorption properties of the electrocatalyst, must be considered.
Current and future challenges
The utmost urgency and global dimension of the energy challenge demand a drastic acceleration of the materials development process, from discovery and design to device level integration and deployment. From among the influencing features, listed as 1–7 above, a minimal set of descriptors (or features) should be devised to steer the selection and development of catalyst layer materials and guide materials integration, assembly, and fabrication towards CLs with optimal mass activity, power performance and stability. This minimal set should guarantee the most rapid and resource-efficient discovery and development process. A new breed of future labs is emerging to accomplish this mission [180–182]. They are self-driving laboratories, comprising autonomous closed-loop discovery systems that blend robotic platforms for synthesis, fabrication and characterization together with high-throughput screening (HTS), assisted by machine learning [7], model-driven data analytics [183], as well as physics-aware modelling and high-performance computing (HPC).
In the field of electrocatalysis, a crucial, albeit often overlooked aspect in this endeavour is that materials should be evaluated and compared (e.g. in HTS) under conditions, for which they are expected to perform and last in the electrochemical device. This corresponds to closing the loop in figure 12 between modelling and diagnosis at the device level (top in figure 12) that provide the locally prevailing conditions and AI-enabled forays in HTS of materials ('1 pm position' in figure 12). In the context of electrocatalysis, knowledge of the local reaction environment (LRE) for a given materials combination under relevant operating conditions is a crucial prerequisite for the rational selection and accelerated design of electrocatalyst materials and CLs. The LRE is the outcome of a complex interplay of electronic structure effects, surface-transforming reactive processes, electrolyte phenomena, and transport processes that demands physical modelling and HPC to be used at various structural levels [184].
Theory and computation of electrocatalytic interfaces must account, in a self-consistent manner, for coupling and correlation effects among electronic, ionic, and solvent subsystems involved [185, 186]. As a basic capability, they should yield the relation between free surface charge density, σM, and electrode potential, φM, not only close to the potential of zero charge but in the potential ranges that are relevant for reactions of interest, such as the reduction or evolution of oxygen or the reduction of CO2. Dependences of this fundamental charging relation on pH and electrolyte composition should be captured as well. What's more, the interface response is dynamic; it involves charge transfer and polarization effects, coupled to transformations in chemisorption state and solvent configuration.
Advances in theory and computation to meet challenges
Deciphering the local reaction environment
The ideally suited approach to tackle this challenge would be using first principles methods based on Kohn–Sham density functional theory (KS-DFT) that should allow for the explicit control of φM, thus being grand-canonical [187–189]. Suitable approaches should yield self-consistent solutions for the (sub-)nanoscale distributions of electric potential, pH, and ion concentrations as well as chemisorption state and solvent properties. Albeit, thus far approaches based on KS-DFT are not able to simulate systems, in particular the dynamically fluctuating electrolyte side, at sufficient length and time scales [190]. However, molecular simulations are anticipated to receive a boost with further advances in hardware designs, e.g., multi-GPU processing [191]. The transition to the era of exascale supercomputing [192, 193] might provide much needed capabilities to expand the reach of quantum mechanical simulations to thermodynamically relevant scales. Moreover, the emergence of machine learned interatomic potentials will help accelerate simulations and more effectively bridge scales [194–196].
To guide advances in simulations, self-consistent approaches with all subsystems accounted for but simplifying assumptions made in their treatment will continue to be of great value. The first in the line of development of such approaches revealed the non-monotonic surface charging behaviour of a platinum electrode [197], and, thereby, solved an old experimental puzzle [198]. Guided by this development, a simulation approach was adopted that hybridizes KS-DFT for the metal region with the effective screening medium reference interaction site method for the electrolyte side [199]. Two very promising results were obtained with this approach: (i) when correctly set up, especially when treating the near surface water layer explicitly as part of the quantum region, it provides realistic electrolyte distribution functions in the interface and yields interfacial water density profiles that agree with AIMD simulations and (ii) it reproduces, as a first among simulation approaches, the non-monotonic metal charging relation of the partially oxidized Pt(111)/electrolyte interface, in agreement with the analytical theory and experiments. There is thus convergence between theory and simulation to build on, but still a long way to go in terms of bringing realism into simulations. As a major limitation, in either approach the control variable to define the interface state is not the electrode potential, but the surface coverage by chemisorbed oxygen as a proxy variable. Due to the unique relation between electrode potential and oxygen coverage, this approach is viable for the Pt electrode. However, it is not generalizable to a wider range of interface problems, since proxy variables to replace the electrode potential with are not a priori known. A reactive DFT-based approach is needed to close this gap.
A further step on the theory side is the development of a hybrid density-potential functional [186]. Variational analysis of this functional yields a grand-canonical model of the electrochemical double layer (EDL). It describes metal electrons at the level of Thomas–Fermi–Dirac–Wigner (TFDW) theory and treats the electrolyte solution classically at the mean-field level, taking into account electrostatic interactions, ion size effects, and nonlinear solvent polarization. The model uses parametrizable force relations to describe the short-range forces between metal cationic cores, metal electrons, electrolyte ions, and solvent molecules. Partial charge transfer in the presence of ion specific adsorption is treated in Anderson–Newns theory. With these features, the model represents a viable framework to study EDLs under the constant potential condition, as needed for deciphering multifaceted EDL effects in electrocatalysis. The approach reproduces the capacitive interface response of the Ag(111)–KPF6 system. Future refinements should account for fluctuation effects and correlations [200, 201]. Moreover, limitations of the simple TFDW theory need to be overcome with more advanced functionals [202].
Accelerated computational protocol for extended simulation of interfaces
In the future, KS-DFT-based first principles simulations of electrochemical interfaces, microkinetic models parametrized with energy parameters from KS-DFT calculations, and machine learning (ML) will be combined to study reactive processes at extended time and length scales. ML models of interatomic potential energy and force fields based on deep neural networks have the potential to overcome the accuracy vs. efficiency dilemma molecular simulations [203]. Deep potential molecular dynamics (DPMD) simulations with precision comparable to KS-DFT-based simulations increase the length and time scales of ab initio molecular dynamics with a computational cost that scales linearly with system size [204]. DPMD simulations are required to examine the influence of structural composition on the surface-controlled coupled dynamics of water and ions. These elements can be combined together into multistep computational protocols for determining the structure and composition of the most stable interface structures for varying electrode potential and pH, and then use those structures to conduct MD simulations of dynamic processes at the interface.
Infuse interface simulations with a sense of realism
In typical journal articles on CL structure and function, it can often be read that the 'catalyst is in contact with ionomer'. What does 'contact with ionomer' mean or imply? The wording as such is a problem and the misconception behind the wording is challenging to overcome. In a well-functioning CL under relevant operating conditions, the catalyst is not in contact with ionomer, but with water in a confined region that is bound on one side by catalyst and support and on the other side by an ionomer skin layer, cf Figure 13. These water-filled nanogap regions with width < 2 nm are asymmetric and heterogeneous. Supported nanoparticle systems result in heterogeneous surface charging on the metal side of the gap. The ionomer skin exhibits a high charge density due to fixed but flexible anionic charges. Deep-potential MD simulations with proper treatment of surface charging, dipolar, and polarization effects can be used to determine the stability of such a nanoprotonic gap. Compared to fixed interface and charge configurations considered in References [205, 206], interface properties at the catalyst–support boundary can be continuously tuned by φM—the interface will 'breath water in or out', which should be exciting 'to see' in a future simulation! Potential and proton density distributions and the electrocyclic activity in the gap will respond to these variations. Theoretical and DPMD-based studies will be of great value for studies of these phenomena.
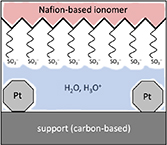
Figure 13. Heterogeneous and asymmetric nanostructure, account-ting for key structural features at interfaces in PEFC catalyst layers.
Download figure:
Standard image High-resolution imageConcluding remarks
Catalyst–electrolyte interfaces are ubiquitous in electrochemical energy science. Detailed assessment of dynamic processes involved in desired and undesired electrocatalytic processes hinges on knowledge of the local reaction environment at these interfaces. Advanced self-consistent theoretical approaches and computational approaches accelerated with machine learning will be needed to decipher this environment for realistic interface configurations and in operando conditions. Understanding the conditions that prevail in the water-filled nanoprotonic gap region between catalyst/support and ionomer skin layer will be crucial to steer the design and rapid screening of catalyst layer materials and to guide their assembly into an optimally functioning and stable electrode.
Acknowledgments
The author acknowledges funding of research presented in this article from the Helmholtz-Gemeinschaft Deutscher Forschungszentren e.V. (HGF), Program-oriented Funding (PoF IV), under the Research Program Materials and Technologies for the Energy Transition (MTET).
10. Modelling and machine learning of fuel cell
Ryosuke Jinnouchi
Toyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan
E-mail: e1262@mosk.tytlabs.co.jp
Status
Proton exchange membrane fuel cells (PEMFCs) are promising power sources for automobiles. Several automakers have commercialized fuel cell vehicles with the required power density and durability [207], and further extensions of applications are expected for heavy duty transportations, such as trucks, buses, trains, ships and so on [208]. For wider applications, however, higher energy conversion efficiency, longer durability and lower cost are desired [209]. These demands are demonstrated by the performance and cost target set by New Energy and Industrial Technology Development Organization (NEDO) in Japan [210]. Figure 14 shows the target current–voltage (IV) curve of a single cell for heavy duty transportations in 2030 and the curve of the second-generation Toyota MIRAI (Gen2-MIRAI). The NEDO roadmap reports that the applications require the significantly higher efficiency than Gen2-MIRAI as well as a higher maximum operation temperature of 378 K and longer operation time of 50 000 h. The target IV curve was decomposed to the target properties of constituent materials by using a multi-physics simulation predicting an IV curve of a single cell from properties of materials. The resulted target properties of the cathode catalyst for oxygen reduction reaction (ORR) and polymer electrolyte are shown in the same figure. The simulation indicates that the target IV requires the high catalytic activity, gas diffusivity of catalyst layer (CL), proton conductivity of electrolyte and durability. Accordingly, advanced electrode and electrolyte materials are necessary.
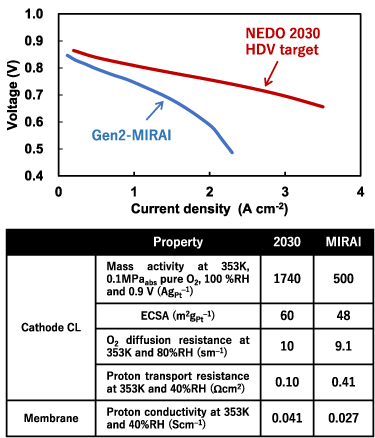
Figure 14. IV curves of the target in 2030 set by NEDO and Gen2-MIRAI. The table summarizes the target properties of the cathode catalyst layer (CL) and proton exchange membrane. Data were taken from [210].
Download figure:
Standard image High-resolution imageCurrent and future challenges
Modelling and simulations have provided essential information on understandings and designs of PEMFCs and constituent materials. As illustrated by the NEDO roadmap [210], the multi-physics macroscopic simulation [211] solving diffusions, conductions and reactions can determine the performance of the single cell from the properties of materials and vice versa. To get ideas of high-performance materials, microscopic simulations are necessary. The time- and length-scales of relevant reactions and transport phenomena span over 0.1 nm to 1 m and 1 fs to days (or even years), and a single method cannot solve the problems. Thus, a variety of simulations suited to each scale have been developed as illustrated in figure 15. The continuum theory simulation [212] is a powerful method to solve the heterogeneous reaction field in the 3D cathode CL that is a porous medium composed of Pt nanoparticles supported on carbons and polymer electrolyte. Coarse-grained molecular dynamics (MD) simulations have been used to predict the mesoscale porous structure [213], and full-atomistic classical MD simulations assuming empirical interatomic potentials [214] have been used to predict atomic scale structures and transport coefficients required for the continuum theory. Potential-dependent catalytic rate constants of electron transfer reactions can be obtained by first-principles (FP)-based models of electrified interfaces [215] and can be used in the FP-based microkinetic catalytic reaction models [216]. FP method is also a powerful tool to predict thermodynamic stability (Pourbaix diagram) of materials [217]. These FP methods have been used to clarify reaction mechanisms of advanced catalysts and to explore active and stable catalysts [216]. The growth of machine-learning (ML) technologies and databases have extended the applicability of these simulation methods. The FP-database was, for example, used to identify acid-stable oxides in the cathode environment [218], and a convolutional neural network model was used to accelerate the screening of ternary Pt alloy catalysts [219]. ML-based image recognition technologies also enabled accurate reconstructions of CL structures from microscopy images, which are required for the continuum theory simulations [220]. There are, however, a lot of challenges in simulations of complex practical materials. Difficulties mainly arise in computations of distribution functions, transport coefficients, potential-dependent reaction rates and interatomic potentials, which need to be transferred from one scale to the other in the multi-scale simulation framework. Empirical interparticle interactions of coarse-grained MD methods often involve significant errors, and the resulted properties can be unreliable. The same problem also happens in interatomic potentials of classical MD methods particularly for reactive systems. Although FP methods accurately predict the atomic interactions, they are too expensive to realise sufficient statistics required for accurate predictions of thermodynamics and kinetics.

Figure 15. Multi-scale simulations of electrode and electrolyte materials. Thermodynamic and kinetic properties are transferred from one simulation to the other, enabling predictions of desired properties.
Download figure:
Standard image High-resolution imageAdvances in science and technology to meet challenges
Emerging ML technologies are the key to surpass the previous limitations of the modelling and simulations toward accurate and robust bottom-up approaches starting from FP methods. ML interatomic potentials (MLPs) were shown to realize orders of magnitude faster computations of interatomic interactions retaining near FP-accuracy [221], and state-of-the-art active-learning schemes accelerated the time-consuming trainings containing preparations of FP datasets [222]. Several MD simulations using MLPs were conducted on anhydrous electrolytes [223] and water/Pt interfaces [224, 225] and exhibited promising results. ML-algorithms were also demonstrated to systematically construct interparticle interactions of coarse-grained model that can accurately compute free energies of solvated molecules [226]. Because MLPs for complex multi-element systems need large FP datasets, their applications are still limited to simple systems. In this rapidly growing field, however, many advanced MLP algorithms are investigated, and accuracy and efficiency continue to improve [227]. In addition to the MLP algorithms, efficient and robust statistical sampling algorithms combined with MLPs are also necessary in order to accurately predict the thermodynamics and kinetics of electrolyte and electrode materials. Free energies of reaction intermediates, for example, are essential descriptors of the catalytic activity, but most of FP-based methods compute the free energies by assuming harmonic vibrations of atoms [216] although interfacial solvent molecular motions are anharmonic. The same difficulties also appear if one tries to compute potential-dependent activation free energies beyond the descriptor-based approach [228]. This severely limits the applicability of the FP method to many significant electrolyte effects on the activity and durability of catalysts [229, 230]. Recent studies [231, 232] demonstrated that MLPs can be used as surrogate models that enable efficient thermodynamic integrations to compute free energies of hydrated ions and adsorbates. Although the examined systems were simple aqueous systems, the algorithm combined with accurate MLPs will provide quantitative predictions of free energies of complex heterogeneous materials.
Concluding remarks
A wide variety of simulation methods have been developed and have provided indispensable information, such as breakdown of overpotentials of a single cell [211], volcano plot of ORR [216] and bottleneck of transport phenomena in cathode CL [214], for designs of PEMFCs and advanced materials. The wide distribution of PEMFCs, however, demands further innovative materials. For the discoveries, unexplored materials and systems need to be examined by simulations as well as experiments. MLP-aided statistical sampling of dynamically fluctuating electrode/electrolyte interfaces is expected to provide a platform of new discoveries because several experiments [228, 229] reported significant electrolyte effects and because the systems have not been sufficiently examined owing to challenges in the conventional simulations. ML-aided seamless connections from the FP method to the coarse-grained MD is also expected to provide a platform because the mesoscopic self-organization of porous cathode CL involves many unclarified questions. We believe that the new simulations will pave the way to discoveries of guiding principles for innovative materials.
11. Modelling electrochemical interfaces and reactions with GPAW: grand canonical ensemble DFT approaches
Marko M Melander1 and Georg Kastlunger2
1 Department of Chemistry, University of Jyväskylä, Jyväskylä, Finland
2 Department of Physics, Technical University of Denmark, Lyngby, Denmark
E-mail: marko.m.melander@jyu.fi and geokast@dtu.dk
Status
Grand canonical ensemble density functional theory (GCE-DFT) is a theoretically rigorous way to simulate electrochemical interfaces under constant electrode potential and electrolyte activity conditions directly mimicking experimental conditions as depicted in figure 16 [188]. GCE-DFT explicitly regards the electrode potential in simulations and complements the widely adopted computational hydrogen electrode (CHE) method [155]. In particular and unlike CHE, GCE-DFT can be directly applied to simulate constant potential reaction kinetics [233], decoupled electron proton transfer reactions and non-Nernstian pH shifts [234, 235]. Thus, a combination of CHE and GCE-DFT is able to describe any thermodynamically-stable and transition state within complex reaction mechanisms at arbitrary potentials and pH [236].

Figure 16. Left: Illustration of a GCE-DFT simulation with a fixed electrochemical potential of electrons ( ) and solvent (
) and solvent ( ) to control the electrode potential and solvent properties, respectively. Right: Simplified workflow of the current realization of calculating GCE-DFT energies within GPAW. Note that in contrast to fixed charge DFT calculations two convergence criteria need to be fulfilled, namely the convergence of the electronic workfunction (
) to control the electrode potential and solvent properties, respectively. Right: Simplified workflow of the current realization of calculating GCE-DFT energies within GPAW. Note that in contrast to fixed charge DFT calculations two convergence criteria need to be fulfilled, namely the convergence of the electronic workfunction ( ) and the maximal atomic forces in the DFT cell (fmax). Reproduced from [242]. CC BY 4.0.
) and the maximal atomic forces in the DFT cell (fmax). Reproduced from [242]. CC BY 4.0.
Download figure:
Standard image High-resolution imageIn recent years, several practical realizations of GCE-DFT have been implemented in the GPAW code [237]. GPAW is an open-source, Python-based DFT code using the projector augmented wave (PAW) method and allows calculations within real-space grid (FD), LCAO [238] and plane wave (PW) basis sets. The Python environment together with Numpy [239] and Scipy [240] facilitates implementation, while numerical performance is guaranteed by external C/C++/Fortran libraries. Unlike PW-based GCE-DFT implementations [241], GPAW works in real-space within FD and LCAO bases; this allows direct utilization of natural 2D periodic boundary conditions (PBCs) for the electrode and electrostatic potential [188, 233, 242] difficult to achieve with PW codes [243]. The 2D PBCs effectively build a reference electrode within the simulation cell [188, 242] provide correct decay of the electrostatic potential needed for treating continuum electrolytes, and allow the simulation of asymmetric slabs/electrodes. All these features combined make GPAW a flexible environment for GCE-DFT development.
Constant potential calculations can currently be performed either by iteratively fixing the work function [233], Fermi level or the electrode inner potential [242], following the algorithm outlined in figure 16. Cell charge neutrality is maintained by the solvated jellium method [233] which offers a computationally stable and fast method with a correctly behaving electrostatic potential. Also Poisson–Boltzmann electrolyte models have been implemented [188] but these are currently not available in the main branch because of numerical instabilities due to the underlying finite-difference Poisson solver. The solvent is treated as a linear dielectric continuum [244] although a classical DFT model of water is also available [245] but not yet tested for electrodes. GPAW's GCE- DFT methods automatically provide the Legendre-transformed total GCE energies. Thus, they can directly be combined with e.g. nudged elastic band calculations to obtain constant potential reaction kinetics or constrained DFT [246] to build GCE-DFT diabatic states [247] for the simulation of Marcus-like electron and proton transfer kinetics.
Current and future challenges
GPAW already achieves out-of-the-box stable and efficient GCE-DFT calculations for a wide variety of systems and the main challenges are related to improving upon current numerical methods and approximations. Like in most solid state codes, only a linear dielectric model is currently available and the parametrization is limited to aqueous systems and small molecules—there are no parameter sets for metallic or other electrode surfaces. Also going beyond the linear dielectric model is expected to improve the solvent description and the surface capacitance in particular. Combining more general dielectric and electrolyte models with improved parametrizations for surfaces would hopefully also enable the simulation of more complex solvents, including models for complex aqueous electrolytes [248] or the modelling the capacitance and layering of ionic liquids [249]. Another challenge is going beyond the dielectric continuum models and treat the electrolyte with more refined methods based on the statistical theory of classical liquids [250], such as reference interaction site method (RISM) and classical DFT approaches.
As mentioned above, the current dielectric continuum model uses the finite-difference Poisson solver which is known to perform badly for elongated system such as slabs with extend vacuum or dielectric regions. In default GPAW calculations this was recently resolved by implementing a Fourier and Fourier-sine transform-based FastPoissonSolver, which not only makes slab calculations faster but also numerically more stable. In currently unpublished work, we have extended this solver to the dielectric continuum Poisson solver and observed greatly improved performance. In the future, we expect this to stabilize the convergence of the generalized Poisson–Boltzmann electrolyte models. Further improvement in stability and efficiency could be gained by moving from iterative fixed potential calculations to a scheme where the constant potential condition is achieved within a single self-consistent field cycle. We expect this to be particularly important for GCE constrained DFT calculations where an additional iterative loop is needed [247] as well as GCE-DFT molecular dynamics simulations.
Advances in science and technology to meet challenges
The combination of (GCE-)DFT and classical liquid theories is well-documented but cur- rent implementations [251–253] have typically been hard-coded in the DFT packages and require significant implementation work. On the other hand, it was recently demonstrated [245] that GPAW can be directly interfaced with an external classical DFT code with minimal changes to both codes. We therefore propose that GPAW could be rather naturally interfaced with already existing, open-source RISM [254] or classical DFT implementations [255] rather than re- programming them; this task should be greatly facilitated by GPAW's flexible and modular Pythonic structure.
The Pythonic structure is also expected to facilitate interfacing GPAW with machine learning methods such as on-the-fly generation [256] of data-driven force fields. An outstanding challenge is treatment of the electrode potential in machine learning methods and currently there are no schemes for achieving this efficiently. A brute-force parametrization of explicitly potential-dependent force fields is readily achievable by reparametrizing at each potential, a substantial computational burden. More promising schemes could be achieved by learning e.g. the electrostatic potential [257], which is directly related to the electrode potential [242], or the charge response kernel [258].
Finally, porting GPAW to support GPUs will make GCE-DFT calculations (and also machine learning integration) significantly faster. While there is a legacy implementation supporting GPUs [259], this is outdated and cannot be used with current versions of GPAW. There is, however, an active on-going development to make GPAW work with CUDA and HIP architectures [260].
Concluding remarks
We have provided an overview of simulating electrochemical systems with GCE-DFT as implemented in GPAW. We have highlighted the features which make GPAW a versatile and flexible platform for developing and using GCE-DFT methods. In general, the theoretical foundation of simulating the electrode–electrolyte interface under constant electrode potential is set and we can already address electrochemical thermodynamics and kinetics as an explicit function of the electrode potential. In the future, focus should be directed at developing better electrolyte and solvent models, improving efficiency and stability, and integrating GCE-DFT with other simulation approaches, such as machine learning methods.
Acknowledgments
M M M acknowledges the funding by Academy of Finland (Project # 338228, CompEL). G K thanks the European Union's Horizon 2020 research and innovation program (Grant # 851441, SELECTCO2) and the Villum foundation (Grant # 9455, V-Sustain). Both authors acknowledge the GPAW developers for their continuous efforts to maintain and improve GPAW.
12. Constant Fermi-level molecular dynamics: recent achievements and future challenges
Assil Bouzid1 and Alfredo Pasquarello2
1 Institut de Recherche sur les Céramiques (IRCER), Centre Européen de la Céramique, 12 Rue Atlantis, Limoges, 87068, France
2 Chaire de Simulation à l'Echelle Atomique (CSEA), Ecole Polytechnique Fédérale de Lausanne(EPFL), CH-1015 Lausanne, Switzerland
E-mail: assil.bouzid@cnrs.fr and alfredo.pasquarello@epfl.ch
Status
Achieving a detailed understanding of the electrochemical processes occurring at solid/liquid interfaces is of paramount importance towards the design of efficient and durable solar cells, energy conversion, and storage devices. These processes are usually driven by an applied bias potential that determines the amount of excess charge (electrons or protons) at the interface, which in turn influences the double layer structure and the electrochemical properties of the interface. At this level, atomic-scale modelling provides powerful tools to access the intimate details of the interface structure in close correlation with its overall electrochemical properties. Several modelling schemes have been proposed to simulate metal/water interfaces through either implicit [261–264] or explicit but static [265, 266] solvents, either at fixed excess charge [267, 268] or by varying the bias potential [243, 269–273]. Among these modelling schemes, constant Fermi-level molecular dynamics (CFLMD), initially developed by Bonnet et al [270] and then further extended by the present authors [274–277], have enabled the study of electrochemical processes at metal/water interfaces under bias potential. In practice, within the CFLMD scheme (see figure 17), the studied system is connected to an external potentiostat at fixed Fermi level that acts like an electron reservoir or a fictitious external electrode. The dynamical control of the Fermi level is ensured by considering the electronic charge of the system as a fictitious dynamical variable subject to inertia such that the extended system is driven by a grand canonical potential. This technique is complemented with a proper alignment method that enables one to reference the applied bias potential to the standard hydrogen electrode, thereby allowing for a direct comparison of the modelling results with experiments. In the case of the Pt(111)/water interface, CFLMD have enabled the study of the structural reorganization of the electrical double layer as a function of the applied bias potential, whereby a bias-dependent water organization has been demonstrated [274]. In addition, this scheme yields a potential of zero charge and a double layer capacitance in excellent agreement with experimental measurements [274, 275]. Another interesting feature of CFLMD lies in its ability to spontaneously inducing chemical reactions when sufficiently strong bias potentials are applied. In the case of the oxygen evolution reaction (OER) at the Pt(111)/water interface, CFLMD have enabled access to the organization of adsorbed species like H2Oads and OHads at the metal surface prior to the OER reaction providing a description in which they have been found to arrange in a hexagonal lattice with an irregular alternation. The OER reaction has then been found to proceed through a hydrogen peroxide intermediate leading to a reduction of 0.2 eV of the reaction overpotential when compared to the conventional OER mechanism [276].
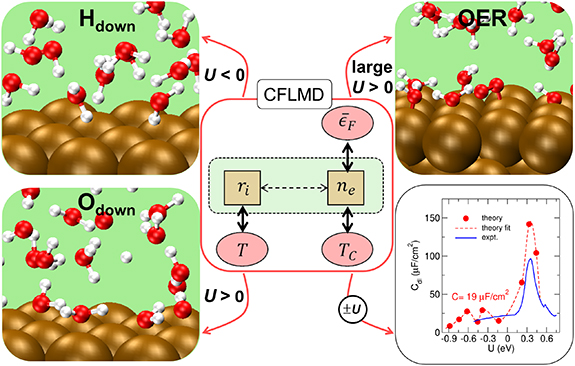
Figure 17. Applications of the CFLMD scheme. Central panel: schematic illustration of the CFLMD scheme. A system of particles described by a set of atomic positions ri
and a total electronic charge ne
is connected to an external potentiostat at a fixed Fermi level  acting like a fictitious external electrode. The electronic charge ne
is considered as a dynamical variable with inertia Me
, and the extended system is driven by a grand canonical potential [270, 274]. The atomic degrees of freedom are coupled to a thermostat set to a temperature T, and the charge dynamics are coupled to a separate thermostat set at a temperature TC
[270, 274]. Top (bottom) left panel: Representative snapshot illustrating the structure of the water double layer at Pt(111)/water interface when the electrode potential U is set at a negative (positive) value [274]. We note that in the case of negative bias, the Pt surface was intentionally not covered by H (see [274]). Top right panel: Representative snapshot illustrating the product of the oxygen evolution reaction (OER) at Pt(111)/water interface (formation of O2) upon application a high positive potential [276]. Bottom right panel: Double layer capacitance Cdl as a function of the applied electrode potential U [274]. Adapted with permission from [274]. Copyright 2018 American Chemical Society.
acting like a fictitious external electrode. The electronic charge ne
is considered as a dynamical variable with inertia Me
, and the extended system is driven by a grand canonical potential [270, 274]. The atomic degrees of freedom are coupled to a thermostat set to a temperature T, and the charge dynamics are coupled to a separate thermostat set at a temperature TC
[270, 274]. Top (bottom) left panel: Representative snapshot illustrating the structure of the water double layer at Pt(111)/water interface when the electrode potential U is set at a negative (positive) value [274]. We note that in the case of negative bias, the Pt surface was intentionally not covered by H (see [274]). Top right panel: Representative snapshot illustrating the product of the oxygen evolution reaction (OER) at Pt(111)/water interface (formation of O2) upon application a high positive potential [276]. Bottom right panel: Double layer capacitance Cdl as a function of the applied electrode potential U [274]. Adapted with permission from [274]. Copyright 2018 American Chemical Society.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Despite the success of CFLMD in studying electrochemical processes at metal/water interfaces, its application to complex systems is still hindered by several intrinsic limitations. First, similar to all MD based simulations, long equilibration times are required to ensure equilibrium and to minimize the deviation from ergodicity. This comes with a considerable computational cost. In addition, the modelled physical system in the case of metal/water interface corresponds to a system of a single half electrode and does not account for the presence of a counterelectrode as in experiments. Given that periodic boundary conditions are usually applied in the simulation, a constant electric field across the water layer cannot be sustained and achieving a proper description of the electrostatic potential becomes an issue. Furthermore, in experiments, the electric field in the water region due to the extra charge at the metal surface is screened out by counterions of the electrolyte, but at present a proper treatment of such ions would require prohibitively large system sizes in the simulation. In practice, a uniform neutralizing background is used, which effectively screens out the potential evolution across the liquid region and artificially reduces the charge located on the electrode [274]. Second, the CFLMD is currently mainly applied within standard semilocal density functional theory (DFT). While this framework provides a cost-effective tool for performing molecular dynamics simulations of the metal/liquid interfaces at fixed bias potential, it does not satisfy the piece-wise linearity condition of the total energy upon electron occupation as expected from exact DFT. Through the application of Janak's theorem, one can see that this practically results in single-particle energy levels varying with electron occupation, implying that resonances between adsorbate energy levels and the Fermi level are spuriously broadened upon varying the bias potential [277]. The use of hybrid functionals could overcome such issues, but their prohibitive computational cost currently prevents their systematic use in molecular dynamics simulations. Finally, modelling chemical reactions by varying the Fermi level within the CFLMD requires the use of unrealistically high bias potentials [276] to overcome the energy barriers of the targeted reaction during the accessible timespan of simulations. This requires the injection of large amounts of excess charge in the system, which then in turn undermines the comparison between the bias potential set for making the reaction viable in the simulation and the observed value in experiment.
Advances in science and technology to meet challenges
Almost all grand canonical schemes currently employed to address solid/liquid interfacial properties at constant electrode potential suffer from the same aforementioned limitations. Lifting these intrinsic limitations would therefore represent a major step forward towards achieving a realistic and quantitative modelling of electrochemical processes at solid/liquid interfaces. We here provide several possible methodological and technical advances that could contribute to the achievement of a successful grand-canonical scheme for modelling aqueous interfaces at constant bias potential. First, accounting for the occurrence of counterions in the liquid solution could be achieved through auxiliary classical molecular dynamics. In fact, large-scale classical simulations of solid/liquid interfaces with counterions physically present in the solution can be performed in order to determine the electrostatics induced by these counterions upon the charging of the electrode. This would then provide an average electrostatic potential that could be transferred to the ab-initio model. Hence, such a procedure would allow one to effectively account for the screening due to the counterions in the liquid solution during the ab-initio molecular dynamics at fixed potential. This would imply that, prior to any first-principle simulation, one carries out a classical MD simulation in the same conditions of excess charge in order to determine the electrostatics due to the counterions. This procedure can be improved even further by resorting to machine-learning (ML) techniques. In particular, for a given system, several large-scale classical MD simulations at various excess charges in the presence of counterions could be used to train a ML model able to predict the electrostatics of counterions at a given fixed bias potential. Such a model could then be coupled with the DFT description and provide on the fly a correction of the average electrostatics in solution upon variation of the bias potential. Such an approach would be of particular interest for dealing with chemical reactions requiring the bias to be varied in a continuous fashion. Second, to satisfy the piece-wise linearity condition, one could resort to DFT + U schemes in which the parameter U is suitably set to comply with the generalized Koopmans' condition. For higher accuracy, one should assume the higher computational cost of hybrid functionals. Piece-wise linear hybrid functionals can be constructed in a cost-effective way by imposing the generalized Koopmans' condition on localized electronic states associated with defects or with hydrogenic-like potential probes [278]. Third, the last limitation related to the occurrence of high energy barriers when modelling chemical reactions could be overcome by combining grand-canonical schemes at constant bias with advanced sampling methods to model rare events, such as the metadynamics scheme [279] or the thermodynamic integration method [280]. Finally, the implementation of the proposed extensions in an accessible and efficient computer code is key towards their wide adoption by research communities.
Concluding remarks
In summary, despite significant progress in modelling electrochemical processes at solid/liquid interfaces achieved in the last decade, substantial advances still lie ahead to reach a powerful and successful methodology. The advances suggested in the present roadmap contribution would enable a significant step forward in that direction.
13. Density functional theory in classical explicit solvents (DFT-CES): efforts towards understanding the unseen, buried electric double layer
Seung-Jae Shin1,2, Minho M Kim1 and Hyungjun Kim1
1 Department of Chemistry, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea
2 Department of Materials Science and Engineering, Yonsei University, Seoul 03722, Republic of Korea
E-mail: linus16@kaist.ac.kr
Status
Electric double layer (EDL) is ubiquitously formed at electrochemical interfaces. Because an electrochemical reaction is governed by electron transfer (ET) across the EDL region, one can deduce the intimate connection between the EDL structure and electrochemical properties, although the details have not yet been unravelled [281].
A solid structure can be well-characterized by means of diffraction or surface-science experimental techniques. Whereas, it is extremely difficult to resolve the three-dimensional atomic arrangement of a liquid structure such as that of an EDL. Therefore, to obtain the necessary atomic/electronic level information of EDL structures, the need for a reliable and predictable molecular modelling method is emphasized.
From the modelling viewpoint, it is always important to choose a simulation method with appropriate length- and time-scales. As the Debye length spans from 0.3 nm to 3 nm at the conventional electrolyte concentration (0.01 M–1 M), and the EDL relaxation time is measured to be in the order of nano-to-micro seconds [282], one may argue that compared to the scales provided by classical molecular dynamics (MD), those provided by ab-initio molecular dynamics (AIMD) may be insufficient to investigate the EDL structure and dynamics. However, a quantum mechanical (QM)-level of understanding on the electronic structure of the electrode, such as a location of the Fermi level for defining an electrochemical potential, is also essential to predict the potentiodynamic polarization behaviour. Therefore, a multiscale approach combining a QM-level description for the electrode region and a classical MD-level description for the electrolyte region deems to be optimal.
A conventional multiscale QM/MD method simultaneously propagates the dynamics of both regions. Consequently, the QM-region dynamics, as a bottleneck, strongly limits the achievable time scale. Particularly for the EDL system, the typical electrode in the QM region consists of numerous atoms and electrons. Assuming a weak coupling between liquid electrolyte dynamics and electrode phonon, we employed a mean-field coupling between these two regions—iterative QM optimizations and MD decouple time-scales in these two regions (figure 18(a)), thus enabling a sufficiently lengthy dynamics simulation for the electrolyte [99, 283, 284]. To ensure high fidelity, the interaction parameters across the two regions are further optimized based on QM energetics, which has been validated to reproduce a reliable interfacial tension [285] and water contact angle [286]. This first-principles-based multiscale simulation method, namely the density functional theory in classical explicit solvents (DFT-CES) has been developed by our group to be applied to investigate in the structural investigation of EDL.

Figure 18. Current status of DFT-CES in describing the EDL. (a) Iterative scheme of mean-field coupled DFT optimization and nanoseconds MD simulation. Reprinted with permission from [284]. Copyright 2018 American Chemical Society. (b) Theoretical reproduction of camel-shaped behaviour of EDL capacitance using minimal counter-ion (MCI) description. DFT-CES predicts the potential (E) dependent surface charge density (σ) shown in the left panel, and its derivative defines the differential capacitance (C = dσ/dE) shown in the right panel. (c) Full local density profiles of water (background colour), ion (blue and pale-yellow), and electrode charge (yellow) at cathodic interface (left panel) and anodic interface (right panel). Reproduced from [287]. CC BY 4.0.
Download figure:
Standard image High-resolution imageCurrent and future challenges
By including a minimal number of counter ions to compensate the electrode charge, the DFT-CES simulations succeeded to capture the key features of the camel-shaped behaviour of EDL capacitance (figure 18(b)), and elucidated their molecular origins [288]. The anodic hump originates due to electrosorption of anions, and the cathodic hump is associated with an unprecedented phase transition behaviour [287]. This minimal counter-ion (MCI) description satisfies the electroneutral condition within the simulation cell by only including ions with one type of polarity. In this context, it corresponds to the dilute-limit concentration of the electrolyte, and can be compared with the Poisson-Boltzmann-type description of the Gouy–Chapman-type models. However, owing to the MD-description of the electrolyte, a mean-field approximation is not required for handling the ion–ion interaction. It should be noted that the MCI description includes both Helmholtz and diffuse layers (figure 18(c)); thus, the predicted capacitance should be compared to the entire EDL capacitance.
Despite of the success of MCI description in dilute limit cases, the EDL structure formed at a higher concentration is more relevant to the actual electrochemical conditions. In the concentration regime of 0.1 M–1 M, it is apparent that ions with both polarities coexist in the EDL region, and their concentration is modulated by the bulk concentration. However, a complication arises as the local concentration of the ions at the interface differs from the bulk concentration, and our understanding is typically limited to the latter. Mathematically, the electroneutral condition only determines the difference of cation and anion numbers, but excess number of ion pairs in the EDL region must change depending on the bulk ion concentration. Therefore, we need to develop a computational algorithm that can modulate ion-pair concentration in the EDL region depending on the bulk electrolyte conditions.
Furthermore, a more advanced force-field (FF) is required. The DFT-CES employs the FF lacking the electronic polarization effect with using fixed-charge model. In the case of water, although the dielectric screening is mostly attributed to the orientational polarization of water dipoles, there still exists a non-negligible contribution from the electronic polarization. Also, it has been known that conventional FF cannot fully capture the interaction modes between the electrode and water [289]. Thus, a more advanced FF description that can model the complicate electrode-electrolyte interaction and possibly chemical reactions, will help increase the accuracy level and capability of DFT-CES.
Lastly, the biggest challenge is related to how the electrochemical reaction can be modelled. As the QM-region structure can be optimized, DFT-CES, per se, can provide adsorbate binding energies and surface reaction energetics with including a full-atomic consideration of EDL structure [290]. More challenging quest will be related to how the ET rate can be simulated and linked with the electrochemical reaction properties.
Advances in science and technology to meet challenges
From the thermodynamic viewpoint, the local ion concentration in the interface region can be considered to be in equilibrium with the bulk electrolyte, i.e., an ion reservoir. Therefore, similar to various thermostatting methods to couple the MD simulation cell with 'heat reservoir', we need to develop a computational algorithm to couple the interface simulation with 'ion reservoir', namely a 'chemostatting' method.
By considering a grand canonical ensemble, one possible method is the hybrid Monte Carlo (MC)/MD, which incorporates MC steps during MD to create or annihilate ions according to the chemical potential of ion pairs in the reservoir [291]. However, here the simulation control parameter is the chemical potential, which cannot be directly dictated from the ion concentration value. Instead, a computational method to directly modulate the ion concentration can be designed by including a sufficiently thick electrolyte slab that can include a bulk electrolyte region in the same simulation cell (figure 19(a)) [292].
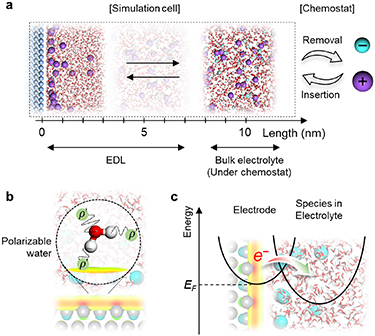
Figure 19. Future challenges to expand the functionality of current DFT-CES. (a) 'Chemostatting' method to control the electrolyte concentration at the interface depending on the bulk concentration. (b) Advanced FFs (e.g. polarisable FFs) can improve the accuracy level of electrolyte description. (c) To describe an ET process, quantum-dynamic behaviour needs to be simulated in a full or an approximated manner (e.g. Marcus theory).
Download figure:
Standard image High-resolution imageTo include an electronic response of the electrolyte phase, an effort to couple advanced FF techniques with DFT-CES needs to be endeavoured. Polarizable FFs [293–295] can account for the electronic polarization effect of molecules and ions in the electrolyte phase, which is missing in the current DFT-CES (figure 19(b)). It should be noted that coupling the reactive FFs [296, 297] with DFT-CES will help achieve a more realistic simulation. During aqueous electrochemical processes, water molecule is often multifunctional—it serves not only as a dielectric solvent (which is the current description of DFT-CES), but also as a reactant, proton donor/acceptor, reaction promotor, etc. By enabling the O–H bond dissociation/association of water, the simulation can provide means to explore and identify various roles of water during aqueous electrochemical reactions.
ET process is fundamentally a quantum-dynamic phenomenon, occurring at a much shorter time scale than the EDL relaxation (figure 19(c)). Therefore, to obtain an adequate computational description of ET process in the EDL, another scale of simulation must be integrated into the DFT-CES. Mixed quantum–classical techniques, such as a surface-hopping [298], are regarded as efficient and reliable methods for simulating quantum dynamics. To enable such methods within the DFT-CES framework, we need to develop a computational procedure for constructing potential energy surfaces and computing interstate couplings, particularly using the solid-state wavefunctions [299]. Moreover, because the diabatic surfaces are better suited for describing the ET process [300], development of a quantum dynamics algorithm based on diabatic representation might be required.
Concluding remarks
Despite its central importance in electrochemistry, structural investigation of the EDL is challenging for both theoreticians and experimentalists due to its high complexity, system-heterogeneity, and concealment in buried space [301–304]. Being fully equipped with solid-state electronic structure theory and a reliable liquid-structure simulation, DFT-CES provides atomically resolved information of the EDL structure, and its reliability is validated by comparing the predicted and experimental EDL capacitances. Compared to other methods, DFT-CES is characterized by its ability to simulate full length- and time-scales of the electrolyte phase (in comparison with the AIMD), and by all-atom-level accurate description of the EDL structure (in comparison with the continuum dielectric description). However, current DFT-CES is limited only for the dilute-limit concentration of the electrolyte, and it lacks some important quantum–mechanical phenomena that occurs at the classically-described electrolyte region. Based on recent achievements in the computational chemistry, e.g., computational statistical mechanical models and mixed quantum–classical simulations for quantum dynamics, we aim to develop novel computational methods for the DFT-CES in near future. By overcoming the current limitations of DFT-CES using new methods, we expect to better understand the molecular grammar of EDL structure, which governs the complex electrochemical phenomena.
Acknowledgments
This work was supported by the Samsung Science and Technology Foundation under Project Number SSTF-BA2101-08.
14. Implicit solvation models for electrochemical interfaces
Kathleen Schwarz1 and Ravishankar Sundararaman2
1 National Institute of Standards and Technology, Gaithersburg, MD, United States of America
2 Rensselaer Polytechnic Institute, Troy, NY, United States of America
E-mail: kathleen.schwarz@nist.gov and sundar@rpi.edu
Status
Implicit solvation modelling simplifies the simulation of liquid-containing systems relative to fully atomistic approaches by eliminating the need to sample the space of all configurations of liquid molecules. Such techniques have a long history in liquid-phase solvation for predicting homogeneous catalysis [305]. Here, they can be parameterized to extensive solvation free energy data and approach the 0.04 eV (1 kcal mol−1) standard of chemical accuracy for solvation of neutral solutes in aqueous and several non-aqueous solvents [306]. Implicit solvation models have become increasingly prevalent for solid–liquid and electrochemical interfaces in the past two decades with the development of transferable models with few parameters [307, 308], which is necessary due to the relative dearth of interfacial data to parameterize them. Combined with the capability to perform electronic structure calculations at fixed electrode potential using grand-canonical density functional theory [156], implicit solvation models have become the workhorse for first-principles electrochemistry.
The treatment of electrolytes using implicit solvation models has advanced rapidly to capture important effects at electrochemical interfaces, covered extensively in recent reviews [157, 241]. Specifically, solvation models to accurately predict the capacitance have been developed that are sensitive to both the sign and magnitude of the solute electric field [309, 310]. These new solvation models (figure 20) include nonlinearity in both the dielectric and electrolyte response to the electric field, with separate solvent-only and electrolyte regions. As a surface charges, the solvation model cavities must also respond to the solute electric field, especially accounting for the sign of the electric field [311, 312]. However, a vast majority of current first-principles electrochemistry calculations employ older and simpler solvation models missing many of these critical features of newer models, which need further software adoption and benchmarking.

{kind=link}
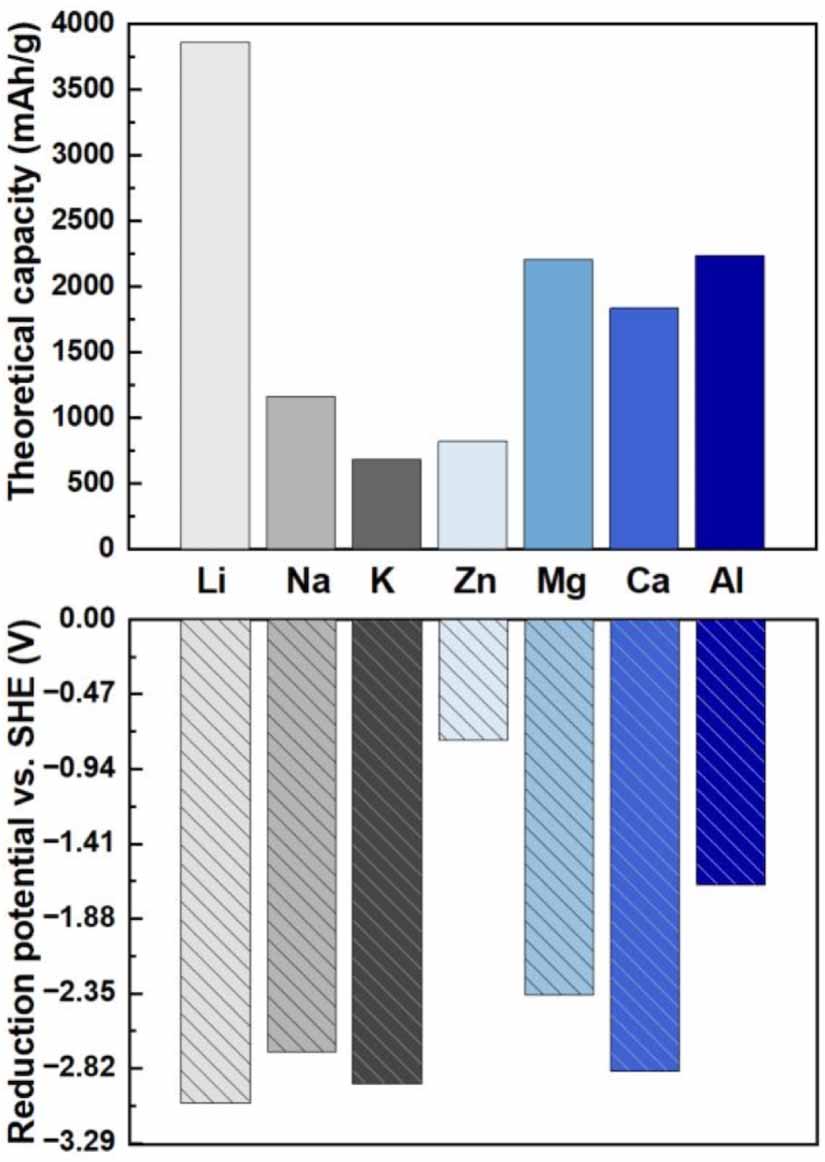{kind=link}
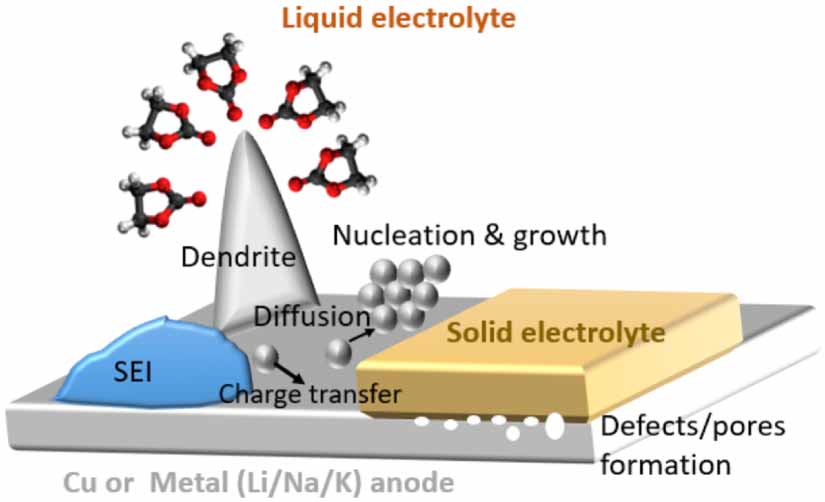{kind=link}
{kind=link}
{kind=link}
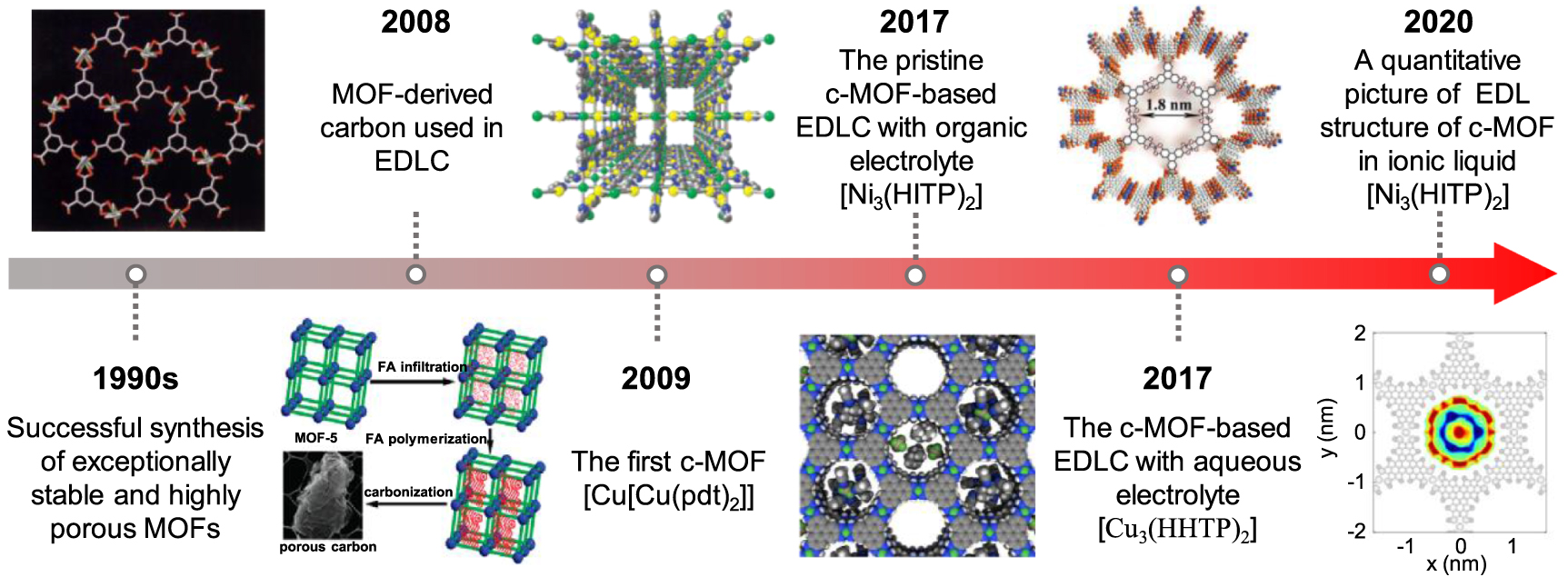{kind=link}
{kind=link}
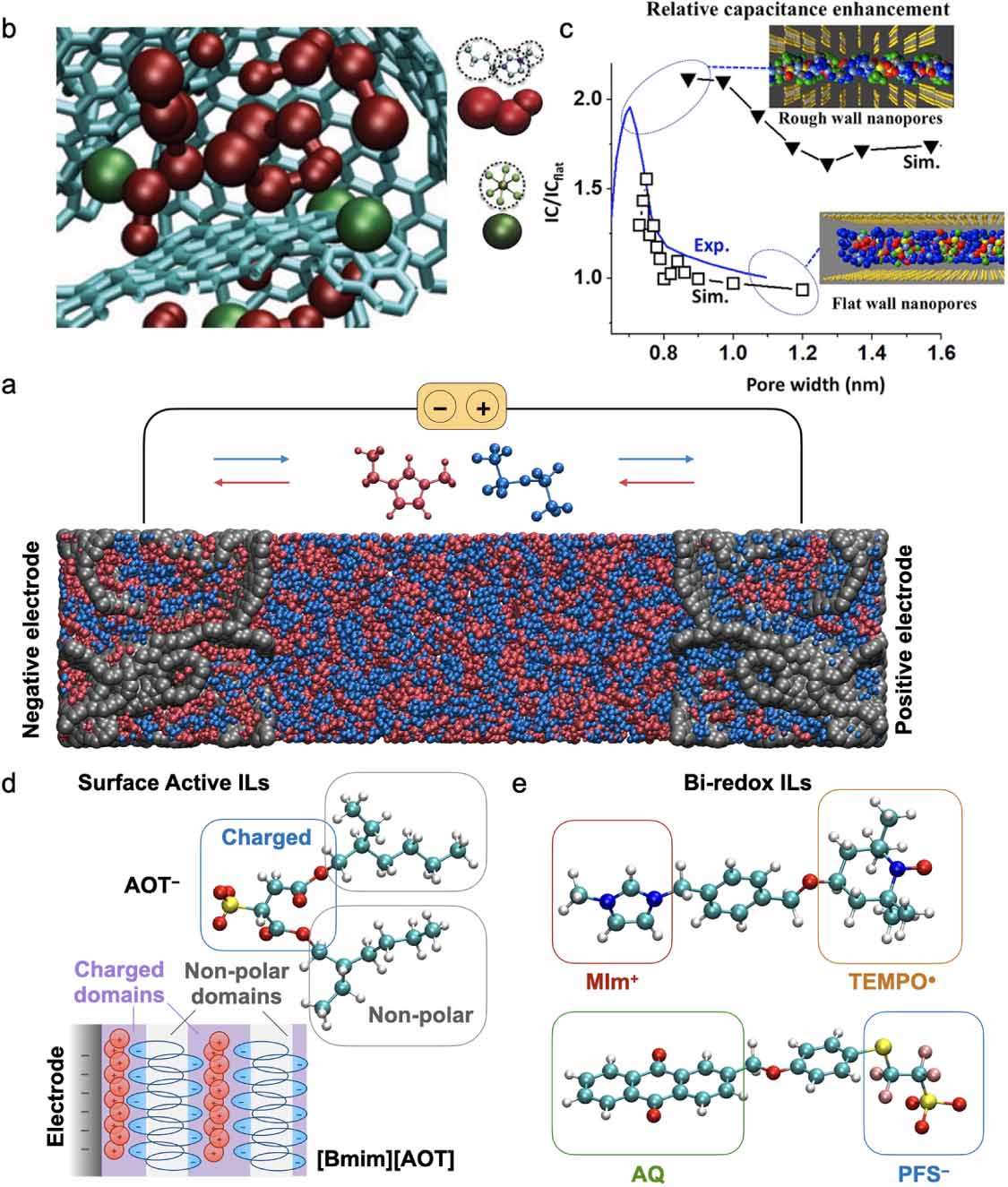{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 20. Advanced implicit solvation models for the electrochemical interface approximate the charge response of solvent and electrolyte ions outside the solvation cavity(ies). Describing electronic response in the electrode–electrolyte gap region, charge delocalization with solvent molecules and chemisorption of ions requires improvements in implicit solvation models or reliable hybrid implicit-explicit approaches, while accurate energy level alignment and charge transfer at the interface require electronic structure beyond density-functional theory.
Download figure:
Standard image High-resolution image{kind=link}
In parallel, advances in explicit solvation using ab initio and machine-learned-potential molecular dynamics present a unique opportunity for improving implicit solvation. (See [313] for a detailed review). Explicit interface simulations can provide critical data on interface energetics, missing from experiments, to improve parametrization of implicit models [314]. Finally, while explicit solvation can be applied for benchmarking and detailed studies of some systems, implicit solvation models for electrochemistry remain necessary to facilitate systematic modelling of reaction mechanisms, to simulate larger/more complex interfaces and to interpret and identify distinct physical effects within explicit solvation predictions.
Current and future challenges
While implicit solvation models have advanced to capture key aspects of idealized electrochemical interfaces, they typically do not account for many common effects at realistic interfaces (figure 20). Broadly, these can be separated into limitations in the solvent structure, electron–solvent interactions and electronic structure.
First, solvation models typically describe idealized non-adsorbing solvent and electrolyte, neglecting effects like ion pairing in the bulk, ion packing effects near the interface at high charges, and ion and solvent chemisorption at the interface. Future solvation models need to capture electrolyte-specific effects, potentially leveraging liquid-structure models that describe atomic-scale liquid structure [315, 316].
The interaction between implicit solvation models and the electronic system is typically limited to electrostatic interactions with energy corrections for secondary effects (e.g. dispersion) [305]. This imposes a strict and artificial partitioning between solute and solvent electrons, precluding effects such as the change of electron distribution at electrode surfaces from vacuum to electrochemical environments (figure 20). In particular, electron delocalization and charge transfer between the electrode and adjacent solvent molecules may play an important role in determining the capacitances of electrochemical interfaces, especially those involving hydrophilic electrodes such as Pt [317]; this is excluded in implicit solvation models by construction. Future solvation models may need to approximate such electronic effects, or work robustly in hybrid solvation schemes with explicit molecules in a first solvent shell.
Finally, the accuracy of first-principles electrochemistry is not limited only by solvation models. While it is difficult to precisely pin down solvation errors for specific electrochemical predictions, table 1 provides order of magnitude estimates for errors in solvation energies and the charging energy of interfaces based on typical capacitance accuracy. Overall, these errors span the 0.04–0.4 eV (1–10 kcal mol−1) range. In comparison, errors in surface and adsorption energies in electronic structure methods can be even larger, especially for the most commonly-used semi-local density functional methods (such as with the PBE exchange–correlation functional reported in table 1). Higher-level electronic structure methods such as the random-phase approximation (RPA) for correlation energies can substantially reduce these errors, but need to be more computationally efficient, robust and compatible with solvation models to become widespread for electrochemical predictions.
Table 1. Improving accuracy of first-principles electrochemistry requires advances in both solvation and electronic structure. Implicit solvation errors in solvation energies and interface charging energies are comparable to electronic structure errors in adsorption energies in vacuum. (Surface energies and charging energy estimated for a ∼ 1 nm2 area, to be comparable with adsorbate calculations at typical coverages considered.).
| Quantity | Typical accuracy |
|---|---|
| Implicit solvation Solvation energy: neutral Solvation energy: ions Surface charging energy | 0.04–0.1 eV [306, 311] 0.1–0.4 eV [308, 311] 0.04–0.1 eV [313] |
| Electronic structure PBE adsorption energies RPA adsorption energies PBE surface energies RPA surface energies | 0.4–0.8 eV [318] 0.1–0.2 eV [318] 0.2–0.3 eV [318] 0.1–0.2 eV [318] |
Advances in science and technology to meet challenges
Implicit solvation models remain vital in first-principles simulations of electrochemical systems, but the accuracy of electrochemical predictions is limited by challenges in solvent structure, electron-solvent interactions and electronic structure, as outlined above. Recent advances in multiple fields have introduced new opportunities to address each of these areas, as we discuss next.
First, advances in liquid-structure theory, based on classical density-functional theory as well as integral equation theory of liquids, hold the key to capturing electrolyte-specific effects in solvation [315, 316]. Early demonstrations of such approaches to electrochemical systems show promise in capturing non-trivial charging and double-layer structure effects [316, 319], but require substantial benchmarking and improvements in robustness and ease of use to supplant current implicit models. This necessary benchmarking for liquid-structure theory, and more broadly for implicit solvation model development, has become easier and faster with improvements in ab initio, classical and machine-learned-potential molecular dynamics.
The interface between the electronic and implicit methods is arguably the most challenging area that future solvation methods need to address. Capturing electron delocalization and charge transfer between solute and solvent requires fundamental modifications to the formulation of solvation methods. For example, this may require electronically-aware solvation models that are capable of exchanging electrons with the solute treated with a grand-canonical electronic structure method. Alternatively, hybrid approaches combining electronic structure, molecular dynamics and implicit solvation for different spatial regions could also capture such effects. Such hybrid methods need improvements in robustness and ease of use, potentially including automatic partitioning of the simulation between different methods [313].
Finally, with increasing accuracy in the next-generation of solvation models, advances in electronic structure methods that can be readily combined with such solvation models will also become necessary. Electronic structure methods such as RPA are promising alternatives to density functional theory (DFT), as they can treat adsorbates and metals with comparable accuracy (in contrast to hybrid-functional DFT), and correctly predict their energy level alignment. However, these methods are still computationally very expensive, difficult to converge, and require more sophisticated interfacing [320] with implicit solvation models than addition of solvation energies from DFT-level calculations [321]. Their computational cost makes their application in explicit solvation prohibitive, underscoring the need to advance beyond-DFT + implicit solvation approaches. Recent developments in machine learning RPA energies using DFT inputs [322] have the potential to make such techniques more accessible for application in first-principles electrochemistry.
Concluding remarks
Implicit solvation models provide a computationally efficient way to describe the solvent and electrolyte in simulations of the electrochemical interface. Current electrochemical solvation models can accurately model idealized electrochemical interfaces, but require advances to capture electrolyte-specific effects, liquid structure and electronic interactions with the electrode. Opportunities also exist in extending solvation models to new environments, such as electrodes and non-aqueous solvents that are important for batteries and electrosynthesis [306], and to treat the impact of solid environments such as surface films of metals [323]. In parallel, improvements in high-quality experimental data on the energetics of solvated and charged interfaces would be invaluable for further development of solvation models for electrochemistry. Finally, a concerted improvement of beyond-DFT electronic structure methods interfaced with next-generation implicit solvation models is necessary for robust and higher-accuracy prediction of electrochemical processes [313].
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Acknowledgments
Ravishankar Sundararaman acknowledges support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award #DE-SC0022247