

Abstract
In this work, we have systematically studied the stability, electronic structure and magnetic properties of the pristine, four defect states case of blue phosphorene and the six heteroatoms doping in blue phosphorene by first-principles calculations. In our findings, both defects and heteroatoms doping can regulate the band gap of blue phosphorene and the transition from indirect to direct band gap can be dramatically tuned by DV1BP, DV2BP and Al, Si atoms substitutional doping in blue phosphorene. The presence of defects and heteroatoms doping effectively modulates the electronic properties of blue phosphorene, rendering the defect-containing phosphorene semiconducting with a tunable band gap. Spin–orbit coupling can be induced by introducing SV-, DV- defects in blue phosphorene. The results provide theoretical guidance for future bandgap regulation and magnetism, defective and substitutional doping blue phosphorene may have potential electro-optical and electromagnetic applications.
Export citation and abstract BibTeX RIS
Introduction
Two-dimensional materials (2D) has entered a period of rapid development since the identification of graphene [1, 2]. It is well-known that 2D materials have widely studied for their inherent excellent properties. However, certain defects such as the zero bandgap [3, 4] and the nonmagnetic of stanene, graphene and so on [4–9] limit their applications in nanoelectronics and spintronics. As a member of the two-dimensional materials, blue phosphorene has high carrier mobility [10], tunable bandgaps [11], which has been reported in experimentally by molecular-beam epitaxial growth [12–15]. Meanwhile, it is obvious that blue phosphorene with defects such as large band gap, non-magnetic, and indirect band gap, which inhibit its applications in optical electronics, spintronics and so on [16–18]. Based on these defects, it is essential to solve these problems of bandgaps and magnetism.
One approach for inducing the magnetic moments of 2D materials is to introduce defect and embed metal atoms in blue phosphorene. Previous studies have shown that the magnetism of stanene can be tuned effectively via embedding transition metal in the stanene monolayer [5, 19, 20]. Magnetisms can be induced by transition metal atoms adsorbing on graphene and defects [21–23], while the pristine graphene appears to be non-magnetics. The same effect also appears in other two-dimensional materials for example black phosphorene [24–27], MoS2 [28–33], arsenene [34, 35] and so on. On the other hand, the indirect transfer to the direct band can be modulated by embedding metal atoms into the blue phosphorene. For instance, Xu et al studied the transformation from indirect to direct bandgap when O2, −OH, −COOH, and −CN adsorbing on blue phosphorene [18]. Chen et al investigated the changes of electronic structure based on pristine and Cu decorated InN monolayer [36, 37]. Similarly, the defects of silicene open direct bandgaps and improve the conductivity [38]. From the above developments, it can be observed that defects are very effective in tuning the magnetic and electronic properties of two-dimensional materials. Most electronic properties, and physical applications of 2D materials are always affected by defects. More importantly, innovative devices may be generated by defects. Typical point defects in black phosphorene and silicene already have been investigated, which extremely alter the electronic structure [25, 38].
In this work, density functional theory (DFT) calculation is employed to calculate electronic properties, stability and magnetic properties of blue phosphorene with vacancy defect and heteroatoms substitutional doping. First, blue phosphorene is a non-magnetic 2D material with indirect band gap, and limit its applications of optoelectronics, nano-electronics and spintronic. Then we systematically explored the electronic structure and stability of various defects, such as single vacancy (SV), two types of double vacancy (DV1 and DV2), six vacancy (SIXV) case. Finally, we introduced a variety of heteroatomic doping (e.g. Li, Na, Al, Si, Fe, Co, Sn) with monoatomic substitutions (P97ATOM1), two types of biatomic substitutions (P96ATOM2-1, P96ATOM2-2), and six atomic substitutions (P92ATOM6) in blue phosphorene. We have systematically studied the stability, magnetic and electronic structure of heteroatom substitution doping in semiconductor blue phosphorene. In our work, the electronic structure and magnetic properties of the two-dimensional blue phosphorene can be effectively controlled.
Computational methods
In our present analysis, we apply SIESTA-3.2 (Spanish Initiative for Electronic Simulations with Thousands of Atoms) package to calculate electronic properties of 2D materials based on DFT [39, 40]. The generalized gradient approximation and Perdew–Burke–Ernzerhof (PBE) exchange correlation function are adopted for structural optimization and structural relaxation [41, 42]. The energy cutoff is set for 200 Ry and the basis is set for double zeta plus polarization orbitals [42]. All atomic positions are fully relaxed until residual force and total energy are converged to 10−6 eV and 0.03 eV Å−1, respectively. The unit cell lattice parameter of blue phosphorene is a = b = 3.278 Å, which are in good accordance with experimental results [43]. A large 7 × 7 supercell of blue phosphorene including 98 P atoms with a vacuum layer ∼25 Å in the Z direction in order to prevent interaction between adjacent interlayers. The k-point is set 15 × 15 × 1 for structural relaxation and calculation of electronic properties. We use hybridization calculations and PBE calculations of SIESTA software, which show that the PBE calculations fit well with the results from previous calculations, so in the following we use PBE for all of our electronic structure calculations as shown in table S1 (available online at stacks.iop.org/NANO/32/135702/mmedia).
Results and discussion
The electronic structure of pristine blue phosphorene
At the first step, we have applied the first principles to calculate the electronic structure of the pristine blue phosphorene. As shown in figure 1, the P atoms are placed in a plane-hexagon lattice, where there are two P atoms at 1 × 1 supercell. Figure 1(a) shows the optimized structure of 7 × 7 × 1 supercell blue phosphorene, including 1 × 1 supercell is surrounded by a purple diamond shape, the relaxed lattice constant, buckling parameters and the length of P−P bond of the 1 × 1 units cell is 3.278 Å, 1.234 Å and 2.261 Å, respectively, the obtained simulation results were in very good accordance with the experimental results [43]. The band structures of pristine blue phosphorene spin-up and spin-down is presented in figures 2(a) and (c), and it is obviously that the lowest point of the conduction band and the highest point of the valence band correspond to different K points. As shown in figure 2, the consistency of the spin-up and spin-down trajectories is found to be non-magnetic. In conclusion, it is shown that blue phosphorene is a non-magnetic semiconductor with indirect bandgap, and the band gap is 2.03 eV. These characteristics have limited the study of blue phosphorene in some areas such as optoelectronics, nano-electronics and spintronic, so vacancy defects and heteroatom-doping have been introduced to improve their capabilities in our study.

Figure 1. The top and side views of optimized structures: (a) pristine blue phosphorene, (b) SV-(5∣9), (single-vacancy, SV blue phosphorene), (c) DV-(5∣8∣5), (double-vacancy, DV1 blue phosphorene), (d) specific manifestations of the four defects (e) DV-(9∣4∣9) (double-vacancy, DV2 blue phosphorene), (f) SIXV-(18), (six-vacancy, SIXV blue phosphorene).
Download figure:
Standard image High-resolution image
Figure 2. The band structure of (a) spin up, (c) spin down with blue phosphorene, (b) the DOS (density of states) of pristine blue phosphorene.
Download figure:
Standard image High-resolution imageDefect states in the blue phosphorene
It is very important to investigate defect states for the developments and applications of semiconductor materials. In order to imitate the vacancy defect of blue phosphorene, we consider the four models that includes single vacancy (SV), two types of double vacancy (DV1 and DV2), six vacancy (SIXV) case. The optimized structure of the defect is shown as figures 2(b)–(c) and (e)–(f), which is different from previous defect states. In our works, single vacancy is formed by removing one P atom and the SV defect is found to be an unstable structure after removing one P atom, which is subject to Jahn–Teller distortion, P1 and P2 atoms form a chemical bond on the blue phosphorene surface with a bond length of 2.58 Å, so one five-atom ring and one nine-atom ring are formed at the defect site (see figure 1(b)). Meanwhile, two types of defects are considered: DV1 by deleting two adjacent P atoms while the DV2 is formed by missing the two atoms on opposite sides of the hexagon. DV1 defects form two five-atom rings and one eight-atom ring, all dangling bonds are saturated by forming the 2.52 Å P1–P2 bond (see figure 1(c)). The remaining four atoms of DV2 form a quadrilateral after optimization, the bond length of P1-P2, P2-P3 is 2.32 Å, 2.35 Å, respectively, so DV2 possess two nine-atom rings and one four-atom ring (see figure 1(e)). The SIXV by removing the six atoms that make up the hexagon in the 7 × 7 blue phosphorene (see figure 1(f)). The charge density (figure S2 and table S2) shows that P-P bonds of SV, DV1 have weakly chemical bond. It can be found that the initial defect is very unstable and produces a new stable structure after optimization.
Stability and magnetism of the SV, DV1, DV2 and SIXV-blue phosphorene
To test the stability of the vacancy defects with blue phosphorene, we calculated the formation and binding energy. The formation energy  and the binding energy
and the binding energy  were defined as [44]:
were defined as [44]:
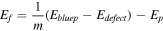
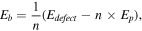
where 
 and
and  are the total energy of pristine blue phosphorene, the energy of SV, DV1, DV2 and SIXV, the energy of one P atom, respectively. The
are the total energy of pristine blue phosphorene, the energy of SV, DV1, DV2 and SIXV, the energy of one P atom, respectively. The  and
and  are the number of defect atoms and the P atom number of the blue phosphorene supercell with SV, DV1, DV2 and SIXV as shown in table 1 and figure S1. The greater the absolute value of the binding and formation energies of the defect-blue phosphorene, the more stable the structure is. Based on the equation, the calculated formation energy of blue phosphorene with SV, DV1, DV2 and SIXV are −5.68 eV, −4.63 eV, −5.47 eV and −4.29 eV, respectively. The results indicate that the blue phosphorene with defects is a stable structure.
are the number of defect atoms and the P atom number of the blue phosphorene supercell with SV, DV1, DV2 and SIXV as shown in table 1 and figure S1. The greater the absolute value of the binding and formation energies of the defect-blue phosphorene, the more stable the structure is. Based on the equation, the calculated formation energy of blue phosphorene with SV, DV1, DV2 and SIXV are −5.68 eV, −4.63 eV, −5.47 eV and −4.29 eV, respectively. The results indicate that the blue phosphorene with defects is a stable structure.
Table 1. Structural parameters of the optimized blue phosphorene including band gaps, magnetic moment M ( ) and binding energy (
) and binding energy ( ).
).
| Defect | Band gap (eV) Up Down | M (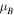 ) ) |
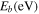
| Properties | |
|---|---|---|---|---|---|
| Perfect | 2.026 62 | 0 | −3.331 | Indirect | |
| SV-(5∣9) | 0.9197 | 0.9283 | 0.999 992 | −3.307 | — |
| DV-(5∣8∣5) | 1.2575 | 0 | −3.304 | Indirect | |
| DV-(9∣4∣9) | 0.5066 | 0 | −3.287 | Indirect | |
| SIXV-(18) | 1.677 | 1.024 | 5.999 974 | −3.268 | — |
Next, in order to discuss the electronic characteristics of blue phosphorene with SV, DV1, DV2 and SIXV defects, we investigate DOS for SV, DV1, DV2 and SIXV systems. Note that in figure 3, red and blue represent spin-up and spin-down DOS, respectively. From figure 3, it can be found that the spin-up are consistent with spin-down band structures in DV1- and DV2- blue phosphorene, and the spin DOS are symmetrical, which indicates that blue phosphorene with DV1 and DV2 possess nonmagnetic ground states. Whereas the structures of SV and SIXV are just the opposite, they have different band structure and different DOS for spin-up and spin-down. This indicates that blue phosphorene with SV and SIXV exist magnetic state. According to the table 1, the band gaps are all smaller after introducing the defect, the bandgap of DV2 is only 0.5066 eV. It is easy to find DV1- and DV2-defects blue phosphorene exhibit an indirectly bandgap. However, SV and SIXV produced magnetic moments of about 1 μB and 6 μB . In order to describe the magnetism of SV and SIXV in more detail, we went on to plot the spin charge density (see figure 4), and it is clear that there is a spin-up charge all around the defect state, considering that it is the dangling bond around the defect that induces the magnetism.

Figure 3. The DOS and band structure of (a) SV, (b) DV1, (c) DV2 and (d) SIXV.
Download figure:
Standard image High-resolution image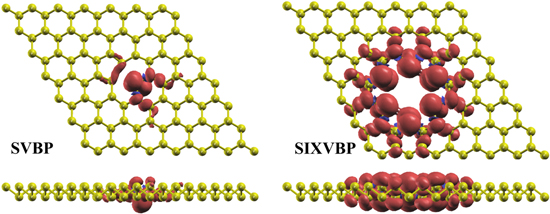
Figure 4. The spin density distributions for SV and SIXV blue phosphorene with magnetic ground state, the isosurface value is taken at 0.0008 e Å−3. Red and blue regions correspond to the spin-up and spin-down electrons, respectively.
Download figure:
Standard image High-resolution imageStability and magnetism of the heteroatom doping in blue phosphorene
To investigate the electronic structure and magnetism of blue phosphorene with heteroatom doping, we plotted the band structure, spin DOS and magnetic moment of heteroatom doping (Li, Na, Al, Si, Fe, Co, Sn) in blue phosphorene. We named the substitution of one P atom, two P atoms, and six P atoms by heteroatoms in blue phosphorene as P97ATOM1, P96ATOM2-1, P96ATOM2-2 and P92ATOM6, respectively, and the pattern of the heteroatom substitution is shown in figure S3. In order to clarify the stability of the defects in blue phosphorene, we calculated the formation and binding energy of heteroatom doping in blue phosphorene. The formation energy  and the binding energy
and the binding energy  are calculated as follows [44]:
are calculated as follows [44]:
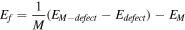
where 

 and
and  are the total energy of heteroatom doping P97/96/92ATOM1/2/6- blue phosphorene, defects-blue phosphorene, the energy of doping atom and the energy of one P atom, respectively. The M and N are the number of doping atoms and P atom in the blue phosphorene supercell. The formation energy is shown in figure 5, and it can be found that heteroatoms substitutional doping in the blue phosphorene are able to achieve a stable structure, the larger the binding energy, the more stable the structure is. Si atoms doping in blue phosphorene has the largest formation energy and are the most stable structures, followed by Al and Fe atoms are doping in blue phosphorene.
are the total energy of heteroatom doping P97/96/92ATOM1/2/6- blue phosphorene, defects-blue phosphorene, the energy of doping atom and the energy of one P atom, respectively. The M and N are the number of doping atoms and P atom in the blue phosphorene supercell. The formation energy is shown in figure 5, and it can be found that heteroatoms substitutional doping in the blue phosphorene are able to achieve a stable structure, the larger the binding energy, the more stable the structure is. Si atoms doping in blue phosphorene has the largest formation energy and are the most stable structures, followed by Al and Fe atoms are doping in blue phosphorene.
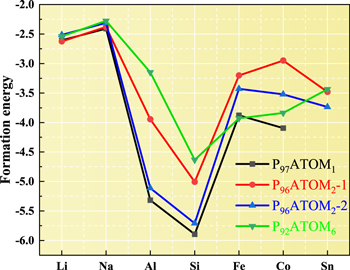
Figure 5. The formation energy of P97/96/92ATOM1/2/6-blue phosphorene.
Download figure:
Standard image High-resolution imageThen we calculate magnetic moments of the heteroatom (Li, Na, Al, Si, Fe, Co, Sn) substitutional doping in blue phosphorene. The numerical results indicate that different atoms can induce different magnetic moments in the blue phosphorene when heteroatom doping in P97/96/92ATOM1/2/6-blue phosphorene. The calculated results indicate that Li, Na, Al, Si, Fe, Co and Sn atoms possess magnetic moments of 1 μB , 1 μB , 1 μB , 2 μB , 4 μB , 3 μB and 2 μB , respectively.
According to the magnetic moments as shown in table 2, the s-orbitals of Li, Na, Si atoms are strongly hybridized with P atoms in P97ATOM1-, P96ATOM2-2-blue phosphorene, the d-orbital of Fe, Co are strongly hybridized with P atoms in P96ATOM2-1-, P92ATOM6-blue phosphorene, which determine the magnetic moments of blue phosphorene. For Li, Na, Al- doping in P97ATOM1- and P96ATOM2-2 blue phosphorene cases, the calculated magnetic moments are 1.999 712 μB , 1.999 816 μB , 1.987 73 μB , 1.997 385 μB , 1.000 001 μB and 1.999 986 μB , respectively. For Li, Na, and Si atoms doping in P97ATOM1-blue phosphorene, Li atom has one valence electron, the P atom has five valence electrons and forms a pair of Li–P bonds after Li atom are doping in a blue phosphorene, but the two nearby P atom is not saturated and thus generates magnetic moment. The same reason for generation of magnetic moment with Na atoms. For Si atom, there are four valence electrons, which form three pairs of Si–P bonds, and one valence electron that is not saturated, so the magnetic moment is generated. For Li, Na, Si substitutional doping in P96ATOM2-2-blue phosphorene, which produces the magnetic moment as same as P97ATOM1-blue phosphorous. The Li, Na atoms is hybridized with surrounding P atom to generate magnetic moments. After the two Si atoms doping in the P96ATOM2-2-blue phosphorene, the remaining two valence electrons of Si are not saturated, and the magnetic moment is generated.
Table 2. Structural parameters of the optimized heteroatom substitutional doping in blue phosphorene including band gaps, magnetic moment M (μB ) and binding energy (Eb ).
| Band gap (eV) | |||||
|---|---|---|---|---|---|
| Atom-defects | Up | Down | M (μB ) | Eb | Properties |
| P97Li1 | 1.4699 | 0.8439 | 1.999 712 | −3.3 | — |
| P97Na1 | 1.367 | 0.927 | 1.999 816 | −3.298 | — |
| P97Al1 | 1.8931 | 0 | −3.327 | Direct | |
| P97Si1 | 1.5457 | 1.2568 | 1.000 001 | −3.333 | — |
| P97Fe1 | 0.6914 | 1.3504 | 3 | −3.317 | — |
| P97Co1 | 0.9382 | 0 | −3.321 | Indirect | |
| P96Li2-1 | 0.4598 | 0 | −3.29 | Indirect | |
| P96Na2-1 | 0.4086 | 0 | −3.285 | Indirect | |
| P96Al2-1 | 1.2561 | 0 | −3.317 | Direct | |
| P96Si2-1 | 1.2512 | 0 | −3.339 | Direct | |
| P96Fe2-1 | 0.6914 | 1.3072 | −6.000 025 | −3.311 | — |
| P96Co2-1 | 0.4563 | 0 | −3.308 | Direct | |
| P96Sn2-1 | 0.8749 | 0 | −3.308 | Indirect | |
| P96Li2-2 | 0.455 | 0.5326 | 1.987 73 | −3.271 | — |
| P96Na2-2 | 0.5652 | 0.453 | 1.997 385 | −3.267 | — |
| P96Al2-2 | 1.723 | 0 | −3.324 | Direct | |
| P96Si2-2 | 1.5026 | 1.1116 | 1.999 986 | −3.336 | — |
| P96Fe2-2 | 0.8068 | 0 | −3.31 | Indirect | |
| P96Co2-2 | 0.6633 | 0 | −3.309 | Indirect | |
| P96Sn2-2 | 0.3175 | 0 | −3.296 | Indirect | |
| P92Li6 | 1.2569 | 0 | −3.224 | Indirect | |
| P92Na6 | 0.9936 | 0 | −3.208 | Indirect | |
| P92Al6 | 0.7208 | 0 | −3.261 | Indirect | |
| P92Si6 | 1.2779 | 0 | −3.352 | Indirect | |
| P92Fe6 | 1.3747 | 0.5944 | 21.999 968 | −3.306 | — |
| P92Co6 | 0.5682 | 0.5001 | −13.999 982 | −3.318 | — |
| P92Sn6 | 0.5444 | 0 | −3.278 | Indirect | |
For Li, Na, Si doping in P96ATOM2-1- and P92ATOM6-cases, the calculated magnetic moments are 0 μB . The reason is that Li and Na atoms hybridize with each two Li atoms and two Na atoms doping P96ATOM2-1-, P92ATOM6-blue phosphorene, and there is no interaction with the nearby P atom resulting in the Li–Li bond, and the Na–Na bond is saturated. After the substitutional doping of the Si atom, the two valence electrons of Si interact with the two P atoms, and the two remaining valence electrons of Si are paired, thus both Si atoms are saturated with zero magnetic moment. For Li-, Na-doping in P92ATOM6-blue phosphorene, Li and Na have only one valence electron, and the ring corresponds to exactly six Li/Na atoms make up a pair of Li–P bonds and Na–P bonds with six P atoms, so no magnetic moment is produced. For Si atoms, there are four valence electrons, which combine with the adjacent P atoms to form Si–P bonds. Other two valence electrons of Si hybridize with the neighboring Si atoms, and finally one valence electron remains for each Si atom, constructing three pairs of Si-Si double bonds.
For Al atoms doping in the blue phosphorene, the three valence electrons of P form an Al–P bond with three valence electrons of Al atoms, in other words, the three valence electrons of Al replace the three valence electrons of P to reach saturation and do not produce magnetic moment. Sn atoms are similar with Al substitutional doping in the blue phosphorene. For Fe doping in P97ATOM1-blue phosphorene, three Fe–P bonds are formed and the remaining three valence electrons are not saturated. For Fe atoms doping in the P96ATOM2-2-blue phosphorene, two Fe–P and one P–P bonds are formed, and the remaining valence electrons are not saturated. For Fe doping in P92ATOM6-blue phosphorene, the same reasoning as above, no saturation is reached. For Co substitutional doping in P97ATOM1-blue phosphorene have no magnetic moment, the reason is that Co-P bond is formed. For Co doping in P96ATOM2-1-, P96ATOM2-2-blue phosphorene, no saturation is reached. Therefore, substitutional doping of Li, Na, Si, Fe and Co atoms into the blue phosphorene can be an effective technique to induce magnetism, which expand the applications of blue phosphorene in spintronics fields.
In order to further investigate the magnetic properties in the Li, Na, Si, Fe and Co atoms doping in blue phosphorene, the spin density distribution is shown in figures 6 and 7. In figure 6, Li, Na, and Si atoms are doping in P97ATOM1- and P96ATOM2-2-blue phosphorene, resulting in spin polarization. From Li, Na doping in P97ATOM1- and P96ATOM2-2-, spin-up electrons are wrapped around the Li atom, in line with the above analysis. The result shows that the formation of Na's spin density is consistent with Li atoms. For Fe doping in blue phosphorene, the spin-up density surrounds Fe atoms, which is consistent with the above analysis. The substitutional doping of two Fe atoms as shown in figure 7(a), the spin-down density is generally greater than that in spin-up density. The Co atom is also surrounded by spin-down charges as shown in figures 7(c). Figure 7(b) is the opposite of figure 7(a), Fe atoms have more charge in the spin-up direction than in the spin-down direction, thus showing an overall spin-down. Overall, the spin charge density diagrams are consistent with the magnetic moments.
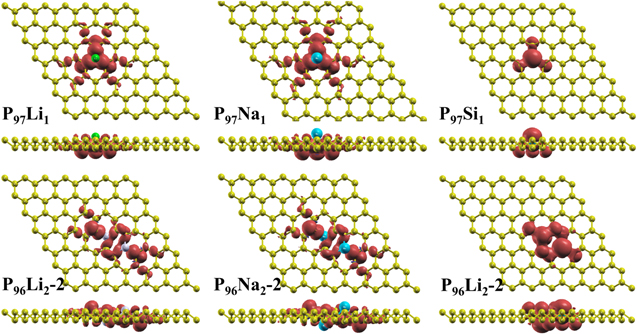
Figure 6. The spin density distributions for Li-, Na-, Si-substitutional doping in P97ATOM1-, P96ATOM2-2-blue phosphorene with magnetic ground state, the isosurface value is taken at 0.0008 e Å−3. Red and blue regions correspond to the spin-up and spin-down electrons, respectively.
Download figure:
Standard image High-resolution image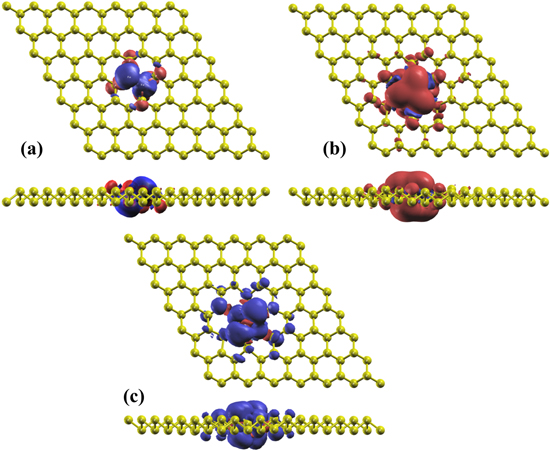
Figure 7. The spin density distributions for (a) P96Fe2-1 (b) P92Fe6 and (C) P92Co6, the isosurface value is taken at 0.0008 e Å−3. Red and blue regions correspond to the spin-up and spin-down electrons, respectively.
Download figure:
Standard image High-resolution imageElectronic structures of the heteroatom doping in blue phosphorene
Now we turn to investigate the electronic characteristics of heteroatom substitutional doping into blue phosphorene. Pristine blue phosphorene has an indirect bandgap of approximately 2.026 eV, as shown in figure 2. It can be seen from the Li, Na, Si, Fe atoms doping in P97ATOM1-case that the spin-up and the spin-down are asymmetric, which indicates that the Li, Na, Si, Fe substitutional doping in P97ATOM1- system has a magnetic ground state as displayed in figure S4 and which is agrees with the above magnetism calculations. In figures S4 and 8 (P97Al1), it can be found that the spin-up and spin-down are symmetric for Al, Fe substitutional doping in P97ATOM1, which is agree with the above magnetism calculations. For the nonmagnetic Al, Co doping in P97ATOM1-, it is displayed indirect bandgap and P97Al1 is displayed direct bandgap. The reason is that hybridization occurs after Al substitutional doping in P97ATOM1- and the band gap is reduced by nearly 0.2 eV compared with the pristine blue phosphorene. After hybridization it changed from an indirect bandgap to a direct bandgap.
For the heteroatom substitutional doping in P96ATOM2-1, Fe atom have magnetism properties due to the asymmetric of DOS with spin-up and spin-down (see figure S5). For the magnetic Li, Na, Co and Sn substitutional doping in P96ATOM2-1 systems, they still possess semiconductors properties which are similar to above analysis. In the band structure of the P96Al2-1 system, the CBM and CVM are located at G point, showing a direct band gap of 1.256 eV; the conduction band minimum and valence band maximum are located at M point in the band structure of the P96Al2-1 system, showing that the direct bandgap is 1.25 eV. The doping of Co atoms results in a smaller bandgap, and the bandgap is 0.4 eV, which has a conduction band minimum and a valence band minimum at the G point. The reason is that other atoms (Al/Si/Co) occurs orbital hybridized with P atoms (see figure 8 P96Al2-1, P96Si2-1 and P96Co2-1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. The DOS and band structure of P97Al1, P96Al2-1, P96Co2-1 P96Si2-1 and P96Al2-2.
Download figure:
Standard image High-resolution image{kind=link}
For the Li, Na and Si doping in P96ATOM2-2 case, figure S6 shows that the spin-up and spin-down density are asymmetrical because of the presence of magnetism. For the Al, Fe, Sn and Co atoms substitutional doping in P96ATOM2-2 case, the spin-up and spin-down density are symmetrical. In the figure 8, it can be found that the spin-up and spin-down are locate on G points for the P96Al2-2. After Fe, Co atoms are doping in P92ATOM6-blue phosphorene, the orbits of spin-up and spin-down in the DOS are inconsistent, showing magnetic properties as plot in figure S7. Other heteroatoms substitutional doping in blue phosphorene shows the non-magnetic state ascribe to the asymmetry of spin-up and spin-down state density. Atomic intercalation induces the magnetic properties of blue phosphorene, and the bandgap characteristics of blue phosphorene is found to change after some atoms are substitutional doped. The results further demonstrate the important effect of heteroatom substitutional doping on the electronic structure of P97ATOM1-, P96ATOM2-1-, P96ATOM2-2- and P92ATOM6-blue phosphorene.
Conclusion
First principles calculations were carried to investigate the electronic structure, stability and magnetic properties of the pristine blue phosphorene, SV-, DV1-, DV2-, SIXV-, heteroatom (Li, Na, Al, Si, Fe, Co, Sn) doping in P97ATOM1-, P96ATOM2-1-, P96ATOM2-2- and P92ATOM6-blue phosphorene. First, we calculated the electronic structure of the pristine blue phosphorene, the results show that blue phosphorene is a non-magnetic 2D material with indirect band gap, and limit its applications of optoelectronics, nano-electronics and spintronic. Then we systematically explored the electronic structure and stability of various defects, and the calculated binding energy reveals the stability of all structures. The DOS and spin density reveal that SV, SIXV occurs magnetic after introducing the vacancy defects, which indicates that the defects respond to spintronic devices well. In addition, it can be found that Li, Na, and Si atoms can effectively regulate the magnetic properties of P97ATOM1, P96ATOM2-2-blue phosphorene, and Fe atom can alter the magnetic properties of P97ATOM1, P96ATOM2-1 and P92ATOM6-blue phosphorene after incorporating spin polarization. In particular, Co atom also induces the magnetic properties of P92ATOM6, which indicates that these atoms are potential tools for inducing magnetism. It is also revealed that both defects and atomic insertion can regulate the band gap of blue phosphorene in our founding. In particular, Al atoms have an incredible effect on three defects, P97ATOM1-, P96ATOM2-1-, and P96ATOM2-2-, and they all change from indirect band gap to direct band gap. With the assistance of the DOS, considering the strong interaction between Al and P atoms, the electronic characteristics of the defective blue phosphorene are greatly changed. The results of P97Al1, DV1, DV2, P96Si2-1 provide theoretical guidance for future bandgap regulation and magnetism, such as the transition from an indirect to a direct bandgap of blue phosphorene, defective and substitutional of heteroatom in blue phosphorene may have potential electro-optical and electromagnetic applications.
Acknowledgments
This study is supported by the National Natural Science Foundation of China (Grant No.11564008), and the Natural Science Foundation of Guangxi province (Grant No. 2017GXNSFAA198195), and the Shanghai Supercomputer Center.