


Abstract
The size-dependent exciton dynamics of one-dimensional aggregates of substituted perylene bisimides are studied by ultrafast transient absorption spectroscopy and kinetic Monte-Carlo simulations as a function of the excitation density and the temperature in the range of 25–90 °C. For low temperatures, the aggregates can be treated as infinite chains and the dynamics is dominated by diffusion-driven exciton–exciton annihilation. With increasing temperature the aggregates dissociate into small fragments consisting of very few monomers. This scenario is also supported by the time-dependent anisotropy deduced from polarization-dependent experiments.
Export citation and abstract BibTeX RIS
Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
In future, supramolecular organic structures will play an important role in photonic applications such as organic solar cells [1] or optoelectronics [2, 3]. In these devices, the behavior of excitons is crucial for their performance [4, 5]. Aggregates of chromophores [6] are attractive building blocks for such applications since they can host mobile excitons and energy can be transferred along them via exciton diffusion [7–9]. This also holds for J-aggregates of substituted perylene bisimides as was observed by single-molecule spectroscopy [10]. Recently, we showed that excitons on aggregates of the perylene bisimide N,N'-di[N-(2-aminoethyl)-3, 4, 5-tris(dodecyloxy)benzamide]-1,6,7,12-tetra(4-tert-butylphenoxy)-perylene-3,4:9,10-tetracarboxylic acid bisimide (PBI-1, see scheme

Scheme 1. (a) Structure formula of the investigated dye (PBI-1) and (b) structural model of a small aggregate consisting of six monomers [13]. The aggregates are stabilized by the interaction between the π-systems of the central perylene chromophore of adjacent monomers and the formation of hydrogen bonds between oxygen atoms and the NH-groups of the terminal substituents.
Download figure:
Standard imageTo investigate the behavior and mobility of the excitons and their interaction with each other, we study the time-dependent exciton population and the dynamics induced by exciton–exciton annihilation with ultrafast pump–probe absorption spectroscopy. Our previous studies were restricted to very long aggregates, which were treated as infinite chains [11]. This poses the question of what are the circumstances under which this assumption is justified and what happens in the case of short aggregates. Here we apply our methodology to aggregates with different lengths and compare our results with a hopping model that uses the kinetic Monte-Carlo technique and can treat chains of finite length.
Concentrations above 10−5 M of PBI-1 in MCH are used, as the dye forms J-aggregates at room temperature in this concentration regime but dissociates at higher temperatures. This can be seen from the temperature-dependent absorption spectra shown in figure 1 for a PBI-1 solution in MCH with a concentration of 0.2 mM. At low temperatures the first absorption band is narrower, more intense and red-shifted compared to the corresponding band at high temperatures as is expected for the lowest excitonic transition of a J-aggregate compared to its counterpart in the monomer [18]. Accordingly, temperature is a suitable parameter to control the size of the aggregates [19, 20].
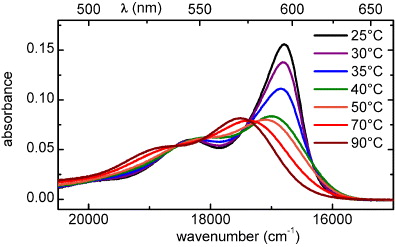
Figure 1. Absorption of PBI-1 in MCH (c = 0.2 mM) for different temperatures.
Download figure:
Standard imageIn the following, the applied experimental techniques and modeling approaches are introduced and then temperature-dependent ultrafast transient absorption experiments are presented and compared with Monte-Carlo simulations. From the results, we conclude on the mobility and interaction of the excitons and the dissociation behavior of the aggregates.
2. Experiment
The transient absorption measurements are made with a pump–probe setup equipped with a non-collinearly phase matched parametric amplifier [21] delivering excitation pulses at 530 nm with pulse durations of 35 fs after compression with a prism sequence. A white light continuum [22] generated in a 4 mm thick calcium fluoride substrate [23] serves as the probe and is dispersed after the sample by a fused silica prism and spectrally resolved detected by a photodiode array. The setup is pumped by a regenerative Ti:sapphire laser system (CPA 2001; Clark MXR) which provides ultrashort pulses at a wavelength of 775 nm and a repetition rate of 1 kHz. A combination of two wire-grid polarizers and an achromatic half-wave plate allows us to adjust independently the energy and polarization of the excitation pulses. The delay time between the pump and the probe is scanned with a motorized linear stage equipped with a retroreflector in the beam path of the pump light. The excitation pulses are focused to a spot diameter of 220 μm at the sample, while the probe diameter is two times smaller there.
The sample solution is filled into a fused silica cell with a layer thickness of 1 mm. The cell is placed in a matching copper block which is electrically heated and which has two holes for the entering and exiting laser beams. Two thermocouples are used to measure the temperature and to provide a feedback signal for the temperature control unit. The temperature can be determined with an accuracy of 2.5 °C.
3. Theoretical models
3.1. Diffusion approach
By integrating a suitable spectral interval of the transient spectra, as will be described below, one obtains a signal which is proportional to the density of the excitons. In a first approach to understand the decay dynamics of this signal at different temperatures, it is assumed that it can be decomposed into two main contributions. The first contribution is from the mainly annihilation-driven decay of the excitons on the aggregates, which can be described by the differential equation [7]

where n(t) is the time-dependent exciton density, τ1 is the intrinsic lifetime of the excitons and γ(t) is the annihilation rate. The factor of 1/2 within the second term takes into account that one of two interacting excitons survives the annihilation process. According to our previous work on PBI-1 aggregates, the exciton motion on the molecular chains is diffusive and restricted to one dimension. In this case γ(t) depends on the diffusion constant D, the lattice constant a and the molecule density N0 according to the equation

Equations (1) and (2) hold strictly only for infinite chains. Integration of equation (1) leads to [24]
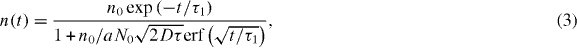
where n0 is the initial exciton density in the sample directly after the pump pulse. The second contribution to the signal is caused by the radiative decay of excited PBI-1 monomers or very small aggregates, which appear at higher temperatures and do not experience exciton–exciton annihilation since they host only one excitation. Their de-excitation is described by a single exponential decay with a time constant τ2. Thus, the full time evolution of the integrated optical density I(t) reads
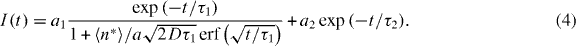
The scaling factors a1 and a2 weight the two contributions at a given temperature and 〈n*〉 denotes the number of excited excitons per molecule.
3.2. The kinetic Monte-Carlo approach
In order to take into account the finite size of the one-dimensional (1D) aggregates, kinetic Monte-Carlo simulations [25] are used to model the exciton kinetics. An ensemble of aggregates is constructed with a total number of considered molecules Nmol by equally partitioning them into Nagg linear aggregates. Initially, Nex molecules are excited randomly according to the fraction Nex/Nmol = 〈n*〉 of optically excited molecules calculated from the experimental data according to [11]. In the subsequent kinetics, the effects of exciton hopping, exciton annihilation and radiative decay are taken into account by respective rates. For the exciton hopping between neighboring sites, we use the hopping rate assuming Förster transfer. The rate kmn from site m to site n is given in the slow modulation limit by [26]
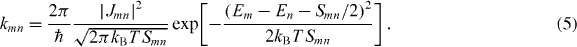
Here Em denotes the site energies that, in principle, could be different for different sites owing to energetic disorder. In the here considered case, energetic disorder is neglected, i.e. the site energies can be set to zero. Smn = Sm + Sn is the combined Stokes shift for the transitions at site m and n and Jmn is the Coulomb coupling between the transitions. The monomer Stokes shift is Sm = 1150 cm−1 [14] and the Coulomb coupling Jmn will be treated as a parameter when fitting the experimental kinetics; only nearest-neighbor coupling is assumed. Exciton–exciton annihilation is considered by introducing the bimolecular rate kann which applies whenever two excitons are on the same site. Here a fixed value of kann = 10 ps−1 is used. Finally, the population of each site is depleted according to a radiative decay rate krad of 0.27 ns−1 [11].
In the kinetic Monte-Carlo simulations, we have used 10 000 realizations for the initial conditions according to Nex. Specifically, we have used Nmol = 2000 and three concentrations, i.e. Nex = 58, 110 and 178 corresponding to excitation densities 〈n*〉 of 0.029, 0.055 and 0.089. They result from the experimentally applied excitation powers of 0.58 × 1015, 1.16 × 1015 and 1.88 × 1015 photons cm−2 per pulse, respectively. Note that in case the number of molecules was not a multiple of the aggregate size the nearest integer value for Nmol has been used. Further, we excluded the initial double excitation of a site. During the simulation the total exciton population with respect to all molecules is monitored. The Coulomb coupling and aggregate size are systematically varied so as to obtain the best fit to the experimental data.
After obtaining an optimal set of parameters for fitting the experimental decay curves, we have extended the model to include interchain hopping with a rate kint. This allows us to scrutinize the initial assumption of strictly 1D exciton transport.
4. Experimental results
Figure 2 shows transient spectra measured after optical excitation of PBI-1 dissolved in MCH with a concentration of 0.2 mM for temperatures of 30 and 90 °C at five different delay times. The energy per pump pulse is 120 nJ and the pump and probe polarizations are set to the magic angle 54.7° between each other. In this way, pump-induced anisotropies do not influence the transient absorption signal. Further measurements were carried out at intermediate temperatures. In all cases a strong negative optical density change is observed, which can be attributed to a combination of ground state bleach and stimulated emission from the populated electronic state. The spectral widths and positions correlate nicely with the ground state absorption at the respective temperature (see figure 1). The excited state absorption (ESA) is overlaid by the ground state bleach and the stimulated emission; for example, for the long aggregates the ESA leads to an almost zero differential optical density in the range below 575 nm.
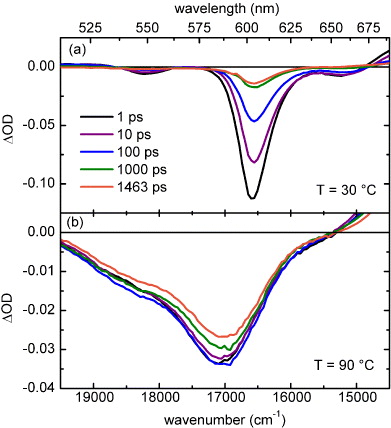
Figure 2. Transient absorption spectra for different delay times after optical excitation of 0.2 mM PBI-1 dissolved in MCH: (a) at 30 °C and (b) at 90 °C.
Download figure:
Standard imageAt a temperature of 30 °C the negative signal decays non-exponentially on a time scale of a few hundreds of picoseconds, while at 90 °C the signal decay is much less pronounced and occurs mostly after 1 ns. The changes in the spectral shape of the transient spectra are in both cases small, indicating that the signal evolution is dominated by a simple relaxation of the population of the electronically excited S1 state back to the ground state. To obtain a measure of this population, i.e. the number of excitons, and to eliminate the influence of the small spectral changes, the negative band of the differential optical density is integrated. The time-dependent integrated optical density is shown in figure 3(a) for three different temperatures.

Figure 3. (a) Integrated transient absorption signal due to optical excitation of PBI-1 in MCH (c = 0.2 mM, 〈n*〉 = 0.055, see text) for three different temperatures (symbols). The solid lines are model functions fitted to the data, which consist of two contributions, exciton–exciton annihilation and a single exponential decay. (b) Amplitudes of the two contributions as a function of the temperature. Black refers to exciton–exciton annihilation and red to the single exponential decay. The solid lines are a guide to the eye.
Download figure:
Standard imageTo analyze the behavior of the integrated signal, equation (4) is fitted to the data (solid curves in figure 3(a)). The fraction of initially excited molecules in the sample 〈n*〉 = n0/N0 can be obtained from the experimental data as described in [11]. Taking into account saturation effects leads to an excitation density 〈n*〉 of 0.055 per monomer for the applied pulse energy. The lifetime of the excitons on the aggregates τ1 = 3.6 ns and of the monomers τ2 = 4.1 ns are known from our previous work [11] and the lattice constant is assumed to be a = 0.48 nm [12]. Accordingly, the scaling factors a1 and a2 and the diffusion constant D are the only free parameters. If we assume that at low temperatures, almost all PBI molecules are integrated in aggregates, the diffusion constant for the long J-aggregates can be determined to be D = (0.9 ± 0.3) nm2 ps−1, leaving the scaling factors as the only free parameters. Therefore, the fractions of monomers and aggregates in the sample can be obtained. At this point it is important to note that the J-aggregates are still treated as infinite chains within this model. Accordingly, we cannot make any prediction about the actual aggregate size at a given temperature. The diffusion constant found deviates somewhat from the one reported in [11]. This is probably due to uncertainties in determining the initial exciton concentrations and the influence of the exponential contribution, which does not appear in the original model.
Figure 3(a) shows that the model described by equation (4) can reproduce the exciton dynamics for all temperatures very well. At low temperatures, the annihilation-driven decay dominates the dynamics, indicating that most of the dye molecules are aggregated (amplitude a1 in figure 3(b)). With the chosen parameters the model also fits measurements with other excitation densities and thereby different annihilation dynamics like the ones with excitation densities of 0.029 and 0.089 presented in figure 5. As the temperature rises, amplitude a1 declines, whereas the contribution of the radiative decay of the monomers a2 rises. The sum of the amplitudes slightly decays with increasing temperature, indicating that the ratio of the amplitudes is not a strict measure of the population ratio. The reasons are probably because the transition strength of the integrated band is somewhat different for the excitons on the aggregates and electronically excited monomers and because the ESA is contributing differently to the integrated signal for both species [11, 14]. The change from annihilation-dominated dynamics to a single exponential decay occurs within a temperature window of about 25 K around 55 °C. Therefore, one may conclude that upon heating the PBI-1 aggregates break up into monomers without undergoing any intermediate state. Measurements at elevated temperatures with different excitation power show only a weak dependence of the dynamics on the excitation density (data not shown). This supports our conclusion because only the annihilation dynamics on the aggregates depends on the excitation density, and owing to the fact that the monomer fraction rises with temperature, the energy dependence gradually disappears.
To test our notion, polarization-dependent measurements were carried out. A common measure of the polarization dependence is the transient anisotropy parameter r(t) that can be obtained by changing the relative orientation of the pump and probe polarization from parallel polarizations yielding the signal Sig||(t) to perpendicular polarizations yielding Sig⊥(t) [27, 28]:

At low temperatures, a fast decay of the transient anisotropy during the first 20 ps is observed (see figure 4). This fast decay is caused by the exciton–exciton annihilation on the J-aggregates, which are present at this temperature. Due to the strong excitation energy dependence of the annihilation probability [11], the signal for parallel polarized pump and probe beam Sig||(t) decays much faster than Sig⊥(t), leading to an accelerated decay of the transient anisotropy. It is noteworthy that already the mobility of the excitons on a chain with twists and kinks can lead to an anisotropy decay. In our earlier investigation [11], the anisotropy decay was studied at room temperature as a function of the excitation density and it was found that the decay accelerates with increasing excitation density. At very low excitation levels it takes place on a time scale of several hundreds of picoseconds, whereas for the experiment in the present study it is about ten times faster. At very low intensities the anisotropy decay seems to be dominated by the effect of excitons diffusing along a twisted and buckled chain, whereas at the presented excitation level the annihilation dynamics is primarily responsible for the anisotropy decay. A similar behavior of the anisotropy was also observed by time-resolved fluorescence spectroscopy for aggregates of a cyanine dye [29]. Interestingly, the fast anisotropy decay strongly slows down at intermediate temperatures (see figure 4), where also the single exponential decay dynamics replaces annihilation. This implies that the excitons lose their mobility, and the influence of annihilation on the anisotropy decreases drastically. Furthermore, we see that the transient anisotropy decays slightly faster at high temperatures than at intermediate temperatures. If we assume that the anisotropy decay in both cases is mainly caused by rotational diffusion of the excited species, the dynamics can be modeled by a single exponential decay starting at a value of r(t = 0) = 0.4 [30]:

where τrot is the time constant for the rotational diffusion process that is connected to the hydrodynamic volume V of the rotating species by the Stokes–Einstein relation [30]

where η(T) is the temperature-dependent viscosity of the solvent MCH and kB is the Boltzmann constant. Figure 4 shows that the rotational diffusion model is in good agreement with the measured data for high as well as for intermediate temperatures. Calculating the hydrodynamic volume of the rotating species allows us to make at least qualitative statements upon the size of the species. For a temperature of 55 °C a value of V = (9.43 ± 0.22) nm3 and for 80 °C a value of V = (7.12 ± 0.16) nm3 is calculated, indicating that the rotating species at intermediate temperatures is not a monomer but a very small PBI aggregate, probably a dimer. In this case it is unlikely that two excitons reside on the same aggregate and annihilation plays no role.
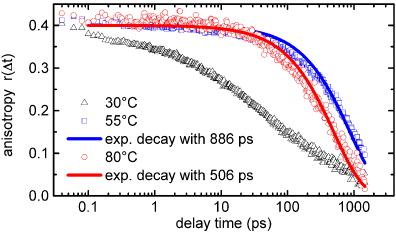
Figure 4. Time evolution of the anisotropy parameter r(t) for different temperatures. The curves for 55 and 80 °C are fitted with a single exponential decay.
Download figure:
Standard imageA crude approximation for the hydrodynamic volume of the rotating species can be done by assessing the size of the PBI molecules from the density ρ = 1.5 g cm−3 of comparable perylene bisimides given in [31]. The single-molecule volume can be estimated by

where M = 2383 g mol−1 [12] is the molar mass of PBI and NA is the Avogadro constant. Equation (8) yields a single molecule volume of V = 2.64 nm3, but it has to be mentioned that the measured hydrodynamic volumes include an additional solvent shell. For simplicity, the PBI-1 molecules are treated as spheres. To account for the solvent shell, the radius of the single PBI-1 hard sphere is increased by the thickness rMCH = 0.38 nm of one MCH monolayer, which is calculated from the structure of the MCH molecule. Recalculating the volume of the whole sphere including the shell we arrive at a volume of 7.94 nm3 , which is in proximity to the hydrodynamic volume of 7.12 nm3 measured at 80 °C. To calculate the hydrodynamic volume of a small aggregate, we repeat the above procedure after replacing V by NV for an aggregate consisting of N molecules. Thereby, we arrive at a volume of 13.04 nm3 for the PBI-1 dimer which is close to the hydrodynamic volume of 9.43 nm3 measured at 55 °C. This consideration seems to approve our above statement that at high temperatures the sample mainly consists of PBI-1 monomers whereas at intermediate temperatures very small aggregates are present.
5. Comparison with the hopping model
Although equation (3) is strictly valid only for an infinite chain, the average aggregate length is expected to shorten with increasing temperature [19]. The limited size should influence the dynamics since the number of excitons which can interact with each other decreases with decreasing aggregate length. To quantify the effect of the aggregate length and to test the applicability of the diffusion model, a hopping model based on a Monte-Carlo approach is used to simulate the exciton dynamics. In figure 5 the measured exciton populations are compared with Monte-Carlo simulations and the analytic diffusion model for three different excitation densities between 2.9 and 8.9% of the chromophores. The fitting has been performed for an excitation density of 2.9%, which gave using the parameters from section 3.2 a value for the coupling matrix element of Jmn = 120 cm−1, which is in good agreement with our previously measured value of 110 cm−1 [11]. Note, however, that this value has to be considered as an effective coupling since it reflects the underlying hopping model, i.e. either equation (5) or the diffusing equation. The bare electronic Coulomb coupling has been calculated to be −514 cm−1 in [13]. Furthermore, we found that beyond an aggregate size of 80 monomers there is little size dependence of the kinetics. The thus obtained parameter set is used for the higher excitation densities as well.

Figure 5. Comparison between the hopping model (lines) and the experimental data at 30 °C (symbols) for different initial excitation densities 〈n*〉.
Download figure:
Standard imageThe hopping rate khopp = 2 kmn = 5 ps−1 calculated by equation (5) can be compared with the experiment. The diffusion constant

is directly related to the hopping rate and the distance a between two hopping sites. If the distance is taken as the monomer separation (see above) the fitted annihilation model corresponds to a hopping rate khopp = (7.8 ± 2.7) ps−1. This is in reasonable agreement with the Monte-Carlo simulations.
The deviations between the Monte-Carlo simulations and the analytic model increase as expected with decreasing aggregate length (see figure 6(a)). If the number of hopping sites is less than 45 the differences are so pronounced that they should be clearly seen in the experiment. From this we conclude that the number of intermediate aggregates that show significant annihilation but cannot be treated as infinite chains is low. In other words, the long aggregates encountered at low temperatures dissociate with increasing temperature into small fragments containing at most a few monomers.

Figure 6. Experimental data for 〈n*〉 = 0.055 at 30 °C compared to (a) the hopping model for different aggregate sizes and (b) the hopping model with an additional interchain hopping rate kint.
Download figure:
Standard imageThe results of the extended two-dimensional Monte-Carlo model with an interchain hopping rate kint, given a fraction of the optimal intrachain rate kmn, are shown in figure 6(b) for the case of intermediate excitation density. Apparently, including interchain hopping leads to strong deviations from the observed kinetics. Even if the jump rate between aggregate chains is several orders of magnitude smaller than the hopping rate along the aggregates a significant discrepancy from the experimental results emerges. This observation gives evidence that the excitons cannot jump between the aggregates and the aggregates are well described as 1D chains along which the excitons can move. In this case and as long as annihilation is negligible, a 1D diffusion length  of 80 nm can be estimated from the lifetime τ1 of the excitons that corresponds to 170 PBI-1 molecules.
of 80 nm can be estimated from the lifetime τ1 of the excitons that corresponds to 170 PBI-1 molecules.
6. Summary and conclusions
The size-dependent exciton dynamics of 1D PBI-1 aggregates is studied by ultrafast transient absorption spectroscopy and kinetic Monte-Carlo simulations as a function of the temperature and excitation density. For low temperatures the aggregates can be treated as infinite 1D chains and the dynamics is dominated by diffusion-driven exciton–exciton annihilation. With increasing temperature the aggregates decompose into small fragments consisting of very few monomers. The chances that two excitons on one such fragment are excited are very low and annihilation processes can no longer occur. Aggregates of intermediate size seem to be not contributing significantly. Polarization-dependent experiments support this scenario since the anisotropy decay at intermediate temperatures points to orientational relaxation of entities with a volume of several monomers. At high temperatures the anisotropy decay accelerates somewhat, indicating an increasing fraction of monomers.
Acknowledgments
Financial support from the German Science Foundation via the collaborative research center SFB 652 'Strong correlations and collective effects in radiation fields: Coulomb systems, clusters and particles' is gratefully acknowledged.