



Abstract
A common method to assess the performance of (super resolution) microscopes is to use the localization precision of emitters as an estimate for the achieved resolution. Naturally, this is widely used in super resolution methods based on single molecule stochastic switching. This concept suffers from the fact that it is hard to calibrate measures against a real sample (a phantom), because true absolute positions of emitters are almost always unknown. For this reason, resolution estimates are potentially biased in an image since one is blind to true position accuracy, i.e. deviation in position measurement from true positions. We have solved this issue by imaging nanorods fabricated with DNA-origami. The nanorods used are designed to have emitters attached at each end in a well-defined and highly conserved distance. These structures are widely used to gauge localization precision. Here, we additionally determined the true achievable localization accuracy and compared this figure of merit to localization precision values for two common super resolution microscope methods STED and STORM.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Most resolution criteria (Rayleigh [1], Sparrow [2], Houston [3]) used today are a variation of a common scientific approach: they try to define a minimum distance d for which the sum of the diffraction patterns of two point objects are imaged like two individual objects instead of just one. This type of arbitrary resolution criterion additionally disregards noise of any kind [4]. Noise due to photon statistics is inherent in fluorescence imaging and inevitably adds some level of uncertainty; additionally detector noise or sensitivity as well as fluorescent background from the sample are examples of noise sources that can further spoil the separation of two point emitters, or worse feign resolution where there should be none [5].
Authors reporting on super resolution microscopy (SRM) methods based on stochastic single molecule emitter switching have traditionally used a totally different resolution measure: the precision with which the position of a single emitter can be estimated. Prominent representatives of stochastic switching nanoscopy are stochastic optical reconstruction microscopy (STORM) [6], and photoactivated localization microscopy [7], and similar additional variants [8]. These methods all work in principle by shutting down most emitters in a random fashion, such that the remaining emitters are essentially single entities that can be individually localized. By repeating this process with a different subset of emitters, a full super resolution image can be synthetically assembled after mathematically fitting all detected events (i.e. extracting individual coordinates and precision values).
In general terms, nothing speaks against using position precision of single emitters as resolution measure for all type of microscopy techniques, including nanoscopy methods based on targeted switching, such as stimulated emission depletion microscopy (STED [9]), reversible saturable optical fluorescent transitions [10], ground state depletion [11] and structured illumination microscopy [12]. Targeted switching nanoscopy in essence does not shut off emitters in a random fashion, but instead separates emitters (single or few) within certain well-defined regions that are moved across the sample, in order to render a super resolved image.
To allow for trustful resolution criteria to be estimated by localization precision one however needs a well-defined phantom. In other words, for all microscopy techniques, the problem in claiming an absolute resolving power lies in testing how well a recorded image truly resembles the specimen. Localization precision merely measures the deviation of the estimated position when the same emitter pattern is imaged many times with different realizations of noise. This means that the figure of merit for resolution (i.e. a precision estimate) is just a measure of repeatability. Localization precision is not necessarily the same as localization accuracy, which denotes the deviation from the true position of the emitters in the sample [13, 14]. In particular stochastic switching methods are known to stray systematically from the true value due to effects of molecular dipole orientation, for example [15–17]. They are by their parallel nature also more susceptible to sample drift than microscopy based on targeted methods. Additional sources for bias are e,g. detector noise and background signal [14]. As a result, repeated measurements on the same fixed dipole (or an estimation of the deviation using statistical methods on a single measurement) may indicate high precision but in fact hide poor accuracy.
Unfortunately, accuracy is experimentally hard to determine from a recorded image, as it requires an a priori knowledge of the sample. In order to get an accurate fix on true positions, one would need a highly repeatable sample (i.e. a phantom) with a well-known structure and several reference points that can be exactly pinpointed. The estimated imaged position of the emitters can then be related to the true position established by the reference points and the quality of the estimation (i.e. the localization accuracy) can be worked out. Presently, biological test samples in use are for instance actin filaments [18] or microtubule [19], since there is at least some degree of a priori knowledge about those structures. Yet, these cytoskeletal structures are far from being ideal test samples in a sense that absolute position accuracy is hard to infer from the structures as fluorescence labeling and biological variability can introduce structural variances.
In contrast, DNA-origami offers a way to manufacture structures on the required length scale with high confidence and repeatability. Actually, the use of the system has already been demonstrated as a molecular ruler for fluorescence microscopy [20–22]. There, the authors have both looked at the localization precision of single spots as well as the distance between double-emitters. However, variation of the distance between multiple nanorods has not been, to our knowledge, used to extract the true localization accuracy or for a comparison of nanoscopic measurements. Also, for the first time we report on localization accuracy for STED.
Here, we show how to make use of DNA-origami, and in particular its repeatability, in order to report localization accuracy instead of just precision measures. By imaging a simple DNA-origami system with fluorophores attached to it, an estimate of true resolution in terms of localization accuracy is generated. This allows better comparison of nanoscopy methods as well as the generation of more unbiased claims of resolving powers. By varying the fluorophores and the specifics of the sample preparation (embedding medium, etc), this approach can be used to characterize the faithfulness for virtually any type of fluorescent microscopy technique.
Results
Our goal is to estimate the localization accuracy of 2D fluorophore positions using a simple well-characterized nanoscale test specimen for two of the most popular SRM methods, STORM and STED. The simplest such sample consist of two point-like emitters separated by a robustly resolvable, highly conserved and uniform distance. For creating such a sample we chose to use DNA-origami to fabricate 140 nm long nanorods, whose design (figures 1, S1 is available online at stacks.iop.org/NJP/19/025013/mmedia), apart from being one of the simplest approaches of defining two positions with a smallest amount of material used, provided a stiff scaffold for the attached fluorophores, reducing the contribution of the sample to the variability of the measured localization accuracy [22]. To provide a sample suitable for both SRM techniques in terms of signal strength and resolvability we designed the nanorods to have twelve–twelve designated single-stranded DNA binding sites spaced approximately 100 nm apart along the rod, where we selectively attached AlexaFluor488- or CAGE 552-modified complementary oligonucleotides, giving us nanorods with two points of emitters separated by approximately 100 nm (figures 1(A), (B)). Although DNA origami's applicability to produce a robust platform for such applications have already been established [20–22], we characterized the nanorods in order to have an estimate of the extent of the sample's contribution to the variance of localization accuracy by measuring the two potential, main sources of variance originating from the sample: the variance of the distance between binding sites caused by distortion of the structure and by the sub-optimal incorporation of anchoring oligos (figure S3). We estimated the contribution of the rod's distortion to the variance of distance by measuring the longitudinal length distribution of the DNA-origami structures on negative-stained transmission electron microscopy (TEM) micrographs (figures 1(C), S5), as it has shown to be a faithful representation of the designed and solution phase dimensions of DNA nanostructures following the same design paradigm as our nanorods by works of other groups [23–27] and by our comparative measurements using Cryo-EM (figure S2). As expected, the rods' measured average length of 141.596 nm closely matched the designed length of 140. 365 nm with the relatively small variance of 16. 638 nm2 (figure S3(A)). We measured the variability originating from the incomplete incorporation of fluorophores to be even smaller, as with the measured 96.057% occupancy we estimated the distance variance to be 0.0521 nm2 (figure S3(B)). Because of the high homogeneity and precision DNA-origami provides the magnitude of the variances originating from the sample, as discussed before, are much lower than one observes in biological samples. This along the fact that the effect of the measured variances would contribute uniformly to the measurements with both methods, and that they were well below the resolution limit of the probed SRM techniques convinced us that our DNA-based structures provide a suitable sample to be used as a reference to compare localization accuracy.

Figure 1. Nanorods fabricated using DNA-origami. (A) Computer rendering of the DNA-origami nanorod with 12-12 addressable labeling-sites at approximately 100 nm distance. (B) 2% agarose gel run with a 1 kb DNA ladder (i), (vi), scaffold ssDNA (ii), empty origami nanorod (iii), 100 nm origami nanorod (iv) and 100 nm origami nanorod labeled with AlexaFluor488 imaging-oligo (v) stained with ethidium bromide and imaged with UV- (a) and 489 nm illumination (b). (C) Field-of-view electron microscopy micrograph of origami nanorods (scale bar 100 nm).
Download figure:
Standard image High-resolution imageFor each nanorod, we estimated the position of the individual emitters at both ends, using a maximum-likehood estimation for STED or centroid estimation on localized clusters for STORM data. For N nanorods this gives us a set of positions  These are samples of normal distributed random variables
These are samples of normal distributed random variables  and
and  with a covariance matrix
with a covariance matrix  which we assume to be diagonal with equal elements
which we assume to be diagonal with equal elements  along the diagonal. The
along the diagonal. The  and
and  are the unknown true positions of each emitter. Now, one would be tempted to estimate the position accuracy using the variance (or standard deviation) of the localization of a single emitter, as it is usually done [28]. However, this is a biased estimator since the position of an emitter in the image may be shifted with respect to its true position in object space [15–17]. We instead choose to use the distance measure
are the unknown true positions of each emitter. Now, one would be tempted to estimate the position accuracy using the variance (or standard deviation) of the localization of a single emitter, as it is usually done [28]. However, this is a biased estimator since the position of an emitter in the image may be shifted with respect to its true position in object space [15–17]. We instead choose to use the distance measure  between two imaged emitters, which has the following advantages: first, we can average over a large number of equally spaced samples from the same population (the position of two emitters is never the same, but the distance is highly conserved) and second, we may use one emitter of each nanorod as a reference point to the other emitter. Simply put, the first emitter defines a coordinate origin for measuring the position of the second emitter and vice versa.
between two imaged emitters, which has the following advantages: first, we can average over a large number of equally spaced samples from the same population (the position of two emitters is never the same, but the distance is highly conserved) and second, we may use one emitter of each nanorod as a reference point to the other emitter. Simply put, the first emitter defines a coordinate origin for measuring the position of the second emitter and vice versa.
In general, the variance of the distance is  but for distances larger than the resolution, we assume that the two localizations are independently resolved and
but for distances larger than the resolution, we assume that the two localizations are independently resolved and  Furthermore, we expect the variances for both labeled positions to be the same on average and thus approximate
Furthermore, we expect the variances for both labeled positions to be the same on average and thus approximate  With this, we can estimate localizability by estimating the variance of the distance and dividing by two. Importantly, since
With this, we can estimate localizability by estimating the variance of the distance and dividing by two. Importantly, since  captures any apparent position shifts due to e.g. dipole orientation, we expect
captures any apparent position shifts due to e.g. dipole orientation, we expect  to be an unbiased estimator reporting on the variance of localization with respect to the true position in object space, i.e. localization accuracy.
to be an unbiased estimator reporting on the variance of localization with respect to the true position in object space, i.e. localization accuracy.
In order to confirm this, we conducted Monte Carlo simulations by artificially generating a large number of images of two-point emitters at specified distances d, estimating their positions from the images [29] and then estimating  from the distances
from the distances  This was compared to the biased position variance
This was compared to the biased position variance  from single emitters obtained by simply taking the localization precision of each emitter position [8]. We also compared this to the usual position estimator [28] given by
from single emitters obtained by simply taking the localization precision of each emitter position [8]. We also compared this to the usual position estimator [28] given by  i.e. the Cramér–Rao lower bound (CRLB) for estimation of a single emitter under Poisson noise. Here,
i.e. the Cramér–Rao lower bound (CRLB) for estimation of a single emitter under Poisson noise. Here,  is the standard deviation of the width of the Gaussian point spread function (PSF) and P is the average number of fluorescent photons collected from one emitter.
is the standard deviation of the width of the Gaussian point spread function (PSF) and P is the average number of fluorescent photons collected from one emitter.
Figure 2 shows the simulation results for the estimated localizability for DNA nanorods imaged with a Gaussian PSF with standard deviation s, i.e. FWHM  The expected number of detected photons from each emitter was set to 100. At this point we have not yet introduced any position bias, i.e. the images of single emitters are not shifted with respect to the true position [15–17]. Also, we have not restricted our analysis to a specific microscopy method yet. For
The expected number of detected photons from each emitter was set to 100. At this point we have not yet introduced any position bias, i.e. the images of single emitters are not shifted with respect to the true position [15–17]. Also, we have not restricted our analysis to a specific microscopy method yet. For 
 is only slightly above the CRLB, confirming that
is only slightly above the CRLB, confirming that  can be faithfully taken to estimate the position precision.
can be faithfully taken to estimate the position precision.  is close to the theoretical bound, too, showing that the accuracy estimated from a single emitter is correct for the case of unbiased positions with large separation. As d approaches 2s,
is close to the theoretical bound, too, showing that the accuracy estimated from a single emitter is correct for the case of unbiased positions with large separation. As d approaches 2s,  and
and  are expected to increase as we are not working in the single emitter regime anymore and the position estimations start to mutually affect each other. However, for
are expected to increase as we are not working in the single emitter regime anymore and the position estimations start to mutually affect each other. However, for  this bias increase is less steep and less influential for the localization (see figure 2(A)). Conveniently, this prevents us from having to use very long nanorods to test for example confocal or widefield microscopes, where large nanoscale DNA structures might lead to an increased spread in emitter separations. For nanorods longer than 200 nm the variance originating from the sample is becoming a more significant factor as at these lengths curling and bending of the structure are harder to avoid, as we have also observed (see figure S4), leading to an associated significantly higher variance of true emitter distance [22].
this bias increase is less steep and less influential for the localization (see figure 2(A)). Conveniently, this prevents us from having to use very long nanorods to test for example confocal or widefield microscopes, where large nanoscale DNA structures might lead to an increased spread in emitter separations. For nanorods longer than 200 nm the variance originating from the sample is becoming a more significant factor as at these lengths curling and bending of the structure are harder to avoid, as we have also observed (see figure S4), leading to an associated significantly higher variance of true emitter distance [22].

Figure 2. Simulations of position accuracy estimation using distance. (A) Position accuracy  estimated using the distance variance between two emitters separated by various distances d. For d down to ∼2.5s,
estimated using the distance variance between two emitters separated by various distances d. For d down to ∼2.5s,  yields accurate position precision in accordance with the single emitter precision
yields accurate position precision in accordance with the single emitter precision  for large d. Also, our estimates come close to the theoretical lower bound for position precision in the applicable distance ranges. (B)
for large d. Also, our estimates come close to the theoretical lower bound for position precision in the applicable distance ranges. (B)  accurately reproduces position bias
accurately reproduces position bias  for total variances lower than the emitter separation. In contrast,
for total variances lower than the emitter separation. In contrast,  only captures position variance due to noise. (C) With high read-out noise levels of ∼10% of the peak intensity (∼10 counts),
only captures position variance due to noise. (C) With high read-out noise levels of ∼10% of the peak intensity (∼10 counts),  still produces correct results. For all simulations, we tested 1000 images, P = 100, pixel size = s/2. In (b), (c) d = 2.5s.
still produces correct results. For all simulations, we tested 1000 images, P = 100, pixel size = s/2. In (b), (c) d = 2.5s.
Download figure:
Standard image High-resolution imageNext, we establish that the mutual distance precision  indeed reports on position bias. To this end, we assume that position bias is normally distributed and we repeat our simulations with the position of each emitter image shifted by a random vector sampled from a normal distribution with variance
indeed reports on position bias. To this end, we assume that position bias is normally distributed and we repeat our simulations with the position of each emitter image shifted by a random vector sampled from a normal distribution with variance  which might in a real sample be caused by drift or fixed transition dipoles, for example. Since the variance of the sum of two independent normally distributed variables is the sum of the individual variances, the correct position accuracy is
which might in a real sample be caused by drift or fixed transition dipoles, for example. Since the variance of the sum of two independent normally distributed variables is the sum of the individual variances, the correct position accuracy is  The comparisons are shown in figure 2(B). Here we used d = 2.5s to stay just in the single emitter regime. As expected, the traditional estimator
The comparisons are shown in figure 2(B). Here we used d = 2.5s to stay just in the single emitter regime. As expected, the traditional estimator  only incorporates deviations due to noise and thus fails to capture the true position accuracy. In contrast, our estimator based on distance
only incorporates deviations due to noise and thus fails to capture the true position accuracy. In contrast, our estimator based on distance  correctly reports position accuracy, even under conditions with high readout noise (see figure 2(C)). Please note that in figures 2(B), (C),
correctly reports position accuracy, even under conditions with high readout noise (see figure 2(C)). Please note that in figures 2(B), (C),  is taken in the usual way for methods using stochastic single molecule switching, i.e. sequentially. Thus,
is taken in the usual way for methods using stochastic single molecule switching, i.e. sequentially. Thus,  is not influenced by the other emitter, in contrast to figure 2(A).
is not influenced by the other emitter, in contrast to figure 2(A).
Next, we applied our method to two common nanoscope methods, namely STED and STORM. To this end, we used DNA-origami nanorods with two groups of point-emitters attached, seperated by approximately 100 nm (figure 1). Figures 3(A) and (B) shows representative fluorescence nanoscopy images and figures S6–7 shows raw overview images of the imaged nanorods for each microscopy method, which allows a visual inspection of the nanoscale structures. From the images, the localization accuracy for  is then deduced. The claimed and expected resolution for the single molecule stochastical STORM image would normally be taken from its generated precision histogram (see figure 3(D)), which has a mode of 8 nm or 64 nm2 localization precision. This is an exceptional value that is a result of the high number of photons recordable from the caged dye. Yet, this figure does not testify on the correlation between recorded image and specimen ex ante and on top, the precision distribution is skewed with a long tail towards poorer localization precision. Only the distance measurements reveal with confidence that the localization accuracy is indeed in this range, namely 70.2 nm2 (figure 3(D)). The localization accuracy for STED (
is then deduced. The claimed and expected resolution for the single molecule stochastical STORM image would normally be taken from its generated precision histogram (see figure 3(D)), which has a mode of 8 nm or 64 nm2 localization precision. This is an exceptional value that is a result of the high number of photons recordable from the caged dye. Yet, this figure does not testify on the correlation between recorded image and specimen ex ante and on top, the precision distribution is skewed with a long tail towards poorer localization precision. Only the distance measurements reveal with confidence that the localization accuracy is indeed in this range, namely 70.2 nm2 (figure 3(D)). The localization accuracy for STED ( nm2) is found to be slightly better and moreover accurately complies with what one would expect using the FWHM
nm2) is found to be slightly better and moreover accurately complies with what one would expect using the FWHM  and the number of photons (P = 8.4) estimated from the STED-images together with the equation
and the number of photons (P = 8.4) estimated from the STED-images together with the equation  nm2. The underlying reason for localization precision and localization accuracy being similar is most likely that STED is known to show little position bias caused by fluorophore orientation [15] and, additionally, since read-out is spatially and temporally correlated, STED is locally not very susceptible to sample drift. For STORM, the similarity between the values shows that we have successfully reduced drift and that we have dipoles that are rotationally diffusing. We note that while the distance estimated with STED (104.1 nm) comes very close to the designed distance (105.5 nm), STORM with an estimation of 98.5 nm seems to slightly underestimate it. We attribute this to the well known, minimizable but not entirely avoidable, phenomena that is inherent to localization-based imaging techniques, namely the occurrence of simultaneous multi-molecular emission events. In the case of our nanorods, this can mistakenly result in single localizations with a position closer to the middle of the rod, leading to a shrinking of the overall measured distance [30].
nm2. The underlying reason for localization precision and localization accuracy being similar is most likely that STED is known to show little position bias caused by fluorophore orientation [15] and, additionally, since read-out is spatially and temporally correlated, STED is locally not very susceptible to sample drift. For STORM, the similarity between the values shows that we have successfully reduced drift and that we have dipoles that are rotationally diffusing. We note that while the distance estimated with STED (104.1 nm) comes very close to the designed distance (105.5 nm), STORM with an estimation of 98.5 nm seems to slightly underestimate it. We attribute this to the well known, minimizable but not entirely avoidable, phenomena that is inherent to localization-based imaging techniques, namely the occurrence of simultaneous multi-molecular emission events. In the case of our nanorods, this can mistakenly result in single localizations with a position closer to the middle of the rod, leading to a shrinking of the overall measured distance [30].

{kind=link}
{kind=link}
Figure 3. Position accuracy estimation for various super resolution techniques. (A), (B) Representative images of nanorods with emitters at ∼100 nm separation imaged with (A) STORM and (B) STED. Note that the position of each emitter in (A) is the average position of a multitude of localizations. (C) Comparison of distance distributions for STORM (left) and STED (right). STORM underestimates the designed distance of 105.5 nm by a small amount, while the distance estimated with STED falls close to it (also see S8). (D) Measured position accuracy and precision for STORM and STED. We show the accuracy (blue) as estimated by  Green bars indicate the localization precision. Bars labeled 'calculated' were determined from the estimated FWHMs and numbers of photons. Additionally, the bar labeled 'reported' is given as the distribution of the precision as reported by the Zeiss STORM software, i.e. including precision degeneration due to background. See figure S8 for more information.
Green bars indicate the localization precision. Bars labeled 'calculated' were determined from the estimated FWHMs and numbers of photons. Additionally, the bar labeled 'reported' is given as the distribution of the precision as reported by the Zeiss STORM software, i.e. including precision degeneration due to background. See figure S8 for more information.
Download figure:
Standard image High-resolution image{kind=link}
Discussion
In this work, we show that true localization accuracy in fluorescence microscopy can be determined from measurement of the distance variation of a sample containing pairs of emitters at an accurate and repeatable separation. In particular, super resolution methods based on stochastic switching are sensitive to position bias, predominantly caused by changes due to drift and fluorophore orientation [14, 30]. We carefully note that while our data shows little such effects, we can safely conclude this only from localization accuracy measurements via the emitter distance, as the very same effects precisely do not manifest themselves in the localization precision (figures 2(B), (C)).
The fact that STED shows approximately the same accuracy comes not as a surprise when considering that 2D-STED on a single emitter results in roughly four times less signal in total for a FWHM decrease by a factor of two. These 'low-resolution' photons are carved away in the outer parts by the STED-donut as the area of effective fluorescence shrinks quadratically with decreasing width. (Note that we assume peak height to remain the same.) These effects counter-act in the equation  resulting in a localization accuracy for a single emitter that is approximately equal for STORM and STED.
resulting in a localization accuracy for a single emitter that is approximately equal for STORM and STED.
With our fluorescently labeled DNA-origami structures, localization accuracy can be unambiguously measured. These nanostructures can be incorporated in specimens as fiducial markers and performance probes. If needed, the accuracy achievable with a particular labeling strategy can be measured by replicating the same situation on the nanorods, i.e. by attaching conjugated antibodies or fluorescent proteins to the DNA structure [31]. For gauging 3D resolution, nanopillars [32] could be used in much the same way.
Finally, we would like to point out that localization accuracy should not be confused with resolution. For stochastic switching methods, the degree of correlation between specimen and image not only depends on localization accuracy; for example, on/off times of the fluorophores have to be carefully balanced in order to guarantee sparsity of emitters in each frame as much as possible. With targeted switching methods, most specimen of interest will contain dense emitter clusters that inherently cannot be resolved to the single emitter limit if they are closer than the width of the PSF. Nonetheless, because localization accuracy reports not only on PSF-size, but also on the photon levels at which it can be achieved, it gives a better idea than PSF-size alone. In order to obtain a full picture of the achievable resolution, nanorod samples with different emitter spacing could be tested in order to check for the emitter separation that just results in an increase in localization accuracy as shown in figure 2(A). In any case, localization accuracy is eminently suitable to analyze day-to-day performance of microscopes using the same technique, and to get an accurate fix on true localization accuracy.
Methods
Simulations
All Monte Carlo simulations were performed in MATLAB. We model the image of an emitter at position  by assuming a Gaussian PSF of the form
by assuming a Gaussian PSF of the form
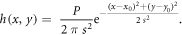
For realistically simulating a dual emitter nanorod, we sum two PSFs at random positions with the constraint that their distance is either fixed or modified by a random bias for each position. We subsequently add Poisson noise and Gaussian noise, if applicable.
Light microscopy
We imaged our test specimen on two different setups. For STED, we used a Leica SP5 gated cwSTED with a 100x/1.4 Oil objective lens. Excitation was at 488 nm; the STED line was 592 nm. Imaging was performed at 100 Hz line frequency for a field of view of 512 × 512 pixels at 4x line averaging. One scan step was 20 nm. STED laser power was set to 60%. Fluorescence was gated between 1 and 6.5 ns. STORM was performed on a Zeiss Elyra PS.1 with a 100X/1.4 oil objective. We excited at 561 nm and simultaneously uncaged at 405 nm. The exposure time was 100 ms and camera gain set to 100.
Position estimation
The procedure for estimating the positions of emitters (for simulated as well as experimental data) depends on the type of microscopy. For STED and our simulations, we take raw images  and perform a maximum-likelihood-estimation by minimizing [33]
and perform a maximum-likelihood-estimation by minimizing [33]
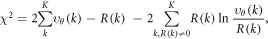
where  is the model function
is the model function  The sum runs over all pixels
The sum runs over all pixels  of the image.
of the image.  are the parameters to be determined. For minimization, we use MATLAB's fminsearch function. STORM data was first processed using the ZEN software (Zeiss) with default settings. Then, we selected emitter pairs in MATLAB and either performed k-means clustering to segment the localizations into two groups for each emitter and used the centroid of each cluster as the position estimation for the corresponding emitter, or performed least-squares fitting on the molecular density.
are the parameters to be determined. For minimization, we use MATLAB's fminsearch function. STORM data was first processed using the ZEN software (Zeiss) with default settings. Then, we selected emitter pairs in MATLAB and either performed k-means clustering to segment the localizations into two groups for each emitter and used the centroid of each cluster as the position estimation for the corresponding emitter, or performed least-squares fitting on the molecular density.
Materials used for DNA-origami nanorod fabrication
Non-modified staple and the 3' AlexaFluor488-modified oligonucleotides were purchased from Bioneer. the 3' CAGE 552-modified oligonucleotides were purchased from IBA GmbH (sequences can be found in table S1). Unless otherwise stated, all other chemicals were purchased from Sigma-Aldrich.
Design of the DNA-origami nanorod
The DNA-nanorod was designed with caDNAno software (cadnano.org). The main-body of the DNA-nanorod is a DNA eighteen-helix-bundle with twelve oligonucleotides in both of the labeling zones, placed approximately 100 nm apart (105, 5 nm), extended at the 5'-end with 21-base long attachement-sites for fluorescent labeling with imaging-oligonucleotides (figure 1(A)). The strand-diagram of the structure of the nanorod (figure S1) along with the sequences of the staple-oligonucleotides used (table S1) can be found in the supplement.
Folding and quality assessment of the DNA-origami nanorods
The DNA-origami nanorods were prepared by mixing 20 nm scaffold (p7560, derived from M13mp18 [23]) and 100 nm of each staple oligonucleotide in a buffer containing 5 mM Tris, 1 mM EDTA (pH 8.5 at 20 °C), and 13 mM MgCl2. Folding was carried out using a PCR cycler by rapid heat denaturation at 80 °C, followed by slow cooling from 80 °C to 60 °C over 20 min, then another cooling step from 60 °C to 24 °C over 15, 5 h. The quality of folded DNA-origami nanorods were assessed with agarose gelelectrophoresis (figure 1(B)) and TEM (figure 1(C)).
Electron microscopy imaging of the DNA origami nanorods
A 3 μl aliquot of the folded nanorods was applied on a glow-discharged, carbon-coated Formvar grid (electron microscopy sciences), incubated for 20 s, blotted off with filter paper, and then stained with 2% (w/v) aqueous uranyl formate solution. EM analysis was performed using a FEI Morgagni 268(D) TEM at 80 kV with nominal magnifications of 22 000. Images were recorded digitally by using the Advanced Microscopy Techniques Image Capture Engine 5.42 software.
Cryo-electron microscopy imaging of the DNA origami nanorods
Cryo-specimens for electron microscopy were prepared using Vitrobot Mk2 (FEI). A 3 μl aliquot of the folded nanorods (in storage buffer (5 mM Tris, 1 mM EDTA, 10 mM MgCl2 and pH 8.5)) was applied on a glow-discharged holey-carbon grid, was incubated for 1 min at the relative humidity of 90%–100% and was frozen in liquid ethane after 2–3 s of blotting and 1 s of draining. Cryo-EM analysis was performed, after the grid was placed into a GATAN 626 cryo-holder, using a FEI CM200 FEG microscope under low-dose conditions with nominal magnifications of 50 000. Images were recorded digitally by using the a TVIPS TemCam F214 charge-coupled device camera.
Purification of the DNA-origami nanorods after folding
The excess of staple oligonucleotides was removed after folding using Amicon Ultra 100 K spin columns (Millipore). The samples were loaded into a pre-wetted spin column and spun at 14 000 g for 2 min at room temperature. The sample was then diluted with storage buffer (5 mM Tris, 1 mM EDTA, 10 mM MgCl2 and pH 8.5) and spun down again. The spinning-washing step was repeated 6 times before retrieval of the samples.
Labeling and purification of DNA-origami nanorods
DNA-origami structures were labeled with imaging-oligos containing 3' AlexaFluor488- or CAGE552-modification for STED and STORM experiments respectively (see table S1 for sequences). The sampled were prepared by mixing 10 nM of DNA-origami structure solution with 4-times molar excess of imaging-oligo solution per attachment-site. The annealing of the imaging-oligos was then carried out with an annealing-program on a thermo-cycler, consisting of 1 h incubation at 37 °C, then a cooling step from 37 °C to 22 °C over 2.5 min, followed by incubation on 22 °C for 14 h, finally a cooling step from 22 °C to 4 °C. The AlexaFluor488-labeled DNA nanostructures were purified from the excess of imaging-oligos via agarose gel-extraction using the pellet-pestle method [34]. The CAGE552-labeled DNA nanostructures were purified from the excess of imaging-oligos via size-exclusion purification with Sephacryl S300HR to avoid exposure of the sample to UV-light.
Immobilization of DNA-origami nanorods on coverslips
The AlexaFluor488-labeled DNA-origami samples were diluted to 35 pM with storage buffer (5 mM Tris, 1 mM EDTA, 10 mM MgCl2 and pH 8.5) containing DABCO, the CAGE552-labeled DNA nanostructures were diluted to 35 pM with standard storage buffer. 20 μl aliquot of the diluted DNA-origami sample was spotted on a glow-discharged, 18 mM coverslip (VWR), incubated for 10 min in the dark at room temperature, then coverslips were washed with storage buffer (5 mM Tris, 1 mM EDTA, 10 mM MgCl2 and pH 7.8) with or without DABCO, blotted off and inverted on a microscopy slide (VWR).
Measurement of site-occupancy of DNA-origami nanorods
AlexaFluor488-labeled DNA-origami nanorods were prepared as described before. The excess of AlexaFluor488-labeled oligos was removed by purification with Amicon Ultra 100 K spin columns (Millipore). The concentration of DNA-nanorods was determined with UV absorption-measurements. The concentration of the AlexaFluor488-oligos attached to the nanorods was determined by measuring the fluorescence emission (Exc.:489 nm, Em.:519 nm) of the nanorods and converting the obtained fluorescence-intensity values to AlexaFluor488 concentration values with the help of a standard curve of AlexaFluor488-oligo concentration. The average occupancy value was then calculated from the concentration of DNA nanorods and the concentration of the attached AlexaFluor488 molecules. All the before described measurements were performed with a BioTEK SynergyMx Plate-reader in triplicates (see figure S3).
Measurement of length-distribution of DNA-origami nanorods
The sample preparation and image-acquisition of DNA-origami nanorods using TEM was performed as described before. The picking and alignment of images of individual nanorods from TEM-images was done by using Xmipp 3.1 software. The lengths of the nanorods were determined from the images of single nanorods with a custom written MATLAB program by first transforming the images using the Canny edge detection algorithm to decrease the noise and subsequently calculating the distance between the farther most peaks in the pixel intensity/pixel position plot of the ROI assigned around the nanorods in the images. The length values of the nanorods with undetected edges were manually discarded, and the length distribution of nanorods was then calculated from the remaining length values (see figure S5).
Acknowledgments
This work was supported by the European Commission under the Seventh Framework Programme (FP7), as part of the Marie Curie Initial Training Network, EScoDNA (GA no. 317110), the Märta and Gunnar V Philipson foundation, the Erling Persson family foundation and by the Swedish Research Council (VR 2015-04198) and 2013-6041.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.