

Abstract
Extracellular calcium has been proved to influence the healing process of injuries and could be used as a novel therapy for skin wound healing. However, a better understanding of its effect, together with a system to obtain a controlled release is needed. In this study, we examined whether the ionic dissolution of the calcium–phosphate-based ormoglass nanoparticles coded SG5 may produce a similar stimulating effect as extracellular calcium (from CaCl2) on rat dermal fibroblast in vitro. Cells were cultured in the presence of medium containing different calcium concentrations, normally ranging from 0.1 to 3.5 mM Ca2+. A concentration of 3.5 mM of CaCl2 increased metabolic activity, in vitro wound closure, matrix metalloproteinases (MMP) activity, collagen synthesis and cytokine expression, and reduced cell contraction capacity. Interestingly, the levels of migration and contraction capacity measured followed a dose-dependent behavior. In addition, media conditioned with SG5 stimulated the same activities as media conditioned with CaCl2, but undesired effects in chronic wound healing such as inflammatory factor expression and MMP activity were reduced compared to the equivalent CaCl2 concentration. In summary, calcium-releasing particles such as SG5 are potential biological-free biostimulators to be applied in dressings for chronic wound healing.
Export citation and abstract BibTeX RIS
1. Introduction
Skin wound healing is a dynamic and complex process that aims to repair injured tissue to maintain body homeostasis. It involves the interplay of multiple cell types, growth factors, cytokines, and a balanced pool of metal ions, such as calcium, zinc and magnesium [1, 2], and progresses through four phases that overlap in time and space: hemostasis, inflammation, proliferation, and remodeling. Upon tissue injury, the coagulation cascade is activated, generating a provisional fibrin matrix which allows the migration of cells to the injured site [2]. In addition, factors released from platelets recruit leukocytes, thus beginning the inflammatory response [3]. Then, growth factors and cytokines released by platelets and immune cells initiate further wound healing processes, including re-epithelialization by keratinocytes and recruitment and activation of fibroblasts. Fibroblasts play a pivotal role in the synthesis and remodeling of the tissue. They migrate and proliferate in the fibrin matrix and generate new tissue made mainly of collagen [4]. In addition, fibroblasts activate into myofibroblasts which contain stress fibers that enable contraction and wound closure [5]. Normally, the healing process progresses overtime but, under certain circumstances, the process stalls, leading to chronic wounds.
Chronic skin wounds, also called hard-to-heal or difficult-to-heal wounds/ulcers, are skin injuries that fail to heal over a period of 3 months [6]. They are considered a major health and economic burden for developed countries and cause a significant impact in the well-being of people suffering from them [7, 8]. Unfortunately, current therapies, which mainly involve the use of dressings with moisture controlled properties, commonly fail to heal many chronic wounds. Although research in protein therapies and skin substitutes is showing promising results, their translation to the clinic is hampered by regulatory and economic constraints [9–11]. Thus, there is an urgent need to develop new, economic, effective therapies that reach the clinic faster.
In the last years, bioactive glasses have emerged as an alternative strategy to biological treatments for tissue regeneration and wound healing purposes owing their antibacterial, angiogenic, and cell stimulating properties [12]. Indeed, recent studies have claimed accelerated wound healing when applying composite materials containing bioactive glasses on skin injuries [13–15]. The release of ions while bioceramics degrade seem to be behind their stimulatory effects, as observed in in vitro studies in which cells are stimulated by media previously incubated with bioceramics [16–18]. However, little attention has been paid to unveiling the individual biological contribution of each of the ionic species.
Calcium is released by most bioceramics and, due to its complex role in skin and the healing process, it could be involved in some of the stimulatory effects observed. Calcium is not only relevant acting as a crucial clotting factor at the initial stages of the healing process, but it also plays an important role as an extracellular regulator and internal modulator of key cell types of the process, including keratinocytes and fibroblasts. While the effect of specific calcium concentrations in the regulation of proliferation and differentiation of keratinocytes has been well described [19, 20], the effect on fibroblasts has been poorly investigated. Indeed, even though studies have revealed that calcium affects fibroblast proliferation, collagen synthesis and morphology, they have not explored the optimal stimulating concentrations [20–23].
Considering the pivotal role of dermal fibroblast in the healing process, from the late inflammatory phase until full re-epithelialization, it is relevant to achieve a better understanding of the effect of different calcium concentrations on the biological response of fibroblasts. Additionally, the effect of the degradation products liberated from calcium-releasing bioactive glasses on fibroblasts has been underinvestigated. High calcium-releasing bioactive glasses have generally been utilized for hemostatic purposes [24, 25], but not for stimulating other healing stages. For wound healing purposes, most of the studies have used silica-based bioglasses with a relatively low calcium content and release [13–15].
One of the most important objectives in tissue engineering is to direct cell behavior by achieving full control of their surrounding microenvironment [26]. Nanotechnology provides efficient tools to introduce suitable signals in biomaterials, allowing us to turn these microenvironments into regenerative milieus. Recently, novel bioactive nanostructured glasses have been designed, named calcium–phosphate organically modified glasses (CaP ormoglasses), that allow a more tunable behavior of the ion release, thus improving their instructive capacity [27–29]. In a previous study, this material showed a controlled calcium release of biologically relevant concentrations able to elicit an angiogenic effect both in vitro and in vivo [30]. With a better understanding of the necessary doses of extracellular calcium to improve the healing of wounds, CaP ormoglass nanoparticles might be promising candidates to be used as off-the-shelf biological-free bioactive system on chronic wounds.
Based on the idea of exploring the potential use of calcium and CaP ormoglasses in the context of chronic wound healing, this study aimed to identify concentrations of extracellular calcium and calcium-releasing ormoglass nanoparticles that stimulate biological responses of skin wound healing carried out by dermal fibroblasts. More specifically, sol-gel produced CaP ormoglasses coded SG5 were characterized and media conditioned with different concentrations of calcium from CaCl2 (Ca–CaCl2) or released by the bioactive glass (Ca-SG5) were tested in vitro on rat dermal fibroblasts to study cell growth, cell migration, extracellular matrix (ECM) remodeling, contractile capacity, and wound healing related gene expression.
2. Experimental
2.1. Calcium–phosphate nanoparticle preparation and size characterization
Ormoglass nanoparticles of oxide based composition CaO:P2O5:Na2O:TiO2 44.5:44.5:6:5 were prepared by controlled hydrolysis sol-gel method at inert atmosphere as described previously [30]. The precursors used were prepared as follows. Calcium (Ca) and sodium (Na) 2-methoxyethoxides precursor solutions were prepared by refluxing metallic calcium (Sigma-Aldrich, 98%) and sodium (Panreac, 99%) respectively in anhydrous 2-methoxyethanol (Sigma-Aldrich, 99%) at 124 °C during 24 h. Phosphorus (P) ethoxide precursor solution was prepared by refluxing P2O5 (Sigma-Aldrich, 99.99%) in absolute ethanol (Panreac, 99.9%) at 78 °C during 24 h. Titanium (Ti) alkoxides precursor solution was prepared by diluting Ti-tetraisopropoxide (Alfa Aesar, 97%) in absolute ethanol. Then, Ti, Ca and Na precursors were added in a balloon with 1 h of vigorous stirring, followed by the addition of previously distilled 1,4-dioxane until a 5% v/v concentration was reached. The last precursor solution, P alkoxides precursor, was added at a control rate of 2.5 ml h−1 at 4 °C with an infusion pump. Then a basic catalyzing aqueous solution with a molar relation 20:0.1:4 H2O:NH3:EtOH and a Ti:H2O ratio of 1:60 was added at 1 ml h−1 and 4 °C. The solution was aged in a closed vial at 70 °C for 4 d under vigorous stirring. After aging, the produced particles were washed with ethanol and centrifuged five times for 5 min at 1000 g. Finally, SG5 particles were manually grinded in an agate mortar, dried at 90 °C, and stored in a desiccator for further use.
The size of SG5 particles was assessed by dynamic light scattering (DLS) on a Malvern Instruments Zetasizer Nano-ZS instrument. The solution used was absolute ethanol containing 1% Tween® 20 (Sigma) to avoid particle degradation and agglomeration. Before the measurement, the solution containing SG5 was homogenized in a bath sonicator and transferred into a quartz cuvette. SG5 morphology was observed with scanning electron microscopy (SEM) (NOVA NanoSEM 230, FEI Company, USA) uncoated on a silicon plate.
2.2. Ion release from calcium–phosphate particles
Ca, P, Na and Ti ion concentrations released by the particles in complete culture medium (CCM) consisting of DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM L-glutamine (Invitrogen), 100 U ml−1 penicillin (Gibco) and 100 U ml−1 streptomycin (Gibco), were measured as follows. Particle suspensions of 1.2 mg ml−1 were prepared and incubated at 5% (v/v) CO2 and 37 °C. After 24 h, suspensions were centrifuged for 10 min at 1000 g. To achieve total degradation, particles were mixed with 5% HCl in the same proportion (w/v) as for the release and were incubated at 37 °C in an automatic shaker (Thermomixer Comfort, Eppendorf) for 24 h. The supernatant was diluted 1/10 in 1% in HNO3 and the Ca, P, Na, Ti levels were measured by inductively coupled plasma optical emission spectrometry (Optima 3200RL, Perkin Elmer).
2.3. Cell isolation and culture
Primary cultures of rat dermal fibroblast were used in all the experiments. These cells were isolated from skin patches from the abdominal area of 10 Wistar rats of 2–4 weeks old. Rats were anesthetized in a chamber with 5% isofluorane and sacrificed in CO2 saturated atmosphere. After shaving their abdomen, skin patches of 1 cm2 were isolated and placed in sterile CCM. Then, patches were cut into pieces of 1 mm2 and were transferred to empty 90 mm Petri dishes. Each dish contained the pieces of two patches separated by approximately 5 mm. Following 15 min, 15 ml of CCM was added per dish and cells from the explants were allowed to grow until 80% confluence. Then, cells were trypsinized (TrypLE TM Express Enzyme, Thermo Fisher Scientific), seeded into T75 NunclonTM flasks (Thermo Fisher Scientific), and allowed to expand until 80% confluence. This population of cells was highly enriched with rat skin fibroblasts. At this point, cells were trypsinized again, pooled, and kept in liquid nitrogen at passage 2 in a solution of 10% DMSO (Sigma-Aldrich) in FBS. All protocols concerning animal care were approved by the Committee on Ethics and Animal Experiments of the Scientific Park of Barcelona (Permit No. 0006S/13393/2011). When needed, cells were thawed from the frozen stock, grown at 37 °C in 5% (v/v) CO2 in CCM, and used within passage 4–6. Medium was changed every 3 d.
2.4. Media conditioning and cell treatment
Media containing calcium from CaCl2 (Ca–CaCl2) or the ion release from the SG5 particles (Ca-SG5) was prepared. Ca–CaCl2 media was obtained by dissolving CaCl2 (>96%, Sigma-Aldrich) in calcium-free DMEM to obtain a solution of approximately 8 mM Ca2+, incubating overnight, and filter sterilizing it (0.22 μm). Ca-SG5 media was prepared as follows. A particle suspension of 0.6 mg ml−1 in calcium-free DMEM supplemented as regular medium was prepared and incubated for 24 h at 37 °C in 5% (v/v) CO2. After incubation, the suspension was centrifuged for 10 min at 1000 g, and the supernatant was collected and sterile filtered (0.22 μm). Calcium released in the media was measured using the quantitative colorimetric method 0-cresolphtalein complexone (Sigma) [31], reading absorbance at 570 nm on the Infinite M200pro microplate reader (Tecan). Finally, media was diluted to prepare specific concentrations needed for the assays. To measure the pH of these samples, an aliquot was kept in the CO2 incubator at 37 °C for 30 min, pH was quickly measured with a Laquatwin pHmeter (B-712, Horiba), and values were compared to a control sample without particle release. In all experiments, prior to addition of conditioned media, cells were rendered quiescent by an overnight incubation in low-serum (2% FBS) and 0.1 mM calcium-containing medium. Then, experimental media was added containing 2% or 10% FBS, depending on the restrictions or needs of each experiment.
2.5. Cell growth
Cell growth in conditioned media containing 10% FBS was studied by measuring metabolic activity and DNA quantification at days 1, 3 and 6. For metabolic activity determination, 2.5 × 103 cells/well were seeded in 48-well plates. At each time point, Alamar Blue (Alamar Blue kit, Thermo Fisher Scientific) was added following manufacturer's instructions and fluorescence was read at Ex/Em wavelength of 530/590. Six replicate wells were used per condition. Increase of cell number overtime was assessed by DNA quantification with the Picogreen dsDNA Assay Kit (Invitrogen). Briefly, 1 × 104 cells/well were seeded in 24-well plates. Four replicate wells were used per condition. At the above-mentioned time points, cells were lyzed in cold Tris-EDTA buffer (Sigma-Aldrich) by three freeze-thaw cycles and quantification of DNA was performed following manufacturer's instructions. Finally, fluorescence was read at Em/Ex 480/520. Each assay was assessed three times. In both assays, fluoresce was measured on the Infinite M200pro (Tecan) microplate reader.
2.6. Wound scratch assay
To determine the effect on cell migration of the different conditioned media, a scratch-wounded fibroblast monolayer model was employed. Prior to cell seeding, a line passing through the middle of each well was drawn on the external side of the plate. Briefly, 4 × 104 cells/well were seeded into 12-well tissue culture plates and cultured to confluence. Four replicate wells were used per condition. The confluent layer of fibroblasts was scrape-wounded using a sterile p200 pipette tip by one linear scratch perpendicular to the drawn line. Wells were washed with PBS to eliminate floating and dead cells, and conditioned media containing 2% FBS was added. Wound closure was monitored by collecting digitized images of the upper and lower area adjacent to the drawn line, immediately after the scratch and 24 h afterwards. Images were acquired with an inverted microscope (Nikon TE200) with a digital camera (Olympus DP72) using the 4× phase contrast objective lens. Reduction of wound areas was analyzed with the ImageJ 1.48i software. The experiment was repeated three times.
2.7. Gelatin zymography assay
The activities of MMP2 and MMP9 of cell culture supernatants were measured by a gelatin zymogram protease assay. Cells (4 × 104 cells/well) were seeded in 24-well plates. Following starvation, conditioned media (2% FBS) was added and, after 24 h, media was collected and centrifuged at 10 000 g for 10 min at 4 °C. Aliquots of supernatants were mixed with 3× non-reducing sample buffer (0.3% SDS; 15% 1M Tris-HCl pH 6.8, 30% glycerol and 0.075% bromophenol blue) and 30 μl were loaded on 8% SDS-polyacrylamide gels containing 0.1% gelatin (Gelatin from porcine skin BioReagent, Type A, Sigma). Then, prepared samples were subjected to electrophoresis under a constant voltage of 125 mV for 90 min. A protein marker (KaleidoscopeTM Prestained SDS-PAGE Standards, Bio-Rad) was run in parallel for molecular weight identification. Following electrophoresis, gels were washed in 2% Triton X-100 (Sigma) for 30 min at room temperature to remove SDS. Gels were then incubated at 37 °C for 18 h in 1× Development buffer (10× Zymogram Development Buffer, Bio-Rad) and stained with 0.5% Coomassie Blue R250 (Bio-Rad) in 50% methanol and 10% glacial acetic acid for 1 d. After destaining, gelatinolytic activities were identified as clear bands against the blue background. To quantify the amount of gelatinase activity, gels were scanned on a densitograph (LAS4000 Imaging System) and the intensities of the digitalized bands were measured with ImageJ software. All density values were normalized to the intensity of the 0.1 mM sample and to the protein level of the cell extract, which was measured with the BCA protein assay (Thermo Fisher Scientific). The experiment was repeated three times including three replicates per condition. For each experiment, two gels were run including different samples.
2.8. Total collagen
Collagen secreted in media by fibroblasts cultured under the different conditioned media was measured using the Sircol Collagen Assay (Biocolor, UK). Cells (2.5 × 105 cells/well) were seeded in T25 flasks with complete DMEM. After starvation, cells were treated with 3.5 ml/flask conditioned media containing 2% FBS for 24 h. Then, media from the flasks was transferred in 2 ml low protein binding microcentrifuge tube, and total collagen was quantified following manufacturer's instructions. Absorbance signal was measured at 555 nm on the Infinite M200pro (Tecan) microplate reader and values were normalized to the protein content from the cell extract measured with the BCA protein assay (Thermo Fisher Scientific). The experiment was carried out three times using triplicates per condition.
2.9. Fibroblast-populated collagen lattice (FPCL)
The stressed-relaxed FPCL model was used as previously described [32]. Briefly, collagen lattices were polymerized in 6-well tissue culture plates. Two hundred microliters of the mix of OptiColTM Rat Collagen Type I (Cell guidance systems) at a concentration of 1.2 mg ml−1 containing 2 × 104 rat dermal fibroblasts were carefully added in each well and allowed to gel for 1 h in a 5% (v/v) CO2 humidified atmosphere at 37 °C. Then, 2.7 ml of the different conditioned media was added. Following 5 days of culture, gels were imaged using a Leica MZ16 F stereomicroscope. Then, gels were detached from the surface of the wells by rimming the lattice with a sterile spatula and images of the floating lattices were captured at 0.5, 1, 2 and 4 h. To quantify contraction, FPCL surface area was measured with ImageJ software and values were normalized to the area of the lattices before being released. The experiment was repeated three times including three replicates per condition.
2.10. Western blotting
The expression of alpha-smooth muscle actin (α-SMA) by fibroblasts incubated in the collagen lattices under different calcium concentrations was quantified by immunoblot analysis. After the last picture acquisition, each gel used in the FPCL experiment was washed in PBS and transferred into microcentrifuge tubes. Cell lysate was obtained by incubating the gels with the radioimmunoprecipitation lysis and extraction buffer supplemented with a protease inhibitor cocktail (Santa Cruz Biotechnology) for 20 min in ice. To ensure complete cell lysis, gels were also sonicated (Ultrasonic Processor UP50H) twice for 5 s at 100% potency in cold. Finally, samples were centrifuged at 10 000 g for 10 min at 4 °C and supernatants were transferred into clean microcentrifuge tubes. Equal sample loading into the electrophoresis was ensured by measuring DNA concentration of each sample with the Picogreen dsDNA Assay Kit (Invitrogen), and 40 μg per sample were mixed with 6× Laemmli buffer. Then, samples were run in SDS-polyacrylamide (10%) gels and transferred to nitrocellulose membranes (0.45 μm, Bio-Rad) by electrophorosesis. Membranes were blocked in 5% skim milk in Tris-buffered saline containing 0.1% Tween® 20 (TTBS) at room temperature for 1 h, and they were sequentially probed with a goat anti-α-SMA polyclonal antibody (1:500 dilution, PA5-18292, Thermo Fisher Scientific) overnight at 4 °C, and then GAPDH (1:500, G8795, Sigma) for 1 h at RT. After three washes in TTBS, membranes were sequentially incubated with secondary antibodies conjugated with horseradish peroxidase: mouse anti-goat (1:1000 dilution, 2354, Santa Cruz Biotechnology) and donkey anti-mouse (1:3000 dilution, SA1-100, Thermo Fisher Scientific) for 1 h in 5% milk-TTBS, and finally developed by ClarityTM Western ECL Substrate (Bio-Rad). The chemiluminescent signal was read with a densitograph (LAS4000 Imaging System), and the intensities of the digitalized bands were measured with ImageJ software. Density values were normalized to their correspondent GAPDH band. Three membranes were analyzed running samples from three different experiments.
2.11. Wound healing RT-PCR array
Differential gene expression of cells treated with 0.1 and 3.5 mM calcium concentration from CaCl2 and from the release of the particles was assessed using the RT2 ProfilerTM PCR Array Rat Wound Healing (PARN-121ZC-2, Qiagen). Cells (8 × 104 cells/well) seeded on 6-well plates were treated with conditioned media (10% FBS) for 24 h, and RNA was extracted using the RNeasy PlusMinikit (Qiagen) following the manufacturer's instructions. RNA (0.5 μg) of each condition were reversely transcribed with a RT2 First Strand Kit (Qiagen) and the cDNA was used with the RT2 SYBR Green Mastermix (Qiagen) in array plates containing a panel of 84 wound healing-related genes, 5 housekeeping (HK) control genes, a rat genomic contamination control and RT-controls. Quantitative RT-PCR was performed with the StepOnePlus RT-PCR system (Appleid Systems). Cycling parameters were 10 min at 95 °C (activation) followed by 40 cycles consisting of 15 s at 95 °C (denaturation) and 1 min at 60 °C (extension). The CT threshold for all the conditions was matched and mRNA expression for each gene was normalized to the HK genes of the array. Relative amounts of RNA for each gene and fold increase were calculated by using the 2 − ΔCT method or the 2 − ΔΔCT method. The genes selected were the ones with, at least, two-fold higher or lower expression than the control sample.
2.12. Statistics
Data are presented as means ± standard deviation (S.D). The results were subjected to one or two-way ANOVA, and statistical differences between the groups were analyzed using post hoc Tukey's test at a significance level of 5%. The statistical software used was GraphPad Prism 6.0 (San Diego, CA, USA).
3. Results and discussion
3.1. Characterization of SG5 nanoparticles and ion release
Particle size was measured with DLS and confirmed with SEM images. DLS indicated that the average size of SG5 particles was 357.3 ± 38.6 nm (figure 1(a)) and SEM images showed particles of this size (figure 1(b)). Because we are interested in studying the effect of the ions released by SG5, the ion profile of the release of the particles after 24 h in CCM was obtained (figure 1(c)). We also measured the ion release after complete degradation of the particles to compare how much of the ion content had been released. The measurement showed that, at the analyzed time point, all the calcium (Ca) and phosphorous (P) had been released from the particles, generating concentrations in the mM range. As expected by the theoretical oxide composition of the particles, the total concentration of P released doubled the total concentration of Ca. On the other hand, titanium (Ti) was only partially liberated and in much lower concentrations, within the μM range. Due to the high concentration of sodium (Na) in cell media, Na released from the particles could not be properly quantified, but measurement of the total release of Na indicated that the maximum that can be liberated is in the mM range.

Figure 1. Characterization of SG5 particles: (a) particle size by DLS, (b) morphology by SEM and (c) ion release of totally degraded particles and ion release in CCM after 24 h. Scale bar of 350 nm.*p < 0.05.
Download figure:
Standard image High-resolution image3.2. Metabolic activity and proliferation of rat dermal fibroblasts
Cell viability and growth was studied by metabolic activity assessment and dsDNA quantification. A wide range of calcium concentrations, from 0.1 up to 7.5 mM, were used, and the effect was evaluated along 6 d. The lowest concentration used, to which the effect of the different concentrations were compared to, was 0.1 mM because it is optimal for keratinocyte growth while does not promote fibroblast proliferation [20]. In addition, two types of solutions were employed: media containing calcium from CaCl2 (Ca–CaCl2) or the ion release from the SG5 particles (Ca-SG5).
Metabolic activity was stimulated with specific calcium concentrations, and differences were detected between Ca–CaCl2 and Ca-SG5 media. Stimulation by Ca–Cl2 was observed on day 3 by the 2.5 and 3.5 mM conditions, but concentrations of 5 mM or higher maintained activity static or even decreased viability (figure 2(a)). On the other hand, stimulation by Ca-SG5 was detected on day 1 by the 3.5 mM-SG5 condition, but a significant decrease was observed on day 6.

Figure 2. Growth of rat dermal fibroblasts after 1, 3 and 6 days of treatment with media (10% FBS) containing different Ca2+ concentrations from CaCl2 and SG5 release. (a) Metabolic activity and (b) proliferation measurement by double-stranded DNA (dsDNA) quantification. Data is expressed as the mean of at least three replicates ± S.D. *p < 0.05 0.1 mM versus all, #p < 0.05 2.5 mM versus 2.5 mM-SG5, ∇p < 0.05 3.5 mM versus 3.5 mM-SG5.
Download figure:
Standard image High-resolution imageThe increase in metabolic activity triggered by Ca–Cl2 and Ca-SG5 at specific time points and concentrations did not correlate with an increased cell growth, as shown by total DNA quantification (figure 2(b)). Indeed, the different calcium concentrations tested did not augment cell growth compared to the lowest concentration used, 0.1 mM, at each time point. These results differ from previous studies which showed that mitogenic activity incremented upon calcium exposition [21, 22]. While these previous studies were performed in low-serum conditions, we used media containing 10% FBS because we wanted to analyze the effect of calcium for a long period of time and in a more physiological environment. Thus, we posit that the high content of growth factors and other proteins from serum in media might have masked the expected calcium-stimulated proliferative effect.
In addition, 2.5 mM and 3.5 mM Ca-SG5 showed a slower growth rate compared to their Ca–CaCl2 counterpart on day 6 (figure 2(b)). This result, together with the previously mentioned on metabolic activity, indicate that long term exposure of fibroblasts to the SG5 release provoked cytotoxicity in the in vitro system. To discard pH effect, we measured the pH of the solutions containing SG5 release and found that the pH of these samples remained equivalent to non-conditioned media (data not shown). Therefore, one or more of the species liberated by the particles caused this effect. Since the method employed for the production of SG5, based on sol-gel without posterior high temperature treatment, favors the presence of non-decomposed organic compounds in the structure of the particles, when released, these could induce some degree of cytotoxicity. However, it should be considered that in an open system such as in an in vivo situation, the cytotoxic effect should diminish, as previously observed [28, 30, 33].
3.3. Effect on cell migration
Fibroblast migration into the newly formed fibrin matrix at the wound site is an essential event for the progression of the healing. To test whether the different Ca2+ treatments affect fibroblast migration, a wound scratch assay was performed (figure 3(a)). The experiment was carried out in low-serum conditions and stopped after 24 h to reduce the effect of cell proliferation. Cell migration was evaluated by measuring the area of the initial wound covered by the cells at the final time point, as shown in figure 3(b).

Figure 3. Migration of rat dermal fibroblast exposed to media (2% FBS) containing different Ca2+ concentrations from CaCl2 and SG5 release. (a) Images of the scratch acquired at 0 h and 24 h and (b) the area of the wound covered by the cells after 24 h. (c) Changes in the invasive behavior of fibroblasts exposed to the different media, represented by the increase in the perimeter of the cell front. Data is expressed as the mean of at least four replicates. *p < 0.05 Condition versus 0.1 mM, ***p < 0.001 Condition versus 0.1 mM; ****p < 0.0001 Condition versus 0.1 mM.
Download figure:
Standard image High-resolution imageAll calcium concentrations, from both Ca–CaCl2 and Ca-SG5, significantly stimulated cell migration compared to the lowest concentration. In addition, the intensity of the effect seemed to be concentration-dependent and the maximum migration was reached using 2.5 mM Ca2+. Similar stimulation of cell migration was found when media with Ca-SG5 was used. Quantification of the increase in perimeter of the cell front also revealed that concentrations of 2.5 mM and above stimulated a more invasive migration behavior (figure 3(c)).
Other studies had shown migration stimulation with calcium on fibroblasts [34] but, to our knowledge, the concentration-dependent behavior had not been reported before. There is not a single proposed mechanism by which extracellular Ca2+ can affect cell migration due the plethora of events in which Ca2+ is implicated and the complexity of cell motility, but it can be linked to the activation of calcium-binding proteins. Studies have shown how increasing extracellular Ca2+ leads to increase in intracellular Ca2+ [35] that results in the activation of calcium-dependent proteins involved in cell migration, such as calpains [36].
3.4. Matrix metalloproteinases (MMP) activity and collagen synthesis
During the remodeling and repair phase of the healing process, fibroblasts play an essential role in transforming the new matrix that is being created. Remodeling requires both degradation and synthesis of new ECM, but in chronic wounds the deposition of new tissue is prevented or impaired by numerous factors [37]. To study the effect that calcium and the ion release of SG5 have on the remodeling ability of fibroblasts, gelatinase activity and collagen synthesis were analyzed.
Gelatin zymography was used to assess MMP2 and MMP9 activity, two key players in ECM degradation. The results show that 3.5 mM Ca–CaCl2 increased the active form of MMP2 compared to the other Ca2+ concentrations (figures 4(a) and (c)). An increase in MMP9 activity was also observed by incubating cells with 3.5 mM Ca–CaCl2 (figures 4(a) and (b)). Increased expression of MMP2 and MMP9 by Ca2+ had been previously described [34], but the clear activation of MMP2 by specific calcium concentrations had not been reported. Interestingly, in the presence of 3.5 mM Ca-SG5, no increase of MMP activity was detected. The additional compounds released by the particles, such as phosphate, may cause this different behavior. This result may be beneficial in the context of chronic wound healing, since MMP2 and MMP9 seem to be abnormally expressed in these type of wounds [38] and are considered partly responsible for the chronic condition [37, 39].
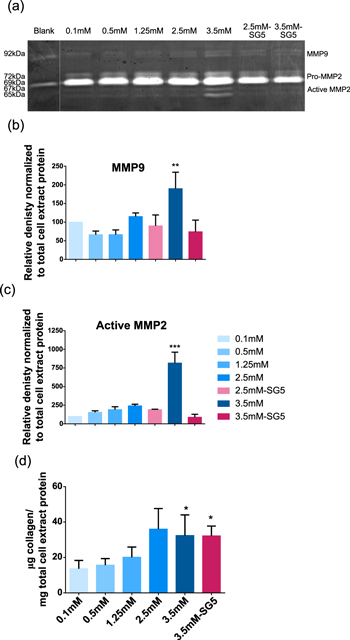
Figure 4. Remodeling stimulation of dermal fibroblasts studied through the analysis of gelatinase activity and collagen synthesis in cell-conditioned media after treatment of cells with Ca–CaCl2 and Ca-SG5. (a) The zymography assay showing bands corresponding to different activation states of MMP2 and MMP9. (b) The intensity of the bands corresponding to total MMP9 and (c) active MMP2 quantified by densitometry. (d) Total collagen secreted in the media by treated cells. Both densitometry and collagen values were normalized to the values of the 0.1 mM condition and to the total protein from the cell extract of each sample. Data is expressed as the mean of at least three replicates. *p < 0.05 Condition versus 0.1 mM; **p < 0.01 Condition versus 0.1 mM, ***p < 0.001 Condition versus 0.1 mM.
Download figure:
Standard image High-resolution imageStimulation of collagen synthesis by extracellular calcium and the ion release of SG5 was evaluated by quantifying total collagen secreted in the different test media. Ca2+ concentrations of 2.5 and 3.5 mM Ca–CaCl2 significantly stimulated collagen synthesis, and a similar effect was observed in the Ca-SG5 condition (figure 4(d)). Although the stimulating effect of calcium on collagen synthesis by fibroblasts has been suggested by others [23, 40], by testing a wide range of calcium concentrations we were able to identify an optimal concentration to promote collagen production by dermal fibroblasts. The de novo deposition of collagen stimulated by both CaCl2 and SG5 could contribute to an improved healing, since expression of the collagen gene in fibroblasts is suppressed in some types of chronic wounds [41].
3.5. Contractile capacity
Fibroblasts play a key feature in the latest events of the healing process by providing the contractile forces that bring the wound edges together. In our study, the stressed-relaxed FPCL model was used to quantify the contractile capacity of fibroblasts cultured with different Ca2+ concentrations and the ionic release of SG5.
Media containing Ca–Cl2 and Ca-SG5 with equivalent calcium concentrations affected fibroblast's contractile capacity similarly (figure 5(a)). Fibroblasts cultured in lower Ca2+ concentrations (0.1 mM and 1.25 mM) were able to contract the collagen matrix faster and in greater extent compared to higher Ca2+ concentrations (2.5 and 3.5 mM). Differences observed among conditions showed a concentration-dependence behavior. Only for the highest calcium concentration (3.5 mM) of Ca-SG5, fibroblasts showed reduced contractile capacity compared to their equivalent Ca–CaCl2 (figure 5(a)). One or more of the molecules liberated by SG5 is triggering these differences. Exposure to either the organic content or the high phosphate concentration released by the particles may be causing this effect.
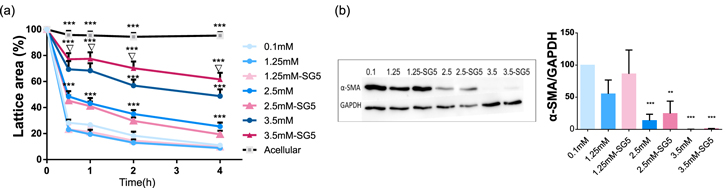
Figure 5. Fibroblast contractile capacity in different calcium concentrations studied in the stressed-relaxed FPCL model. On day 4, lattices were detached and images of the matrices were taken at different time points. (a) Contraction of the lattices represented as the percentage of reduced area compared to the initial size. (b) Immunoblot of the cell lysate against α-SMA and GAPDH and the densitometry analysis of the bands. Data is expressed as a mean of four replicates. *p < 0.05 Condition versus 0.1 mM;**p < 0.01 Condition versus 0.1 mM, ***p < 0.001 Condition versus 0.1 mM; ∇p < 0.05 3.5 mM versus 3.5 mM-SG5.
Download figure:
Standard image High-resolution imageSince expression of α-SMA is known to increase fibroblast contractile activity, [45, 46] α-SMA expression levels were quantified to test whether the differences in contractile capacity could be originated due to differences in α-SMA expression. The immunoblot analysis performed showed correlation between α-SMA levels of expression and fibroblast contractile capacity, unveiling clear differences in stimulation among Ca2+ concentrations (figure 5(b)). At 0.1 mM the expression of α-SMA was the highest among the conditions tested, while at 2.5 and 3.5 mM the expression was significantly lower or even absent (figure 5(b)). SG5 conditioned media stimulated similar levels of expression as their equivalent Ca–CaCl2 concentration (figure 5(b)). According to this result, extracellular Ca2+ would play a crucial role in fibroblasts activation to myofibroblasts. The study of the influence and control of extracellular Ca2+ concentration in the latest stages of the healing process as an strategy to avoid scar formation could be a new concept to explore in the field.
3.6. Expression of wound healing genes
Expression of genes involved in wound healing by fibroblasts exposed to calcium was analyzed by Real-time transcriptase polymerase chain reaction (RT-PCR) using an array of 84 key genes central to the wound healing response. Cells were cultured for 24 h in 10% FBS media containing the low Ca2+ concentration used through the assays, 0.1 mM, or a concentration proved to stimulate many relevant effects, 3.5 mM, either from CaCl2 or from the ionic release of SG5.
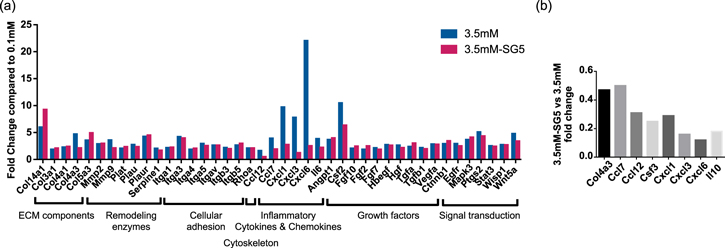
Our results indicate that 38 genes including ECM components, remodeling enzymes, cellular adhesion, cytoskeleton, inflammatory chemokines and cytokines, growth factors, and signal transduction factors were upregulated at least 2-fold in fibroblasts cultured with 3.5 mM Ca–CaCl2 compared to 0.1 mM (figure 6(a)). Even though discrepancies may exist between mRNA and protein levels, the results obtained correlated with the previously exposed biological effects, and provided insight into possible mechanisms generating such effects. The increased expression of mRNA of proteins involved in cell migration in the presence of calcium, such as several integrin subunits and the cytoskeleton regulator Rhoa, was in line with the increased cell migration observed. We hypothesize that these proteins might be partly involved in the stimulation of cell migration reported. In addition, augmented expression of ECM components including different types of collagen as well as gelatinases MMP2 and MMP9 were detected, in accordance with the zymography assay. Even though we did not find increased cell proliferation when quantifying dsDNA, signal transduction factors involved in cell proliferation, such as mitogen-activated protein kinase 3 (MAPK3), were also increased. Thus, extracellular calcium might be triggering specific mitogenic pathways. In addition, increased expression of inflammatory cues, especially neutrophil-recruiting CXC chemokines, were identified, meaning that high extracellular Ca2+ could contribute to the inflammatory state of the wound by increasing expression of inflammatory chemokines and cytokines of, at least, fibroblasts.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Analysis of the gene expression of fibroblasts incubated for 24 h in medium (10% FBS) conditioned with 0.1 mM, 3.5 mM and 3.5 mM-SG5 using a RT-PCR array for wound healing. (a) Genes from the samples 3.5 mM and 3.5 mM-SG5 that resulted in at least a 2-fold change expression compared to the calcium concentration 0.1 mM. (b) Fold expression of the sample 3.5 mM-SG5 compared to 3.5 mM. Abbreviations of the genes analyzed are defined in the supplementary material, table S1 available online at stacks.iop.org/NANO/29/395102/mmedia.
Download figure:
Standard image High-resolution image{kind=link}
Similar results were observed in fibroblasts cultured with 3.5 mM Ca-SG5, for which the upregulated genes were 36 and only one gene was downregulated (Ccl12) (figure 6(a)). Nevertheless, by comparing the expression between 3.5 mM Ca–CaCl2 and Ca-SG5 we found downregulation in the expression of inflammatory cytokines and chemokines in cells stimulated with SG5 (figure 6(b)). Again, exposure to either the organic content or the high phosphate concentration released by the particles may be causing these differences. Since stimulation of inflammatory cytokines in chronic wounds should be avoided [38, 42], the fact that less inflammatory cues are expressed with the ion release of SG5 than with calcium from Ca–Cl2 makes SG5 more suitable for treating this type of wounds.
Overall, these findings suggest that our developed calcium-releasing particles may be used to effectively improve wound healing. However, due to the complexity and plethora of players involved in the wound healing process, a deeper understanding of the effect of the release should be acquired, especially on keratinocytes, inflammatory cells, and angiogenesis. In addition, trustworthy measurements of the levels of calcium in chronic wounds are needed in order to adjust the dose delivered to the optimal one, as it is known that its concentrations can fluctuate through the healing process [1, 43, 44].
4. Conclusions
This study shows that specific concentrations of extracellular Ca2+ and the ionic dissolution of calcium–phosphate-based nanoparticles can stimulate similar relevant wound healing responses of dermal fibroblasts. Extracellular calcium concentrations of 2.5 and 3.5 mM had an impact in metabolic activity, migration, collagen synthesis, MMP activity, contractile capacity and wound healing related gene expression, and the wide range of concentrations tested allowed to detect a clear concentration-dependence behavior in cell migration and contractile capacity. The ionic dissolution of SG5 nanoparticles with Ca2+ concentrations adjusted to 3.5 mM elicited similar responses, indicating that Ca2+ is the main stimulatory molecule of the species liberated by the particles. Nevertheless, other molecules present in the release, such as organic compounds or phosphate, avoided or diminished biological responses undesired in the treatment of chronic wounds that were stimulated by Ca2+, such as increased gelatinase activity or expression of inflammatory factors. Thus, the overall stimulation triggered by the release of the tested bioactive and biodegradable nanoparticles show their suitability for chronic wound treatment. Nevertheless, due to the complexity of the healing process, their effect on other cell types and on a wound model should be studied. This work is a step forward towards the control of the wound spatio-temporal microenvironment at the nanoscale. The use of nanotechnology opens a versatile scenario to generate added-value biomedical devices and will allow the development of novel advanced therapies solutions for wound healing and other medical indications.
Acknowledgments
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO/FEDER) through the project MAT2012-38793 and MAT2015-68906-R, the Spanish Ministry of Education, Culture and Sports with the FPU grant (ref. AP-2012-5310), EIT PoC-2016-SPAIN-03 and the Dermoglass project funded by CaixaImpulse Programme, Obra Social La Caixa and Crowdfunding SUMA-UPC. We acknowledge CERCA Programme/Generalitat de Catalunya. O Castaño acknowledges the support from the Serra Hunter programme.
Conflict of interest
There are no conflicts to declare.