
Abstract
We publish three Roadmaps on photonic, electronic and atomic collision physics in order to celebrate the 60th anniversary of the ICPEAC conference. In Roadmap I, we focus on the light–matter interaction. In this area, studies of ultrafast electronic and molecular dynamics have been rapidly growing, with the advent of new light sources such as attosecond lasers and x-ray free electron lasers. In parallel, experiments with established synchrotron radiation sources and femtosecond lasers using cutting-edge detection schemes are revealing new scientific insights that have never been exploited. Relevant theories are also being rapidly developed. Target samples for photon-impact experiments are expanding from atoms and small molecules to complex systems such as biomolecules, fullerene, clusters and solids. This Roadmap aims to look back along the road, explaining the development of these fields, and look forward, collecting contributions from twenty leading groups from the field.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
INTRODUCTION
Kiyoshi Ueda1, Emma Sokell2, Stefan Schippers3, Friedrich Aumayr4 and Hossein Sadeghpour5
1Tohoku University, Japan
2University College Dublin, Ireland
3Justus-Liebig-Universität Gießen, Germany
4TU Wien, Austria
5Harvard University, United States of America
To celebrate the 60th anniversary of the ICPEAC conference, we publish a series of Roadmaps on photonic, electronic and atomic collision physics. One for each of the three classes of projectile that comprise the breadth of topics encompassed by ICPEAC: I. Light–matter interaction: II. Electron and antimatter interactions; and III. Heavy particles: with zero to relativistic speeds. Each of the Roadmaps is intended to provide an overview of the present status of the field, how it was arrived at and address current and future challenges faced by those working in the broad area of research. As with all IOP Roadmaps, the three articles have been authored collaboratively by leading researchers in the areas and each aims to provide an impression of current trends in the respective field of research.
Roadmap I. Light–matter interaction is dedicated to recent advances in photonic collision physics, one of the major scientific fields covered by ICPEAC. In this area, studies of attosecond electronic dynamics and femtosecond molecular dynamics have been rapidly growing. The natural time scales of electronic dynamics in any form of matter are attoseconds, while those of molecular dynamics are femtoseconds. The advent of new light sources, such as attosecond lasers and x-ray free electron lasers (FELs) with femtosecond pulse durations, are paving the way for such ultrafast science areas. Continuously evolving developments of new light sources such as high-repetition-rate x-ray FELs and super-intense lasers are opening new pathways for new challenges. Relevant theories have also been developing rapidly, helping to design the experiments with new-generation light sources and/or to interpret the experimental results. In parallel to these new directions in photon-impact experiments with the new generation light sources, experiments with established synchrotron radiation sources and femtosecond lasers with cutting-edge detection schemes such as multi-electron coincidence detections and momentum-resolved electron-multi-ion coincidence detections (often called COLTRIMS or reaction microscope) have been continuously revealing new scientific insights that have never been fully exploited. The target samples for these photon impact experiments are also expanding from atoms and small molecules to complex systems such as biomolecules, fullerenes, clusters and solids. The present Roadmap aims at looking back along the road briefly described above, explaining such developments of the fields, and looking forward. For this purpose, we organize the contributions from twenty of the leading groups into five topic sections: topic section 1 on attosecond electronic dynamics, topic section 2 on femtosecond molecular dynamics, topic section 3 on photoionization, topic section 4 on complex systems, and topic section 5 on new light sources for new challenges.
In topic section 1, Burgdörfer, Lemell and Tong discuss recent advances and give a theoretical perspective of attosecond physics, bridging this relatively new field to atomic collision physics, which is a core IPCEAC topic. Pfeifer describes attosecond physics and laser control of electron dynamics in atomic systems, arguing that it serves as a map and compass for navigating future terra incognita of biomolecular and condensed-phase materials. Calegari, Palacios and Martin show the roadmap to attochemistry focusing on ultrafast biological processes, describing the current status of attosecond time-resolved experiments and relevant theory as well as the experimental and theoretical challenges that must be faced to bring the required paradigm shift to the field. Corkum discusses a solid-state perspective on attosecond science, addressing how to control high-harmonic generation (HHG) with synthesized materials and how to use HHG to probe the materials. Sansone, Gryzlova and Grum-Grzhimilo describe the achievements and future directions of attosecond coherent control of electronic dynamics using multi-colour, phase controlled, extreme ultraviolet pulses generated at FERMI, a FEL facility in Italy, from both experimental and theoretical points of view.
In topic section 2, Piancastelli describes how to probe molecular dynamics with time-resolved photoelectron spectroscopy, a technique established at FERMI, and with time-resolved electron-multi-ion coincidence measurements at high-repetition FELs in the future. Weber details how molecular movies are made, taking a series of snapshots of atomic motion in chemical reactions, using XFEL-based time-resolved x-ray scattering from gaseous molecules, and gives perspectives for the planned high-repetition rate hard x-ray FEL (LCLSII-HE). Steinle, Amini and Biegert describe the current status and future challenges for direct imaging of isolated molecules with picometre and sub-femtosecond spatiotemporal resolution, something which is not possible using other techniques at the moment. Berrah and Kukk describe how to study ultrafast electronic and molecular dynamics, specifically local damage at the atomic level, caused by intense X-FEL pulses, and discuss future challenges presented by high-repetition rate FELs. Santra discusses the importance of understanding the interplay between electronic and nuclear dynamics triggered by photoionization of a valence electron, from a theoretical point of view, and considers future challenges for theory.
In topic section 3, Müller looks back on the history of photoionization (PI) experiments on positive and negative ions, describes the latest state of the art experiment at PETRA III, a low-emittance synchrotron radiation facility in Germany, and argues for the use of ion storage rings as one of the most important directions for future PI experiments on ions. Dowek, Lucchese and McCurdy review the experimental and theoretical study of photoelectron angular distributions in the molecular frame (MFPAD) and discuss time-resolved MFPAD studies as probes of nuclear and electronic dynamics in molecules and of attosecond photoionization time delays. Bolognesi and Avaldi look back on the history of photo-double ionization (PDI) on the two-electron systems of He and H2, discuss PDI as a microscope for electron–electron correlations in larger systems and consider time-resolved PDI studies as probes of electron–electron correlations in the time domain. Jahnke, Schöffler and Dörner describe the successful history of COLTRIMS reaction microscope experiments, detail how to get time-resolved information without pump–probe techniques and discuss new challenges of employing these techniques to not only high-repetition XFELs but also high-energy 'orbital angular momentum' photon beams that may become available at new generation ultralow emittance synchrotron radiation sources and open untouched areas of fundamental research. Mairesse, Nahon and Smirnova describe the history and current status of the study of circular dichroism in the photoemission of chiral molecules. Time-resolved measurements with various kinds of light sources, how the dynamical chiral response study of biological molecules benefits from advances in ultrafast laser technology, sample delivery, electron-multi-ion coincidence techniques and theory are considered.
In topic section 4, Schlathölter looks back at the journey destined to gain a fundamental understanding of the molecular mechanisms underlying radiation therapy and discusses the challenges presented by ion traps and storage rings, as well as new light sources such as XFELs and attosecond lasers. Campbell describes the history of the study of electron emission from C60 with various light sources and discusses the use of cryogenic storage rings in combination with laser excitation and photon detection, predicting that it will open a new avenue to fullerene astrochemistry, which will contribute to confronting the challenge of understanding the formation and role of fullerenes in the universe. Rost discusses the study of clusters with FELs and details the unique information that can be retrieved by time resolved experiments, such as non-equilibrium dynamics and decoherence, and considers how advances of theory can inform experiments to address such dynamics.
In topic section 5, Meyer describes the status and challenges at XEF facilities in general and discusses a new horizon promised by super-conducting-technology-based high-repetition FELs with ultrashort pulse durations, combined with advanced detector technology and coincidence techniques. Tanaka describes the status of the Extreme Infrastructure Nuclear Physics (ELI-NP), which produces ultra-intense laser fields with intensities up to 1022–1023 W cm−2 and high brilliance gamma-ray beams with photon energies up to 20 MeV, and details two flagship experiments, the challenge of dealing with strong QED effects in laser-electron collisions with a 10 PW laser pulse and a nuclear resonance fluorescence experiment for 7Li photodisintegration related to nucleosynthesis in the Big-Bang.
We hope that the contributions to this Roadmap introduced above, which are representative of many more related scientific activities, bear vivid witness that photonic collision science has been thriving in the 60 years of ICPEAC history and continues to open new science fields with new light sources.
Acknowledgments
KU acknowledges support from the five-star alliance and IMRAM project. ES thanks SFI and the EU CALIPSOplus programme for support.
ATTOSECOND ELECTRON DYNAMICS
1. Attosecond theory
Joachim Burgdörfer1, Christoph Lemell1 and Xiao-Min Tong2
1Vienna University of Technology, Austria
2University of Tsukuba, Japan
Status
The natural time scale of electronic dynamics in atoms, molecules, and solids and of the formation and propagation of non-stationary electronic states, i.e. 'wavepackets', is attoseconds (1 as = 10−18 s). As recognized since the early days of quantum physics, the time-dependent Coulomb field of a charged particle passing by an atom with high projectile velocity  acts as an extremely short half-cycle pulse of a 'virtual' photon field [1–3] with a broad frequency distribution. When the typical width of its Fourier spectrum
acts as an extremely short half-cycle pulse of a 'virtual' photon field [1–3] with a broad frequency distribution. When the typical width of its Fourier spectrum  (
( linear dimension of the atom) matches the excitation energy of the atom,
linear dimension of the atom) matches the excitation energy of the atom,  the probability for excitation and ionization by the Coulomb field is largest. This celebrated Massey criterion [4] often referred to as the velocity-matching criterion has been one of the corner stones of atomic collision physics, the overarching theme of the ICPEAC conference series now celebrating its 60th anniversary (Sir Harrie Massey was one of the co-organizers of the third edition of the ICPEAC in 1963). Translated into collision times,
the probability for excitation and ionization by the Coulomb field is largest. This celebrated Massey criterion [4] often referred to as the velocity-matching criterion has been one of the corner stones of atomic collision physics, the overarching theme of the ICPEAC conference series now celebrating its 60th anniversary (Sir Harrie Massey was one of the co-organizers of the third edition of the ICPEAC in 1963). Translated into collision times,  is in the tens of attoseconds for outer-shell electrons and can be as short as ∼10 zeptoseconds (1 zs = 10−21 s) for deeply bound inner-shell electrons. Ultra fast dynamics and attosecond physics are, thus, the hallmark of atomic collision physics.
is in the tens of attoseconds for outer-shell electrons and can be as short as ∼10 zeptoseconds (1 zs = 10−21 s) for deeply bound inner-shell electrons. Ultra fast dynamics and attosecond physics are, thus, the hallmark of atomic collision physics.
As major advances in ultrafast laser technology opened up novel opportunities to drive and to interrogate electronic non-equilibrium dynamics with well-controlled electromagnetic 'designer' pulses, the field of attosecond dynamics dramatically expanded its scope well beyond the confines of collision physics [5]. The dream of watching electronic dynamics, charge transfer, bond breaking and making, or electron ejection in real time is becoming reality even though many major challenges still lie ahead to fully realize this dream. These experimental advances also pose major challenges to theory. The accurate description of the time-dependent electronic dynamics driven by strong light fields requires the full non-perturbative solution of the many-electron time-dependent Schrödinger equation (TDSE). On an even more fundamental level, conceptual questions have come to the fore as to which physical observables can be accessed in the time domain that add to and complement information accessible by conventional spectroscopy. Such timing information, often referred to as attosecond metrology and chronoscopy, holds the promise of unprecedented insights into correlated electronic dynamics in atoms, molecules, and solids.
The availability of phase-controlled strong few-cycle IR pulses well correlated with bursts of ∼100 attosecond XUV pulse generated by the same IR pulse through the process of high-harmonic generation (HHG) has enabled the timing of the fundamental photoemission process from atoms, molecules, and solids. In a pump–probe setting the photoemission triggered by absorption by the XUV pulses can be time resolved by the concomitant IR pulse employing either the attosecond streak camera (figure 1; [6]), angular streaking ('attoclock') [7], or the interferometric RABBIT technique [8].
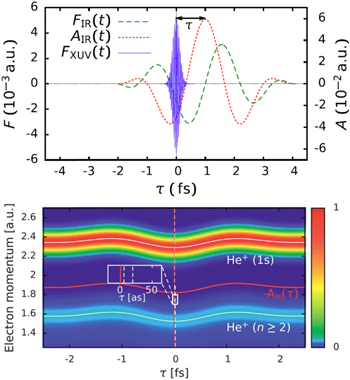
Figure 1. Simulation of attosecond streaking of photo-emission from helium: (top panel) typical temporal profile of streaking fields (IR laser field with λ = 800 nm and FWHM duration of 6 fs, XUV pulse with a FWHM duration of 200 as). (bottom panel) Streaking spectrogram for ionization of a helium atom. The vector potential AIR(τ) is also shown (orange solid line). Comparison with the shift of the spectrogram (grey dashed line) yields the streaking time shift (see inset).
Download figure:
Standard image High-resolution imageOne of the first major breakthroughs of attosecond chronoscopy was the observation of a finite time delay of photoemission relative to the arrival of the ionizing XUV pulse. The Eisenbud–Wigner–Smith (EWS) time delay originally introduced for resonant elastic scattering
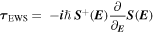
governed by the scattering matrix  became a novel experimentally accessible microscopic observable for photoionization (figure 1; [9]). Delays as short as a few attoseconds could be found for photoionization of rare gas atoms. For photoemission from extended solid-state systems, contributions originating from the primary photoabsorption, the propagation of the excited electron wavepacket to the surface, and scattering during transport could be identified.
became a novel experimentally accessible microscopic observable for photoionization (figure 1; [9]). Delays as short as a few attoseconds could be found for photoionization of rare gas atoms. For photoemission from extended solid-state systems, contributions originating from the primary photoabsorption, the propagation of the excited electron wavepacket to the surface, and scattering during transport could be identified.
Another class of time resolved observables is related to the ubiquitous appearance of quantum beats [10]. A sudden perturbation of the initial state creates a wavepacket composed of a coherent superposition of excited eigenstates of the system
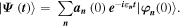
Reduced observables such as the one-particle density  and, more generally, elements of the one-particle density matrix oscillate as a function of time
and, more generally, elements of the one-particle density matrix oscillate as a function of time  that has elapsed between the pump pulse and the subsequent probe pulse. In the field of atomic collision physics, quantum beats have been observed over a remarkably wide range of time scales extending from nanoseconds for fine-structure quantum beats [10] to the zeptosecond time scale for quasi-molecular x-ray emission in energetic highly-charged ion–atom collisions [11]. The recent availability of carrier-envelope phase controlled few-cycle ir pulses and attosecond xuv pulses has opened the door to direct access to the coherent excitation amplitudes
that has elapsed between the pump pulse and the subsequent probe pulse. In the field of atomic collision physics, quantum beats have been observed over a remarkably wide range of time scales extending from nanoseconds for fine-structure quantum beats [10] to the zeptosecond time scale for quasi-molecular x-ray emission in energetic highly-charged ion–atom collisions [11]. The recent availability of carrier-envelope phase controlled few-cycle ir pulses and attosecond xuv pulses has opened the door to direct access to the coherent excitation amplitudes  and beat frequencies. Prominent examples include the beats in coherently excited neutral helium [12], the build-up of a Fano resonance in the time domain (figure 2), the fine-structure beats observed by attosecond transient absorption [14], and charge migration in molecules [15], i.e. oscillations of the electron or hole density across the molecules.
and beat frequencies. Prominent examples include the beats in coherently excited neutral helium [12], the build-up of a Fano resonance in the time domain (figure 2), the fine-structure beats observed by attosecond transient absorption [14], and charge migration in molecules [15], i.e. oscillations of the electron or hole density across the molecules.

Figure 2. Build-up of Fano resonance in the time domain for the photoelectron spectrum. Quantum beats signify the two-path interference between the direct and the indirect pathways. They appear only on ultrashort time scales but are absent in the asymptotic stationary Fano profile. Reprinted figure with permission from [13], Copyright 2013 by the American Physical Society.
Download figure:
Standard image High-resolution imageA third avenue towards encoding and mapping attosecond-time scale information involves interferences dubbed the 'time double slit' or the 'time grating'. The highly nonlinear ionization response to strong IR laser pulses results in the confinement of emission to a small fraction of an optical cycle. Only near the peak field, tunneling ionization effectively takes place giving rise to narrow 'time slits' of sub-femtosecond durations. Repeated emission near different peaks of the same cycle ('intracycle') or of subsequent cycles ('intercycle') form a double slit or a grating in the time domain giving rise to electronic wavepackets with a high degree of temporal coherence [16]. Interferences in electron emission can be viewed as Ramsey interference fringes generated by spatially separated excitation in a molecular beam [17] extended to the (sub)femtosecond scale. HHG itself, the most important building block of attosecond pulse technology, is the consequence of the temporal coherence between successive electron emission bursts every half of a period  of the optical cycle of the IR pulse. Such time-domain diffraction patterns enable access to a multitude of novel observables ranging from orbital tomography [18] to valence-electron wavepackets [19].
of the optical cycle of the IR pulse. Such time-domain diffraction patterns enable access to a multitude of novel observables ranging from orbital tomography [18] to valence-electron wavepackets [19].
Current standard tools for the theoretical description of chronoscopic information include numerical solutions of the TDSE on the single-active electron (SAE) level [20], strong-field distorted-wave Born approximations closely related to the Lewenstein model [21] or the semiclassical three-step model combining tunneling ionization, classical motion of the free electron driven in the IR field and radiative recombination upon return to the atomic core [22]. For two-electron systems, accurate numerical solutions of the TDSE have become available for attosecond XUV and moderately intense IR fields. In the perturbative limit, multi-configuration Hartree–Fock and random phase approximations have been successfully applied. For the many-electron systems such as heavy atoms, molecules, and solids time-dependent density functional theory (TDDFT) has become the workhorse of simulations of the nonlinear attosecond pulse response of matter.
Current and future challenges
For realizing the full potential of attosecond chronoscopic investigations considerable challenges still lie ahead. Present pump–probe settings involve mostly a combination of an ionizing attosecond XUV pulse (or pulse train) and a few-cycle IR pulse. Therefore, coherent excitation of attosecond bound-state wavepackets near the ground state in real time still remains to be realized. The recently developed optical attosecond pulses [23] might overcome this hurdle. Dynamical correlations have so far been mainly accessed in (multiple) ionization processes, most recently for correlated excitation-ionization [24, 25]. With further increase in intensity, attosecond pump–attosecond probe investigations of correlated inner-shell processes as well as of multi-photon multi-electron ionization should become available in the near future. Accurately accounting for such open-shell processes involving multiple continua remains a major challenge to theory.
The development of circularly polarized attosecond pulses provides new opportunities but also new challenges. The trajectory of the ionized electron and, thus, its recollision with the core can be precisely timed by two counter-rotating ω and 2ω driving pulses thereby selectively generating harmonics [26]. Moreover, elliptically polarized high harmonics may allow for inner shell excitation with a net magnetic dipole moment. Probing ultrafast magnetism and relaxation, chirality of molecules, and spin dynamics in materials is just beginning. The additional rotational and spin degrees of freedom accessible pose additional demands on theory as the dynamically relevant Hilbert subspace is considerably enlarged.
Extending concepts of attosecond metrology to more complex targets, most notably to solids, is rapidly becoming one of the key areas of applications. Observing in real time the formation and decay of excitons and plasmons or atomic scale electron and spin transport promises information complementary to what is accessible by conventional spectroscopy. Extensions to the liquid state relevant for applications to biologically relevant materials are underway [27]. Close to single-cycle IR pulses [23] allow for the extreme broadband coherent illumination and the sensitive detection of vibrational and rotational resonances of biomolecules [28]. The ubiquitous presence of dephasing and decoherence in the condensed matter environment destroys the strict Fourier reciprocity of spectral and time information encoded in the wavepacket and emphasizes the importance of direct time domain studies of transport and relaxation. Uncovering the microscopic processes underlying line broadening and dephasing in open quantum systems may, quite likely, become one of the key contributions of attosecond physics to complex systems. Attosecond physics is also expected to become relevant for devices: gas-phase streaking set-ups may soon be replaced by sub-cycle resolved measurements of currents in semiconductor or dielectric structures. HHG in bulk solids and two-dimensional materials has already been successfully demonstrated. Irradiation of nanoscale structures such as nanotips opens up the opportunity to combine attosecond time with nanometer scale spatial resolution [29]. Attosecond transient absorption and reflectance give access to the bulk and surface response of valence and conduction band electrons on the (sub)femtosecond scale [30]. A quantitative description requires, however, a multiscale theory entailing a self-consistent coupling of the nonlinear electronic response to the Maxwell field propagating inside the material and, on longer time scales, to lattice degrees of freedom.
One important area of research awaiting progress as higher intensity attosecond sources become accessible is time-resolved multi-particle break-up. For example, the paradigm case of sequential or non-sequential two-photon double ionization of helium has still not yet been experimentally observed in real time even so time sequencing is at the very heart of this process. Multi-electron emission from larger molecules and solids may shed new light on electron correlations, charge migration, and ultrafast screening of Coulomb holes.
Advances in science and technology to meet challenges
Recent theoretical and computational advances are expected to meet some of the challenges posed by the progress in experimental attosecond physics. The time-dependent multi-particle dynamics calls for a theoretical description beyond the highly successful SAE models. While TDDFT allows the treatment of the many-electron system of extended targets such as C60 or dielectrics on a mean-field level, correlated wavefunction methods such as multi-configuration time-dependent Hartree–Fock have opened up novel opportunities to treat many-electron atoms in ultrashort pulses numerically with high accuracy and in full dimensionality. Extending established methods of quantum chemistry to the time domain such as time-dependent complete active space self-consistent field, time-dependent coupled cluster, and the non-equilibrium Greens function methods and method will expand the arsenal of theoretical tools [31]. Similarly, the time-domain extension of quantum Monte Carlo methods may open new possibilities for extended systems. In between accurate correlated N-electron wavefunction methods that scale rather unfavorably with system size and mean-field methods such as TDDFT which allow for the efficient treatment of large systems but fail to explicitly describe dynamical correlations, the time-dependent two-particle reduced density matrix theory [32] may fill the gap if the challenges of N-representability can be overcome.
Concluding remarks
Attosecond physics is at the core of both atomic collision physics and the development and application of new light sources based on high-harmonic generation. Chronoscopy is increasingly complementing conventional spectroscopy providing information not easily accessible in the frequency domain. Future breakthroughs will likely address relaxation and decoherent dynamics in open and complex quantum systems. Attosecond physics holds the promise to not only observe but to actively control, steer, and manipulate electronic motion with the potential of device applications.
Acknowledgments
JB and CL were supported by the Austrian Science Fund FWF (Proj. Nos. SFB-F41, W1243), XMT was supported by a Grants-in-Aid for Scientific Research (JP16K05495) from the Japan Society for the Promotion of Science.
2. Attosecond physics and laser control of atomic dynamics
Thomas Pfeifer
Max-Planck Institute for Nuclear Physics, Germany
Status
Atomic systems have shaped the development of physics in ways second to none. A key to the discovery of quantum mechanics was the discrete nature of atomic spectral lines. Today, atoms are employed to test quantum electrodynamics, search for the variation of fundamental constants, and to place more and more stringent limits on the existence of new physics beyond the standard model of particle physics.
It thus comes as no surprise that atoms also play a pivotal role in attosecond physics. The reason is obvious: atoms, at least in their ground electronic state, and near closed electronic shells, are excellently understood by theory (section 1). Any departure from their known properties—e.g. in new environments such as in strong fields on attosecond time scales—is thus immediately obvious in experimental observables, such as their spectral features.
Atoms are thus fundamental sensors of dynamical processes on all time scales from attoseconds up to the age of the universe.
High-harmonic generation (HHG), today considered the birth of attosecond physics, was discovered in the 1980s [33, 34], by focusing intense laser pulses into atomic gases while observing surprising plateaus in the extreme-ultraviolet(XUV) spectra.
While providing the key ingredients for attosecond physics—i.e. attosecond pulses coming in trains [35] or isolated bursts [36, 37]—the HHG process itself also gave rise to a whole new field nowadays termed recollision physics [38, 39].
From the first measurement of attosecond pulses in 2001, progress is explosive, leading to the current state of the art with exceptional achievements: direct measurements of visible light wave electric fields by streaking [40], attosecond photoelectron interferometry [41], clocking Auger-[42] and photoionization [43, 44], time-resolved measurements of bound electron motion in rare-gas valence holes [14] as well as doubly-excited states [45]. Almost any of these achievements has now been transferred to fruitful applications in the fields of physical chemistry and condensed-matter physics.
The growth of this field is fueled by technological developments, such as increasingly controlled driver pulses for HHG, including their carrier-envelope phase, but also on the detection side by employing photoelectron/-ion imaging, as well as time-resolved and transient absorption spectroscopy (figure 3).
Figure 3. Fully differential measurement of atomic quantum dynamics driven by multi-parameter controlled light: testing our fundamental understanding of time-domain physics in atoms also sharpens the tools for multi-particle attosecond physics and chemistry.
Download figure:
Standard image High-resolution imageMeanwhile, with yet another technological revolution, namely free-electron lasers (FEL) a new horizon opened: attosecond physics in intense XUV/x-ray fields. For approaching this new physics regime of bright light–matter interaction—where a single atom absorbs multiple high-energy photons on short time scales within a single FEL pulse—atomic systems were crucial [46, 47].
The table here summarizes a few key figures of relevance for attosecond physics nowadays and expected in the near future:
| Properties/Source | HHG | FEL |
|---|---|---|
| Shortest duration | ∼10 as | ∼100 as |
| Pulse energy | ∼1 nJ–1 μJ | ∼0.01–10 mJ |
| Intensity (1 μm focus) | ∼1016–1019 W cm−2 | ∼1019–1022 W cm−2 |
| Photon energy | ∼0.01–1 keV | ∼0.1–10 keV |
The complementarity of the two source technologies is clearly visible.
It should be noted that x-ray FEL pulses in the SASE regime intrinsically exhibit an attosecond-pulsed temporal structure. While this structure is statistical and irreproducible between shots, it can be used for enhanced temporal resolution [48]. While stochastically isolated sub-fs FEL bursts have already been measured, soon seeded and thus fully controlled attosecond pulses will be accessible at FELs. As discussed in section 5, glimpses of the new physics possible in this regime have also meanwhile become available by observing directional emission of electrons from atoms in attosecond controlled FEL waveforms [49].
Current and future challenges
Due to the fundamental structural simplicity of atoms, they ideally allow us to study the emergence of dynamical complexity. When can an atom be described as a quantum system consisting of just two or few levels? When does the coupling to the continuum become important? Can the response of atoms be described by effectively one active electron, or do we need to consider two- or multi-electron effects? And, in the latter case, is it enough to consider a mean-field interaction of a subset of electrons, or does collective, or even correlated, motion of the electrons become important?
For fundamental light–matter interaction, even the most simple case of single-electron ionization remains an active field of research. What is the time delay of photoemission caused by the atomic structure, and the delay due to the combined action of laser and Coulomb fields? Can we define, in a quasi-classical picture, how long it takes an electron to tunnel through a barrier?
What we learn from these questions until now is that electron–electron interaction, giving rise to electron correlation, plays a crucial role and introduces complexity already for the case of two-electron atoms. A challenge in the field is thus to measure, understand and to control correlation dynamics among electrons.
Why is this important? One example: the helium atom's ground-state electronic wavefunction is a perfectly spin-entangled (and thus quantum correlated) state:
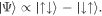
In fact, any atom's ground state provides a fully entangled cluster state. If we can control electron correlation at will, say, with attosecond shaped-pulse sequences, this would allow us to set up a self-learning quantum-computer architecture on an atomic level, with clock speeds near and exceeding PHz frequencies. A far-field vision, perhaps, but so were attosecond light pulses still in 1980, 20 years can mean a lot in science!
On the way there, we need to understand electron correlation dynamics and its manipulation. Two-photon double ionization [50] remains a benchmark experiment to be conducted in Helium and heavier species, and is still only a first step towards multi-photon and strong-field multi-electron ionization and excitation experiments, culminating in the controlled synthesis of atomic wavefunctions, including transiently populated highly-excited and extremely short-lived superpositions of neutral hollow-atom states.
The understanding and full mastery of electron correlation dynamics on an atomic level is also the fundamental key to electron-correlation-driven charge migration [51], which is a major goal in molecular attosecond science (section 3). Full controllability would allow attosecond chemistry to synthesize any stochiometrically allowed molecular structure or even a macroscopic object from a bunch of atoms in the ultimate realization of a molecular 3D printer.
Required are experiments in intense high-frequency fields, as only these are capable of specifically moving electrons with unit efficiency among inner shells. Again, the intense fields of FELs will play a pivotal role in the near future of attosecond physics. What is the dynamics of electrons driven in strong FEL fields, and what fundamental physical mechanisms can we extract to expand from two-electron systems (figure 4) to multi-electron atoms, and from there to clusters (section 18) and larger molecules (sections 9, 17)?
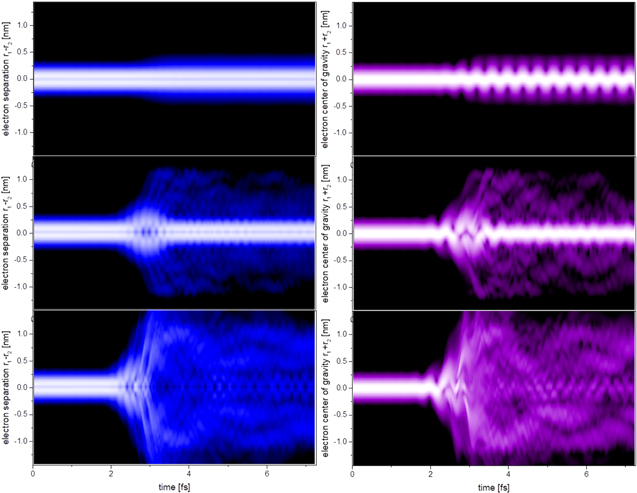
Figure 4. Emergence of dynamical complexity in a two-electron atomic model system, resonantly driven by increasingly intense high-frequency (FEL) pulses (0.8 fs duration): top to bottom: 0.01, 0.1, 0.3 a.u. peak field strength. The squared modulus of the two-electron wavefunction as a function of electron–electron separation r1 − r2 (left) as well as proportional to their center of mass r1 + r2 (right) is shown after integrating over the other coordinate, respectively. With increasing intensity, a complex time-dependent evolution is observed, illustrating the origin of correlated charge migration dynamics on an atomic level.
Download figure:
Standard image High-resolution imageThe near-future technological frontier in attosecond physics is the attosecond-pump–attosecond-probe measurement of electron motion. A fertile testing ground of this technique could be attosecond time-resolved photoelectron emission asymmetry in atoms, Auger spectroscopy, as well as attosecond transient-absorption after the coherent excitation of multi-eV-spaced electronic states in atoms.
Applying and understanding these methods in isolated atoms will calibrate these element-specific techniques, in the best case implemented as two-color as-pump–as-probe. This would allow physical chemists and condensed-matter physicists to specifically address and temporally resolve time-dependent processes in these complex systems (topic section 4) on the electronic time scale [52].
Advances in science and technology to meet challenges
To approach these visions, a continuation of the (r)evolution of light-sources as well as detectors is required.
On the light-source side, intense optical lasers and high-harmonic generation have pioneered the field and have meanwhile matured, allowing the control over pulse shape in space and time, as well as the polarization state of light down to the attosecond domain. The next technological frontiers are more intense drivers for HHG (to enable as-pump–as-probe) and higher repetition rates (for multi-parameter controlled coincidence experiments, see figure 3). In recent years, FELs have entered the scene, achieving more and more controllability over pulse properties and now come with controlled attosecond durations (section 19). While optical laser and harmonic sources still stand out in terms of their shortest pulse durations, coherence and synchronization properties, FEL pulses routinely allow XUV/x-ray-only pump–probe experiments due to their large photon numbers in excess of 1013 per pulse, already supplying intensities on the order of 1020 W cm−2 to explore new regimes of nonlinear optics [53].
Detection systems for atomic quantum dynamics basically come in two major categories: electron/ion detection, and optical spectroscopy. The momenta of several electrons and the remaining ion can be measured in coincidence with Reaction Microscopes/COLTRIMS [54] (section 14). Optical spectroscopy techniques observe absorption, also fluorescence, of targets upon transmission of the light through the target.
These two methods complement each other in a fundamental way: electron detection allows us to observe the final (continuum) state of the samples, while photon detection can reveal the initial (bound) excitation step in the sample and how it is driven by intense light fields.
By making use of both existing detection techniques, and combining them with a multi-parameter control approach for the interacting light pulses, big datasets emerge that capture a major part of the full dynamical information. For example, the impinging light pulses can be varied in frequency, intensity, phase, polarization, and come in sequences with several time delays (figure 3). Two-dimensional spectroscopy [55] was just the beginning. The mechanistic dynamical information on quantum-state coherences (section 10) and populations extracted from such multidimensional measurements on atoms in strong fields will serve as map and compass for navigating the future terra incognita of (bio-)molecular (section 3, topic sections 2–4) and condensed-phase materials (section 4), and for science and technology applications that are yet unconsidered.
Concluding remarks
Atomic physics is the beating heart of quantum dynamics in general, and attosecond science in particular. It provides both a mining and a testing ground for new physics mechanisms, time-domain metrology and control methods. It will play a pivotal role for the future of attosecond physics as the field is pushed forward by the joint thrust of lab-based (HHG) and large-scale (FEL) light sources, and multidimensional detection techniques, allowing the capture of the full set of dynamical information. It will enable precision tests of our understanding of laser-driven quantum dynamics, and give rise to control techniques and applications which are still beyond our wildest dreams. Molecular-scale petahertz-clocked computing could be one of them, as well as laser-directed chemistry on the atomic level by light-field control of multi-electron correlation dynamics in the core and valence shells of any atom.
Acknowledgments
Attosecond physics develops rapidly due the cooperative work of many outstanding junior and senior researchers and their groups, which all deserve recognition. The few references allowed in this section can only reflect a small and non-representative subset of the massive body of researchers and their work in this field.
3. A roadmap to attochemistry
F Calegari1,2, A Palacios3 and F Martín3
1 Center for Free Electron Science and University of Hamburg, Germany
2Institut fur Experimentalphysik, Universitat Hamburg, Germany
3 Universidad Autónoma de Madrid, Spain
Status
Attosecond laser technology has demonstrated its suitability to trace and manipulate electron dynamics in atoms and small molecules. However, application of this technology to more complex molecules, as those involved in biological processes (photosynthesis, radiation damage, etc), is still in its infancy. A common approach to visualize this electron dynamics with few-femtosecond and sub-femtosecond time resolution is attosecond pump–probe spectroscopy [56, 57]. In this method, an attosecond extreme ultraviolet (XUV) pulse is used to excite/ionize the molecule, followed by a time delayed probe pulse to trace the dynamics generated in the first step. Typical probe pulses have frequencies ranging from the near-infrared (NIR) to the x-ray domain, with durations going from few tens of femtoseconds to few hundreds of attoseconds. These experimental capabilities are thus ready to explore the possibility to control chemical reactions by acting at the very early stages of the process, e.g. by guiding electronic motion in such a way that bonds break and form following a desired path.
In pursuing this goal, recent attosecond XUV-pump/NIR-probe gas phase experiments (set up shown in figure 5) performed in aromatic amino acids [15] have captured the occurrence of ultrafast charge dynamics after ionization. In these experiments, the molecule is ionized by an attosecond pulse, creating a delocalized electronic hole that moves all over the molecule on a sub-femtosecond time scale. The interplay between the electronic and nuclear motions dictates the evolution of the hole, which can lead to a final localization of the charge on one specific molecular site or specific molecular fragment. One can thus expect that, depending on the initially created hole, fragmentation will occur leading to different chemical species [58, 59], hence to distinct reactivity. The chemical selectivity that could be attained by using attosecond technology remains yet to be demonstrated.
Figure 5. Scheme for the experimental detection of charge migration in amino acids.
Download figure:
Standard image High-resolution imageThe experimental approach used to track charge migration in amino acids is based on the measurement of the photo-fragmentation yield as a function of the XUV-pump NIR-probe delay [15]. As shown in figure 5, a clean plume of intact and neutral biomolecules is produced by evaporation in the interaction region located in the middle of a mass time-of-flight (TOF) spectrometer. A sequence of isolated XUV attosecond pulses and few-femtosecond NIR pulses is used to initiate and probe the charge migration process. The doubly charged immonium ion (resulting from the loss of the carboxyl group) has been found to show clear oscillatory dynamics, which has been assigned to charge flow between different functional groups of the molecules.
An alternative to pump–probe methods is high-order harmonic spectroscopy (HHS), which has been demonstrated to be an effective tool to track electron dynamics in linear molecules, as e.g. iodoacetilene [60].
Reliable theoretical tools are mandatory to interpret attosecond spectroscopy experiments. However, a full dimensional quantum mechanical treatment of molecular ionization dynamics, accounting for all electronic and nuclear degrees of freedom, is still out of reach by using current computational approaches, except for the hydrogen molecule. Recent theoretical approaches in large molecules have incorporated the effect of nuclear dynamics by using semiclassical treatments in which the motion of electrons is described quantum mechanically and that of the nuclei by using a classical description. Application of these methods to amino acids indicate that electronic coherences can survive for at least 10 fs (see figure 6 and [61]), thus being experimentally accessible. In other molecules, however, electronic coherences have been found to survive 1 or 2 fs [62]. The general consensus is that lifetimes of electronic coherences are strongly dependent on the species under study and the pulse characteristics. Therefore, accounting for nuclear motion seems to be mandatory to correctly describe the effect of electronic coherences on the fragmentation that follows the sudden excitation or ionization of the molecule, even qualitatively.

Figure 6. Evolution of the hole density in glycine after interaction with an attosecond pulse (bandwidth ∼18–35 eV). FNA: fixed nuclei approximation. ED: Ehrenhest dynamics obtained within a TD-DFT approach.
Download figure:
Standard image High-resolution imageBesides the above-mentioned limitations, there is the additional complication of dealing with the electronic continuum of the molecule, which cannot be represented using standard quantum chemistry methods. For this reason, most theoretical approaches for large molecules have ignored the ionization step and have only described the cation dynamics that arises from arbitrarily chosen initial wave packets. On top of that, XUV and x-ray photons can also produce inner-shell vacancies that can subsequently be refilled by outer-shell electrons whose excess energy is then used to ionize other outer-shell electrons. This process, known as Auger decay, is mostly due to electron correlation. While this limitation can be somehow circumvented, e.g. by choosing pumping frequencies such that Auger decay barely contributes, it should be considered in future work, as production of Auger electrons has been recognized as a very important source of damage in biomolecules.
Current and future challenges
One of the most ambitious challenges of attosecond science is to design control strategies that can efficiently be used to induce ultrafast electron dynamics in molecules, in gas-phase or in solution, so that one can selectively break and form chemical bonds, thus leading to new chemical reactions.
On the experimental side, there are still a lot of challenges to extend attosecond technology to complex molecules. First, complex molecules are produced with relatively low densities and the typical photon flux of the attosecond sources is at the limit to provide a reasonable signal-to-noise ratio. Second, direct imaging methods for the electron dynamics still need to be developed and third, control over the electronic excitation is required. This last goal could only be reached by improving the current capability of tailoring attosecond pulses.
On the theoretical side, many efforts are currently made to combine advanced quantum chemistry methods, as multi-reference configuration interaction methods especially designed to accurately treat electron correlation, with scattering theory formalisms appropriate to deal with the ionization continuum [57]. In addition to this, methods going beyond standard classical descriptions of nuclear dynamics, such as Ehrenfest MD or Trajectory Surface Hopping, should be developed. Fully quantum mechanical approaches, which are currently limited to small molecules, may provide further insights on the coupled electron-nuclear dynamics and, more importantly, on establishing if quantum interferences and coherences may indeed have an impact in reactions that are relevant in chemistry or biology and occur at a much longer time scale. The latter also implies going beyond gas-phase chemistry and face the additional challenge of considering the effect of the environment [63].
Advances in science and technology to meet challenges
Recent and future advances in attosecond technology will allow most of the experimental challenges listed in the previous section to be met. In particular, the tremendous progress in the development of ultrafast lasers (OPCPAs and fiber lasers)—already delivering hundreds of watts—allows for driving the attosecond pulse generation process with higher peak intensities and higher repetition rates. At the same time the pulse duration of the free-electron-laser (FEL) sources is approaching the attosecond limit [64]. With the photon flux provided by the FELs, it will be soon possible to perform diffraction imaging of complex molecules with attosecond time resolution, thus providing an effective method to directly imaging and control the electron dynamics.
To meet the above theoretical challenges and establish the mechanisms governing XUV irradiation of medium-size and large molecules one has to develop tools that allow one to describe (i) ionization, including electron correlation terms that determine processes such Auger decay, and (ii) the ensuing coupled electron and nuclear dynamics. Also, as molecules responsible for most biological functions are either in solution or in a hydrophobic environment, not in the gas phase as in most attosecond experiments performed to date, theory should go a step further and consider the effect of such environment.
Concluding remarks
Is it possible to (efficiently) achieve chemical selectivity by using coherent ultrashort light sources? How can electronic coherences affect fragmentation after molecular excitation and/or ionization? Attosecond pump–probe and HH spectroscopies could help in answering these questions by extending the existing experimental methods—which have proven to be successful to attain sub-femtosecond electronic control in atoms and small molecules—in combination with new theoretical approaches able to describe the coupled motion of electrons and nuclei that dictate the chemistry of these systems. If successfully accomplished, this would inevitably pave the way to attochemistry.
Acknowledgments
FC acknowledge the financial support from the ERC Starting Research Grant STARLIGHT No. 637756. AP and FM acknowledge the financial support from the Ministry of Economy and External Trade (MINECO) project FIS2016-77889-R. AP acknowledges a Ramón y Cajal contract from the Ministerio de Economía y Competitividad (Spain). FM acknowledges support from the 'Severo Ochoa' Programme for Centres of Excellence in R&D (MINECO, Grant SEV-2016-0686).
4. A solid state perspective on attosecond and high-harmonic generation
Paul Corkum
University of Ottawa, Canada
High-harmonic radiation from solids has potential applications as a source of XUV radiation and as a means to probe solids.
Status
In general, harmonic radiation is created by the nonlinear interaction of light and matter. When light is a perturbation, harmonics arise as the bound electrons explore their near surroundings, thereby formally making virtual transitions to all excited bound states. This is the condition for conventional harmonic generation.
In the other limit, when light is very intense, near-surface electrons are pulled free of the material, enabling them to move over wavelength-scale distances in response to the light's electric field. This non-sinusoidal electron motion can produce very high harmonics, especially when the light intensity is relativistic.
At intermediate intensity, if the material is irradiated by below-bandgap light, the bound electrons can make a valence-to-conduction-band transition, again creating an oscillating current that will yield harmonics of the fundamental beam.
However, inside a material, these internal electrons are confined to the bands of the solid. Electron motion still creates a time-dependent current that produces the emission, but the harmonic-spectrum results from Bloch Oscillations which are, of course, a signature of the periodic structure of the material. In such cases, the harmonic spectrum encodes the potential structure that the electron feels as it oscillates in the material.
That said, we know from studies of atomic and molecular gases that there is a second, very important mechanism for this intermediate field-driven electron to generate high harmonics. (One might think of this mechanism as a hybrid between the extremes.) Here, the newly ionized electron begins its oscillation in the field, but the oscillation brings the electron back to its origin, where it re-collides with its parent, recombining and thereby releasing its kinetic energy plus the ionization potential as XUV radiation [38]. This source, very important in gases, should also be present in solids where an electron and its correlated hole plays the role of an electron and ion in gases as illustrated in figure 7.
Figure 7. High harmonics generation from an atomic gas as seen from a solid state perspective.
Download figure:
Standard image High-resolution imageIn fact, we now know that 'recollision' is the source of high-harmonic radiation that was discovered in ZnO by Ghimire et al in 2011 [65] and analyzed in the theoretical papers of Vampa [66]. Recollision, (identified by the time-dependent frequency of emission known as the 'atto-chirp') plays a dominant role in many, but not all, experiments [67] where high harmonics are produced from intense mid-infrared radiation that excites transparent solids.
Figure 8. Schematic of an experiment where ZnO with a surface of nano-pyramids is irradiated with 2 µm light from behind. We see a photograph of the diffracted third and fifth harmonic radiation and and an image of the emitting surface. From [68]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageWhile I have described four interrelated mechanisms for generating harmonics of a fundamental beam, I believe that it will be possible to create hybrids between them—between surface harmonics and bulk harmonic radiation, for example—but as yet there are no reported observation.
Can high-harmonic generation become an important source of VUV radiation?
Solids have the advantage of simplicity and flexibility over gases, and the same material can be used multiple times. In addition, attosecond duration pulses can be generated. However, so far we cannot reach nearly as short a wavelength without damaging the solid as we can when we use gases; furthermore, it is not clear if the efficiency of harmonic emission from solids can approach that of gases. That acknowledged, I still predict that attosecond (or high-harmonic) pulses will be commercially generated from solid media at least for photon energy greater than 30 eV.
In gases, high-harmonic photon energy can greatly exceed ionization potential, which implies that there is a maximum medium length beyond which the generated XUV radiation is absorbed by re-ionization.
Equally, in solids, the highest harmonics can have photon energies that are far above the bandgap of the material. However, in solids, the density is much higher than in gases and therefore, the absorption length is very short. In this case, phase-matching equally favours forward- or backward-propagating radiation. Therefore, it is often advantageous to irradiate from the front surface, because high-intensity radiation does not pass through the sample before it creates harmonics.
Solid-state physics has spawned important technical developments that we can exploit to control harmonic radiation
For example, technology allows us to dope materials, and doping will modify the amplitude and phase of harmonic emission [68]—just as we control the amplitude and phase of high-harmonic emission with mixed gases. Doping can be performed with high-resolution ion injection [68], with the solid serving as a fixed template. This unique characteristic of solids may be very important, because it allows us to use the material to create light that naturally focusses. Solids, used as templates on which to write, may free us from the constraints of conventional XUV optics.
But solids also give us an ability to shape materials [68]. Quantum wells, quantum wires, or structured surfaces will allow us to control the electron trajectory, and therefore, to control the efficiency of harmonic conversion [69]. It is even possible to add nano-plasmonic antennas to a solid [70] to significantly decrease the required illuminating power, thereby bringing high-harmonic generation from high bandgap materials within reach of mode-locked oscillators, while also linking high-harmonic generation to modern research on meta-materials. These nanoscale resonant structures will complement build-up cavities for creating XUV frequency combs.
Can high-harmonic generation become an important probe of materials?
Recollision harmonics report the difference between the energies of the two (or more) bands that are involved in high-harmonic emission. In fact, we already know that the band structure of solids can be determined from the two-dimensional (two-color) high-harmonic map [71]. These maps are similar to those used to establish the mechanism of harmonic-generation in solids [66]. Reader might be concerned with disentangling the many bands of a solid, but the requirement of phase-matching appears to select only two (or a very few) bands.
Of course, the band structure of most materials is well known from photo-electron spectroscopy, but photo-electron spectroscopy is not accessible in high-pressure diamond anvil cells, nor can it be used to follow ultrafast dynamics during laser material processing, or materials in hostile environments. It would be a major accomplishment for science if we could time-resolve phase changes driven by lattice rearrangements or by quantum correlations.
Band reconstruction, was inspired by orbital tomography of gas phase molecules. It would be very important if we could transfer the concept of tomography directly from gases to solids. Such a transfer now appears to be possible [72].
While the penetration depth of XUV light in materials is small, it is many monolayers deep. However, at least in carbon, a single atomic layer of material is sufficient to produce measurable high-harmonic radiation [73]. (We know this because high harmonics can be produced from graphene and other 2D materials [73].) If a single layer of material is sufficient, then time-dependent surface chemistry can be probed. I predict that high-harmonic spectroscopy will open a new diagnostic for surface chemistry.
Biological material is often highly ordered, and this order will influence the emission spectrum—as we discussed above. Thus, if thin slices of biological material are used as a substrate, then the structure and composition will be indelibly written on the harmonic spectrum. Furthermore, even amorphous and polycrystalline materials are already used to produce high harmonics [67].
Extreme nonlinear optics offers a new chapter in which, I predict, the short wavelength of the harmonic emission will be harnessed for high spatial resolution and the resulting image will accompany 2 or 3 photon imaging or CARS microscopy.
In closing, we consider electronic circuits. Measurements show that the harmonic spectrum is sensitive to small fields—fields that are on the scale of those used for switching in conventional electronic circuits. It should be possible to obtain 2D images of an operating circuit using real-space optics for measurement up to its limits, and then to use diffractive methods such as ptychography up to the high-harmonic cut-off. It is not immediately clear what the cut-off will be. When MgO is irradiated from the front surface, in my lab we see a cut-off of 45 eV.
5. Attosecond coherent control of electronic dynamics using multi-color, phase controlled, extreme ultraviolet pulses
Giuseppe Sansone1, Elena V Gryzlova2 and Alexei N Grum-Grzhimailo2
1Albert-Ludwigs-Universitӓt, Germany
2Lomonosov Moscow State University, Russia
Status
Coherent control in the micro world is based on the phenomenon of interference of amplitudes of two or several paths from initial state of a quantum system to its final state. Changing phases between the amplitudes controls the final result of the process [74]. Usually coherent control is performed by an optical laser, which is easy to manipulate. Studies of coherent control in the optical domain, both theoretical and experimental, were initiated about 30 years ago. Creation and manipulation of phase synchronized pulses of vacuum ultraviolet and x-ray radiation is a highly complex task because of the lack of the necessary optical elements. Only recently it was first implemented at the seeded free electron laser (FEL) FERMI [75]. Coherent control was realized for the photoelectron angular distributions (PAD) from neon atoms. The two ionization paths were produced by the fundamental frequency (two-photon 'process ω') and by its second harmonic (one-photon 'process 2ω'). The control of the phase between the harmonics, and, consequently, between ω and 2ω ionization amplitudes, was carried out without optical elements by adjustable time delay with a temporal resolution of 3 as of electrons bunch generating the second harmonic. For two parallel linearly polarized ω and 2ω pulses, the asymmetry of the PAD with respect to a plane perpendicular to the polarization direction was controlled by the relative phase. For two circular polarizations, the axial symmetry with respect to the collinear radiation beams would be violated, as predicted by the theory (figure 9).

Figure 9. Formation of typical PADs with time in the Ne(2p) ionization by coherent ω + 2ω linearly (a) and circularly with opposite helicity (b) polarized pulses. For the incoherent pulses the PADs would be constant and symmetric, like in the beginning of the pulse. The evolution is shown for the relative phase φ = 0 between the harmonics. The final PADs for φ = π illustrate their control by the relative phase of the harmonics. For details of the theoretical model see [76].
Download figure:
Standard image High-resolution imageThe realization of coherent control in the extreme ultraviolet (EUV) and x-ray range represents a fundamental milestone in the development of coherent spectroscopy in the EUV and soft x-ray range, which is one of the largely unexplored areas of research in ultrafast phenomena. Advances in this field will be boosted by the implementation in the x-ray regime of spectroscopy techniques already established in the optical domain such as Raman and photon echo spectroscopy. Their demonstration will give new insight into ultrafast phenomena including relaxation processes such as Auger decay and migration of electrons in chemical reactions. One of the main advantage of the soft and x-ray spectral range is the chemical selectivity in the excitation and probing of ultrafast electronic dynamics, a characteristics which is absent for interaction with photons in the optical range. Thus, soft and x-ray femto- and attosecond pulses offer the unique possibility to resolve the ultrafast electronic dynamics in time and in space.
A first demonstration of coherent spectroscopy in the EUV spectral range, based on a four-wave mixing (FWM) process, was reported at the FERMI in 2015 [77]. Two coherent EUV pulses created a grating inside SiO2, which diffracted a third infrared pulse. The FWM signal presented oscillation, which were attributed to the acoustic modes launched in the sample. Recently the technique has been extended to a pure four-wave mixing in the EUV range by the combination of three EUV pulses [78]. The possibility to introduce a delay between the first two pulses will offer an additional control tool towards the implementation of more complex multidimensional spectroscopy schemes [79].
Current and future challenges
The coherent control on the attosecond timescale of electronic processes calls for techniques enabling the manipulation of the amplitude and phase of the spectral components composing the attosecond waveform. This is a formidable challenge as it requires the development of techniques suitable for bandwidth extending over more than tens of eV in the EUV and soft x-ray.
Complementary experimental approaches are currently on going using both table-top high-order harmonic generation (HHG) sources and FELs. They are strongly supported by theoretical works indicating possible implementation schemes and the advantages offered by time-resolved experiments in this spectral range for the observation of ultrafast electronic dynamics [80, 81]. Due to the low conversion efficiency of the HHG process, attosecond pulse generated by table-top sources are still rather weak for the application of schemes based on the combination of different EUV pulses. Phase control of attosecond pulses either by metallic filter [82] or chirped mirrors [83] has been suggested, but both strategies suffer from a limited flexibility and the coupling between amplitude and phase shaping. Traditional approaches, well-established in the femtosecond domain such as pulse shapers based on liquid crystals or acoustic-optical modulators cannot be extended to the EUV domain, due to the lack of suitable materials for modifying the phase in this spectral range. However, alternative approaches such as phase-modulation spectroscopy combined with HHG offer a viable strategy for overcoming these limitations [84].
Rather than controlling the spectral components of the EUV pulses, a less direct, but more promising approach for the control of the relative phase is represented by the steering of the motion of the electron bunch (in the case of FELs) or of the electronic wave packet (in the case of HHG) leading to the generation of the EUV radiation. This was adopted in the experiment described above [75] for controlling the relative phase between the two coherent harmonics. The scaling of this approach to multipleharmonics with complete control of the relative phase between the harmonics should be within reach for seeded FELs like FERMI. The combination of multiple harmonics would also open new perspectives in the synthetisation of trains of attosecond pulses at FELs. The temporal characterisation of the phase differences could be achieved by measuring the photoionisation spectra by the combination of the EUV harmonics and a collinearly propagating infrared field. This approach is well established in the attosecond community for the temporal reconstruction of the attosecond pulse train generated by HHG in gases [8]. The extension to FEL-based harmonics should be possible, even though alternative approaches for overcoming the lack of sub-cycle synchronisation between the EUV harmonics and the IR field must be developed.
In HHG in gases, the electronic dynamics after the tunnelling ionisation process can be controlled by sculpting the driving field. Simulations for determining the optimal wave for HHG were reported [85], as well as the first experimental results based on the combination of different multi-color pulses in the visible and near-infrared spectral range [86]. By shaping the electric field of few-cycle, multi-color pulses (also with perpendicular polarisation) it is possible to precisely control the motion of the electronic wave packet, giving the opportunity to modify the recollision probability and the phase accumulated in the continuum. To which extent this control could be translated into a precise and independent manipulation of the amplitude and phase of the EUV components generated upon recombination remains an open question.
The description of coherent control in EUV poses serious challenges for theory even for isolated atoms. Involving the inner shells, relativistic effects, intermediate and autoionizing resonances, competition of photoionization with the Auger decay call for primordial many-electron approaches for description of the target. Contribution of higher multipoles of the electromagnetic field may be noticeable in the EUV and possible nondipole effects in the coherent control need to be considered. At the same time, a broad interval of field intensities, pulse durations, electron energies,—prohibit application of a universal theoretical toolkit and imply using various theoretical approaches for treating the time evolution. It ranges from different versions of the time-dependent perturbation theory and strong field approximation (the latter rather for EUV + optical pulses) to direct solution of the time-dependent Schrödinger and quantum Liouville equations. Since it is not possible to solve the many-electron equations explicitly, several approximate methods are under development (see also section 1). A variety of time-propagation schemes have been considered too, although most of the examples are still restricted by a model of single-electron moving in a local effective potential.
The direct comparison between experimental and theoretical results is very difficult, because the former are obtained in a reaction volume, with not well defined peak intensities and profiles of the beams and pulse durations. Cases when it is necessary to apply different theoretical approaches for different sites and spectral intervals (for example, near resonances) are not exceptional. Probably this kind of difficulty is the main reason why currently only a very limited number of theoretical groups take on the tasks of direct comparison of their calculations with the experiment.
The coherent control of PADs potentially allows the extraction of new information on the ionization amplitudes and atomic continuum in the spirit of 'complete experiments' [87], not accessible so far by other means, in particular, with incoherent beams. The field would benefit from realization of coherent control on spin polarization and vector correlations of the reaction products. The former may be useful for the possible usage in an ultrafast spin switch.
Advances in science and technology to meet challenges
For the extension of coherent spectroscopy techniques to the soft x-ray at FELs, it will be necessary to further investigate the generation of coherent multi-color schemes already adopted at seeded FEL. Self-seeding approaches deliver pulses with a well-defined spectral bandwidth even though large shot-to-shot fluctuations are observed [88]. Sub-cycle control would also offer new challenges, due to the shorter wavelength of the radiation. The method applied so far for the control of the relative delay with a precision of 3 attoseconds [75] appears to be scalable up to the soft x-ray range, with typical oscillation periods of a few and few tens of attoseconds.
For table-top HHG sources, the development of multi-color, synchronized few-cycle pulses is a prerequisite for sculpting the electric field waveform. Advances in optical parametric amplifiers operating in complementary spectral ranges represent a viable strategy for the synthetization of coherent fields extending from the visible to the near-infrared. These pulses offer not only the possibility to generate more efficiently isolated attosecond pulses, but also to shape the characteristics of the EUV spectrum [89]. The independent manipulation of different spectral range of a supercontinuum generated in a hollow-fiber capillary has already been demonstrated and applied for the generation of isolated attosecond pulses [90]. Control of the EUV properties would require strategies similar to those adopted and well-investigated in the femtosecond domain such as the implementation of genetic algorithms and feedback loops [91].
Concluding remarks
The benefit of the field of coherent control and coherent spectroscopy of electronic processes with a resolution from the complementaries between EUV pulses generated by HHG sources and FELs down to the attosecond timescale will be strongly felt in the upcoming years. Cross-fertilization between these two research fields is expected to give rise to dramatic improvements in our capability to initiate, steer and observe electronic processes in the core and valence shells with unprecedented temporal resolution.
Acknowledgments
The ideas and future perspectives outlined in this report are the result of intense discussions over many years (and many beamtimes) with colleagues and collaborators from the attosecond and free electron laser science communities. We gratefully acknowledge their invaluable contributions.
FEMTOSECOND MOLECULAR DYNAMICS
6. Time-resolved molecular photoelectron spectroscopy at a seeded FEL
M N Piancastelli1, 2
1Uppsala University, Sweden
2Sorbonne Université, France
Status
The investigation of molecular reactions on their 'natural' timescale (femtoseconds to picoseconds) is a sought-after goal not only in chemistry, but also in physics and material science, with possible extensions to biology.
Fundamental questions in ultrafast photodynamics of molecular systems, in particular in photoinduced chemical reactions, are: what is the timescale for the reaction? Is it possible to 'catch' some intermediate species? What is exactly the mechanism? How many steps does it imply? Is it possible to manipulate and control the system by varying some instrumental parameters?
The exciting possibility of observing the evolution of molecules during chemical reactions is often called 'the making of a molecular movie', implying the goal of preparing a movie showing molecular motions with femtosecond up to picosecond time resolution and possibly electronic and/or geometrical structural resolution for all species involved.
The new free-electron laser sources (FELs), with the availability of very intense and very short pulses, have brought the above dream close to reality.
The main avenue in the investigation of photochemical reactions by time-resolved spectroscopic techniques at FELs is a pump–probe sequence, namely laser pumping the ground state of a molecule of interest to a suitable excited state, and probing the intermediate states thus formed with several different methods.
Previous investigation aiming at characterizing ultrafast molecular dynamical processes have been conducted at FELs, primarily with structural techniques such as photoelectron diffraction [92], and x-ray diffraction/scattering [93]. Although structural techniques such as electron or x-ray diffraction are very informative, sometimes a significant change in electronic structure results in a small or even negligible change in geometry. Furthermore, for such techniques lengthy and complex data analysis is required.
A much more direct way to characterize molecular dynamics even in a very short timescale would be to look at valence photoelectron spectra in combination with ion yield spectra, to follow photochemical reaction pathways. Valence photoelectron spectra yield direct information of the electronic structure of the species under investigation and are able to reveal even subtle changes in response to external parameters such as e.g. temperature change or photoexcitation. However, to observe the first stages of a chemical process using photoelectron spectra, picosecond (ps) or femtosecond (fs) time resolution is needed, together with sufficient photon and electron kinetic energy resolution. The most suitable light sources are therefore FELs or high-harmonic generation (HHG). However, up to very recently this relatively simple approach has been hindered by the facts that at FELs the photon energy jitter is generally too large to give sufficiently energy-resolved spectra without heavy data treatment and/or averaging over several time delays, and with simple HHG sources the resolution is masked by the simultaneous presence of multiple harmonics. If harmonic filtering is employed, the resulting intensity is too low to follow subtle changes.
Current and future challenges
A recent very significant step forward in this direction is represented by the newly built FERMI FEL at the Elettra facility, Trieste, Italy [94]. This seeded source provides pulses with negligible photon energy jitter. In this way, photoemission spectra can be measured with sufficient resolution to identify very clearly ionization processes stemming from different electronic states. The seeding scheme used at FERMI also provides an IR pulse which is very accurately synchronized to the FEL pulse, because it is derived from the same Ti:Sa laser that drives the seed chain.
Using the FERMI seeded source as probe in combination with the seed laser as pump, we have performed time-resolved experiments on a prototypical system, acetylacetone [95].
In figure 10 we show valence photoelectron spectra measured for different pump–probe delays. The blue curves in the higher binding energy region correspond to ground-state final states, while in the lower binding energy region spectral features are shown which correspond to the ionization of three different excited states: S2 (light blue), S1 (yellow), and T1 (green).
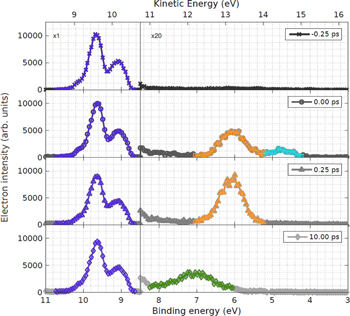
Figure 10. Valence photoelectron spectra of acetylacetone as a function of pump–probe delay. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Nature Communications [95] 2018 CC-BY 4.0.
Download figure:
Standard image High-resolution imageThe time evolution was followed up to 1.5 ps pump–probe delay with 50 fs steps and to 200 ps with larger steps. A clear picture of the evolution of the system is reached, showing that the photoexcitation to the S2 (ππ*) (bright) state is followed by a conical intersection (CI) connecting with the S1 (nπ*) (dark) state, and then the T1 (ππ*) state is reached through ultrafast S1 (nπ*)/T2 (nπ*) crossing which is immediately followed by internal conversion to T1 (ππ*). A minor pathway leading back to the ground state is also identified. Observed fragmentation yielding CHx species is related to the onset of the T1 (ππ*) state formation.
A schematic view of all intermediate states, their crossing paths and possible fragmentation pathways is shown in figure 11.
Figure 11. Schematic view of the photoexcited species which are formed and their time evolution. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Nature Communications [95] 2018 CC-BY 4.0.
Download figure:
Standard image High-resolution imageA second experiment was performed under similar conditions, allowing a real-time study of a ring-opening chemical reaction [96]. In general, it would be very useful to perform electron–ion coincidence measurements, to link a specific electron emission process to a specific ionic species. However, at FERMI the repetition rate available (50 Hz) does not allow this kind of data collection. Therefore a statistical method was used, namely partial covariance mapping, which allowed us to relate specific electronic finals states to specific fragmentation processes.
In our opinion, this approach based on high-resolution valence spectra backed by high-level calculations and ion yield spectra is the ultimate way to shed light on fundamental, basic photo processes such as photosynthesis, photovoltaic energy production, and vision.
Advances in science and technology to meet challenges
While FERMI as the only operational seeded source allows obtaining unprecedented detailed information on molecular dynamics by measuring valence or core photoelectron spectra with a time resolution of 50 fs, a suitable FEL source with negligible photon energy jitter and very short pulses (few fs) does not yet exist. Furthermore, the photon energy range available at FERMI is at the moment limited to reach the carbon K-edge.
To overcome the problem of photon energy jitter, an obvious way would be to use coincidence techniques, to link one particular photoelectron emission to one or more specific ion species production. Coincidence techniques are nowadays available at FLASH, where a repetition rate of hundreds of Hz is possible [97]. Future plans at LCLS, Stanford, California, include the upgrade to LCLS II, with high repetition rate and self-seeding to reduce the photon energy jitter [98]. Also at the European X-FEL the repetition rate will be 27 kHz, sufficient to perform electron-multi-ion coincidence experiments [99].
Another planned evolution for the field is the possibility of going down to the attosecond regime in pulse length. Such opportunity will provide access at FELs not only to nuclear motion, but even to electron motion on its 'natural' timescale. While the attosecond regime is already available at HHG sources, at FELs it will be possible to explore electron dynamics in the x-ray range, with high single-pulse power. Developments in this direction are announced at LCLS II [98] and European X-FEL [100].
Concluding remarks
The possibility of exploring in great detail photochemical processes on their natural timescale is an exciting development nowadays in place at FELs. In particular, the seeded source FERMI provides a negligible photon energy jitter which can be exploited to obtain time-resolved valence and more recently core level spectra with sufficient resolution to characterize reaction intermediate species. Future developments at several FELs include high repetition rate, to allow for electron-multi-ion coincidence measurements, and very short pulses, in the attosecond range, to have access not only to nuclear dynamics but even to electron dynamics.
Acknowledgments
I wish to acknowledge all authors of [95], who participated in the above described experimental and theoretical work at FERMI.
7. Chemical reaction dynamics from time-resolved x-ray scattering of free molecules
Peter M Weber
Brown University, United States of America
Status
Chemical reactions are at the heart of chemistry and form the basis of a wide range of molecular science, engineering and medicine. Yet chemical reactions are difficult to understand on the most fundamental level because they are inherently time-dependent processes of highly coupled, many-body systems. An ideal observation would determine the time dependence of atomic positions within reacting molecules, and the changes in the electron charge densities as chemical bonds break or form. Additionally, for reactions induced by coherent light pulses, important information could be gleaned from the shape of the wave packets in the multidimensional phase space. As a graphical representation of the time-evolving molecular structures, 'molecular movies' could aid the intuitive understanding of chemical dynamics.
Ultra fast time-resolved investigations aiming to explore chemical reactions while they unfold have traditionally employed spectroscopic methods. For all but the smallest molecules, the inversion of spectroscopic data to yield the desired chemical reaction movie is not possible. The aim of ultrafast time-resolved x-ray scattering experiments, as well as the complementary electron scattering, is to derive experimental, time-dependent maps of molecular structures. Beyond the immediate visualization of chemical dynamics useful to synthetic chemists, the molecular movie could reveal previously unseen structural motions associated with reactions, and the excited state structures could serve as benchmarks for the development of computational codes.
While in crystalline phases the molecular structure and the chemical dynamics are constrained by their crystal surroundings, reactions of free molecules in the gas phase can progress uninhibited, offering a direct comparison to calculated reaction pathways. Experimentally, gaseous samples are advantageous because the flowing gas avoids the degradation commonly seen with crystalline samples. Each laser shot hits a refreshed sample, eliminating the restriction to reversible processes. Drawbacks of studies on free molecules include the rotational averaging over the random, or only partially aligned, molecular samples, and their exceedingly low density compared to crystalline samples. The latter is countered by the immense x-ray intensity offered by modern x-ray free electron lasers (XFEL).
Figure 12 shows the experimental concept. A molecular sample is intersected by an optical laser pulse that induces the chemical reaction. The time-evolving system is probed by a subsequent x-ray pulse. Scattering patterns are recorded by a large area detector and read out on a shot-by-shot basis. By binning the recorded scattering patterns according to the delay time between the laser pump and the x-ray probe pulses, a time resolution in the sub-100 fs regime can currently be achieved [101, 102].
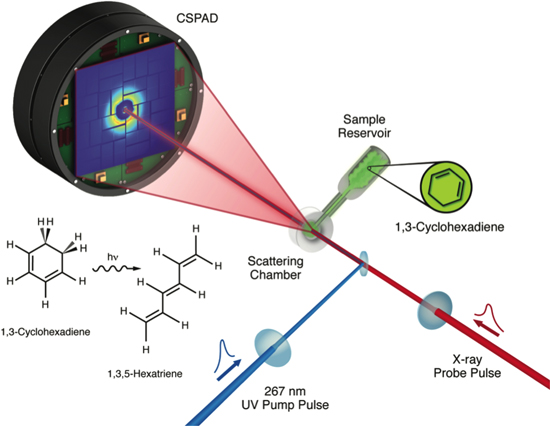
Figure 12. Experimental approach to time-resolved pump–probe scattering of free molecules. An optical pump pulse, often in the UV range of the spectrum, excites the molecules to a reactive state. The transient structures are measured using a time-delayed x-ray pulse. Reprinted figure with permission from [103], Copyright 2015 by the American Physical Society.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Ultra fast time-resolved x-ray scattering experiments have made possible the recording of a movie of the molecular structures while a molecule undergoes an electrocyclic ring opening reaction by bypassing a conical intersection [103]. Another example shows the wave packet motions of a diatomic molecule [104]. Further studies are in progress: movies of molecular vibrations, dissociation reactions, charge delocalization processes and many more. The success of these experiments depends critically on the level of optical excitation: if the pump laser pulse intensity is too high, multi-photon processes may lead the molecular system to a mix of poorly characterized, highly excited or ionized states. To place the molecule with confidence on a single reacting surface the pump intensity has to remain low, which leads to small changes in the scattering signal. Consequently, the dynamic range of the experiment must be such that changes in the scattering pattern of less than 0.1% can be recorded over the entire range of scattering vectors.
Photon energies up to about 10 keV can be conveniently obtained at the Linac Coherent Light Source (LCLS) at the SLAC National Accelerator Laboratory. Using a large area detector scattering vectors up to about 5 Å−1 can then be accessed, allowing for the determination of excited-state structures with precision in the milli-Ångstrom range. Given the excellent signal to noise ratios obtained with pump–probe measurements, this is sufficient for detailed comparison to computed patterns, and thus for the extraction of the structural dynamics. But to characterize the chemical dynamics, one would ideally determine both the center and the shape of the time-evolving wave packets. Simulations show that to image the density distributions associated with traveling wave packets, scattering vectors up to 15 Å−1 need to be accessed, see figure 13 [105].

Figure 13. Contour plots of the calculated modified (elastic) scattering intensity for the ring-opening reaction of 1,3-cyclohexadiene (CHD). Time in fs and momentum transfer q in Å−1, with the modified intensity given as the molecular scattering intensity scaled by the atomic structure factors as defined in [106]. The calculation assumes standard values of the width of the time evolving wave packet. Reproduced from [105]. CC-BY 4.0.
Download figure:
Standard image High-resolution imageTheoretical models are required to accurately describe the scattering of molecules reacting on excited state surfaces. The total scattering signal of an ensemble of free molecules is described by the Fourier transform of electron pair correlation functions in molecules, which are constructed from two-particle density matrices [106]. Thus, a complete description of the total scattering signal consists both of elastic and inelastic components. In addition to the electronic components of the wavefunction, the nuclear rotational and vibrational wavefunctions must be included in the simulation of scattering signals [107]. For polyatomic molecules in excited vibrational and/or electronic states as appropriate for reacting molecules, the simulation of the scattering patterns therefore entails high-level calculations of the respective time-dependent nuclear structures and electronic states. Such a complete description has not yet been obtained.
Advances in science and technology to meet challenges
Next generation XFELs, in particular LCLS-II, will provide higher photon energies and brightness. Photon energies in the 20 keV range would not only allow for more accurate structure determination, but also reveal details of the wave packet motions. Dedicated detectors that capture scattered radiation over a larger range of scattering angles would be important as well.
The higher brightness of LCLS-II is largely a consequence of the increased repetition rate. To take full advantage of the brightness, the experimental scheme must match the detector parameters to the repetition rate so as to enable read-out of individual pump–probe frames together with the shot-to-shot timing information. The high repetition rate coupled with the high photon energy, as may be possible with LCLS-II-HE, would then enable tremendously improved scattering measurements that could truly determine the time evolution of structure and chemical bonding during molecular reactions.
Limits to the temporal resolution at this time result mostly from the duration of the optical laser pulses. Shorter pulses would enable the experiment to resolve higher frequency motions. Moreover, the extraction of spatial density distributions of traveling wave packets requires optical excitation with laser pulses of well-defined coherence parameters. Pulse shaping technologies exist, but have not yet been implemented at the CXI beam line at LCLS where the x-ray scattering experiments are performed. Typically UV photons are required to break chemical bonds, so that the pulse shaping technologies need to be implemented at the UV wavelengths.
Much information is lost in the averaging over the random orientation of free molecules. Alignment of the molecules, using any of the well-established methods, could reduce the information loss [108]. However, given the wide amplitude motions associated with pendular states, the common limitation of alignment schemes to one of the three rotational axes, and the additional experimental uncertainties associated with the alignment parameters, it seems doubtful that the benefit merits the added difficulty. A very different approach would instead record scattering patterns of individual molecules and sort the frames by orientation, similar to existing methods on microcrystals [109]. The high repetition rate of LCLS-II may enable such experiments, opening up an additional exciting frontier of single-molecule, time-resolved chemical dynamics x-ray scattering.
Concluding remarks
Experiments with ultrafast time resolution can probe processes on the inherent time scales of molecular systems. While spectroscopic experiments are fundamentally limited by the time-energy uncertainty, scattering experiments are not: it is possible to simultaneously measure atomic positions and time without fundamental limitation. While technical challenges are substantial, many can be overcome so that much finer details of reaction dynamics could be uncovered. For example, it would be exciting to observe the complicated interplay of electron densities with geometrical structure as a molecule passes a conical intersection. Ultra fast time-resolved x-ray scattering is poised to image, in real time, the atomic rearrangements of molecules during chemical reactions. Foreseeable advances will add the imaging of the electron density distributions, so that it will be possible to watch the time evolution of chemical bonds—a chemist's dream come true.
Acknowledgments
This work was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, under Award #DE-S0017995.
8. Molecular self-imaging with attosecond electron wave packets.
Tobias Steinle1, Kasra Amini1 and Jens Biegert1, 2
1ICFO-Institut de Ciencies Fotoniques, Spain
2ICREA, Spain
Status and background
Unravelling the mechanisms of chemical reactions requires imaging techniques that capture snapshots of the molecular structure on the nuclear (i.e. few femtosecond; 1 fs = 10−15 s) timescale with atomic (i.e. Ångstrom; 1 Å = 10−10 m) spatial resolution. Electron scattering techniques are preferable over photon scattering due to the small de Broglie wavelength (λB) of electrons, providing picometre spatial resolution, and the orders-of-magnitude higher scattering cross section, which permit the imaging of single molecules [110]. Consequently, a wide range of scattering methods are utilized in molecular imaging studies, such as: ultrafast electron diffraction (UED) [111], relativistic electron diffraction (RED) [112], ultrafast electron microscopy (UEM) [113], laser-assisted electron diffraction (LAED) [114], and laser-induced electron diffraction (LIED) [115–118]. Achieving the desired combined space and time resolution has proven difficult as electrons interact with each other, thus limiting the achievable temporal resolution to time-scales on the hundreds of femtosecond range [119]. This problem is elegantly solved in LIED [120, 121], which achieves the ultimate time-limit on the attosecond scale, by utilizing only a single attosecond electron wave packet (EWP). The quantum trajectory of the EWP is entirely determined by the laser field which, after roughly two thirds of a laser cycle[122, 38], drives the EWP back to its origin where it self-interrogates the molecule's geometric structure with attosecond (1 as = 10−18 s) and picometre (1 pm = 10−12 m) resolution. It is worth noting that the entire process of LIED is well-described in the framework of laser-driven electron recollision [122, 38]. Contrary to high harmonic generation, phase-matching is irrelevant for LIED, i.e. the rescattered electron signal is entirely dominated by the long quantum trajectory. An important aspect of LIED is its ability to extract the field-free doubly differential elastic scattering cross section based on the classical recollision model [116, 123], despite measuring inside a strong laser field. A consequence of the strong field and launching the recollision EWP is the triggering of molecular dynamics that can be investigated using an intra-pulse pump–probe scheme with mid-IR (MIR) LIED's intrinsic attosecond timing resolution [124].
An important aspect for the development of molecular imaging is the fact that many different molecular reaction pathways exist which complicate the image analysis. We have shown that this issue can be mitigated [117, 125] through kinematic coincidence detection in combination with MIR high repetition rate sources [126, 127]. MIR recollision [128] is crucial for LIED especially for larger molecules with a lower ionization potential, making it necessary to generate a recollision EWP in the quasi-static regime, and the required 'hard' collision to pinpoint atomic locations. Mid-IR wavelengths are also key to image larger molecular structures with excellent spatial resolution.
Current and future challenges
Figure 14 shows the calculated scaling of the recollision wave packet's transverse extent with driving wavelength together with the de Broglie wavelength of the EWP. The graph indicates that MIR-LIED is in principle able to image molecular structures with large spatial extent; for comparison, the size of the C60 fullerene is roughly 7 Å. The large spatial extent of the recollision EWP occurs, however, with the penalty of decreasing scattering cross section ( ) with increasing wavelength. To overcome this challenge, the repetition rate of the MIR laser must be sufficiently large to acquire enough statistics in a reasonable measurement timeframe. An interesting feature of MIR-LIED is the intermediate kinetic energy regime in which LIED can operate, despite achieving sufficient momentum transfer for structural reconstruction. LIED electron impact energies range roughly between 50 and 200 eV for which hydrogen exhibits appreciable scattering cross section values. Consequently, we have been able to image hydrogen atoms [123, 125] to which UED is typically blind due to the much higher impact energies used in UED. The enticing prospect in LIED is therefore imaging the ubiquitous proton motion, as demonstrated by Wolter et al [117].
) with increasing wavelength. To overcome this challenge, the repetition rate of the MIR laser must be sufficiently large to acquire enough statistics in a reasonable measurement timeframe. An interesting feature of MIR-LIED is the intermediate kinetic energy regime in which LIED can operate, despite achieving sufficient momentum transfer for structural reconstruction. LIED electron impact energies range roughly between 50 and 200 eV for which hydrogen exhibits appreciable scattering cross section values. Consequently, we have been able to image hydrogen atoms [123, 125] to which UED is typically blind due to the much higher impact energies used in UED. The enticing prospect in LIED is therefore imaging the ubiquitous proton motion, as demonstrated by Wolter et al [117].
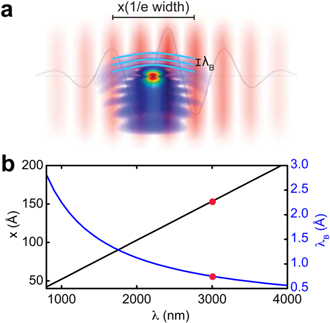
Figure 14. Molecular imaging with an attosecond electron wave packet. (a) An attosecond electron wave packet (EWP; blue) is generated from a molecular target by the electric field of a short laser pulse (red). After propagation during less than an optical cycle, the returning EWP (wave fronts highlighted as blue lines) images its target ion. The lateral spread (1/e width), x, and the de Broglie wavelength, λB, of the EWP's wave fronts are illustrated in panel (a) and plotted as a function of the driving laser wavelength, λ, in panel (b) with respective values highlighted at a driving laser wavelength of 3000 nm (red circles).
Download figure:
Standard image High-resolution imageWith respect to target complexity and size, the gas-phase measurement of larger molecules presents the difficulty of preparing these in a gas-phase molecular jet since larger structures are typically either in liquid or solid form. More importantly, the ionization potential decreases with larger molecules, and an adequate balance must be struck between photoionization to launch the recollision electron and saturating ionization for a given target species.
Future challenges will arise in the LIED of more complex biomolecules, such as azo complexes or peptides. For example, retrieval algorithms are typically based on the quantitative rescattering (QRS) theory with the assumption of atoms being independent from each other (IAM). With the much increased structural complexity and larger molecular systems, the QRS approach may not be tractable since it requires the extrema to be found in a multidimensional solution space. An alternative approach to retrieve structural information is through the Fourier transform (FT) variant of LIED, called FT-LIED, which does not rely on prior structural information or retrieval algorithms. These two straightforward variants of LIED are shown in figure 15.
Figure 15. Variants of LIED. In QRS-LIED (FT-LIED), the attosecond recollision EWP is investigated at fixed (varied) rescattering momentum, kr, and varied (fixed) rescattering angles, θr. The grey shaded area indicates the rescattering region of interest that the LIED method utilizes in the extraction of structural information. In the case of FT-LIED, the Fourier transform of the back-scattered (θr ≈ 180°) attosecond EWP provides an image of the object.
Download figure:
Standard image High-resolution imageAn important further prospect of LIED is the clear possibility to make a full attosecond resolved molecular movie. An obvious choice would be the change of LIED wavelength, but it would only change the time range by a few femtoseconds whilst dramatically changing the recollision dynamics. A better solution could be pump–probe time-resolved LIED in which a UV pulse prepares the molecular state and an MIR pulse images its structure at various different pump–probe delays.
Concluding remarks
MIR-LIED has shown its potential to image isolated molecules with a single electron, thus achieving a combined attosecond temporal and picometre spatial resolution. The strength of the LIED method is access to the first femtoseconds during which the course of a chemical reaction is determined, and we firmly believe that many future investigations will be able to utilize these strengths in pump–probe studies of complex molecular systems.
Acknowledgments
We acknowledge financial support from the Spanish Ministry of Economy and Competitiveness (MINECO), through the 'Severo Ochoa' Programme for Centres of Excellence in R&D (SEV-2015-0522) Fundació Cellex Barcelona and the CERCA Programme/Generalitat de Catalunya. We acknowledge the European Research Council for ERC Advanced Grant TRANSFORMER (788218), MINECO for Plan Nacional FIS2017-89536-P; AGAUR for 2017 SGR1639, Laserlab-Europe (EU-H2020 654148).
9. Molecular dynamics in intense FEL fields
Nora Berrah1 and Edwin Kukk2
1University of Connecticut, United States of America
2University of Turku, Finland
Status
The first x-ray free electron laser (FEL) to reach the inner atomic shells was the Linac Coherent Light Source (LCLS) that has since been used to investigate femtosecond photo-induced molecular dynamics such as H-migration [129], nonlinear physics [130, 131], or ring opening [132] which are important physical and chemical processes [133]. The next x-ray FEL to become available was SACLA in Japan, where, owing to its unique ability to reach deep atomic inner shells, the investigations concentrated on probing the photoinduced dynamics of highly charged systems containing heavy elements [134, 135]. Both of these FELs can deposit, during the few femtoseconds of an FEL pulse, a very large amount of energy into the system; as much as 87 keV into a single fullerene molecule, for example [130]. Such unique experimental conditions trigger also unique molecular dynamics (MD).
Several noteworthy x-ray FEL-based pump–probe investigations of MD have made direct observations of structural changes on femtosecond time-scale. A showcase experiment is the isomerization of acetylene into vinylidene, studied following valence ionization at FLASH FEL. At the LCLS, research with C 1s core ionization focused on determining the onset of the ultrafast deuterium migration in deuterated acetylene (DCCD), converting it into vinylidene (CCDD). That FEL-based work employed x-ray pump–x-ray probe technique with an x-ray split and delay tool. In that experiment, absorption of the first x-ray pulse is followed by Auger decay, initiating isomerization from DCCD2+ to CCDD2+. The motion of D from one C site to the other was captured by the delayed x-ray probe pulse that caused a Coulomb explosion into four ions: C+, C+, D+, and D+. The time evolution of the ion momenta was found to be in the first 12 fs following core ionization [129], demonstrating that non-adiabatic interactions can couple nuclear motion to the electronic relaxation.
An experimental and theoretical investigation of the fs ionization dynamics of C60 at high pulse intensity was carried out to understand the response of a relatively large, strongly-bonded molecule. C60 was ionized above the C K-edge at 485 eV photon energy to reach conditions in which each atom in a C60 molecule, in the x-ray focus, absorbs multiple photons. The core ionization with high-fluence x-ray induces the Auger process, resulting in many photo- and Auger electrons due to the cyclic multiphoton ionization. This process leads to secondary ionization of C60 as well as its fragments ions by the photo- and Auger electrons. The C60 molecule charges up to  and because of the short C–C bond lengths, it fragments via Coulomb repulsion into molecular and C ion charge states distribution from C+ to C6+ at 90 fs pulse duration as shown in figure 16 [130]. Theoretical MD simulations were carried out, as shown by the blue bars in figure 16, revealing that strong secondary effects were responsible for the suppression of the ion yield for the highly charged C ions [130]. Namely, electrons were being recaptured by the highly charged ions. A recent time-resolved experiment was conducted in C60 to investigate further this ionization and fragmentation mechanism.
and because of the short C–C bond lengths, it fragments via Coulomb repulsion into molecular and C ion charge states distribution from C+ to C6+ at 90 fs pulse duration as shown in figure 16 [130]. Theoretical MD simulations were carried out, as shown by the blue bars in figure 16, revealing that strong secondary effects were responsible for the suppression of the ion yield for the highly charged C ions [130]. Namely, electrons were being recaptured by the highly charged ions. A recent time-resolved experiment was conducted in C60 to investigate further this ionization and fragmentation mechanism.
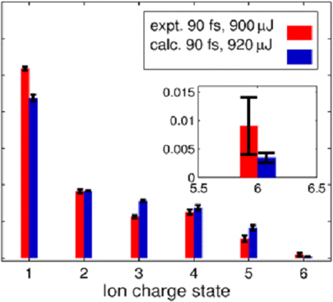
Figure 16. Relative carbon ion yield versus charge state. Experimental (red bars) and model data (blue bars). Reprinted by permission from Macmillan Publishers Ltd: Nature Communications [130], Copyright 2014.
Download figure:
Standard image High-resolution imageThese MD processes were triggered by soft x-ray core ionization in light elements such as carbon. In other MD experiments carried out at SACLA, multiphoton deep core ionization of iodine and xenon atoms led to generation of highly charged states by cascade Auger processes. The following two examples illustrate the ensuing dynamics in a small (di-iodomethane) and a much larger (Xe clusters) quantum system.
In iodine-containing organic molecules such as di-iodomethane, I is an absorption hotspot at x-ray energies above ∼5 keV, leading to positive charge flow from iodine(s) to the entire molecule, concurrent with Coulomb explosion [136]. MD was studied using multi-ion coincident time-of-flight spectroscopy with full 3D momentum resolution. A tailored SCC-DFTB theoretical approach was developed [136] in order to successfully capture the dynamical aspect of the induced Coulomb explosion, reproducing the experimental kinetic energy distributions of fragment ion (figure 17) and various other experimental fingerprints. Similar investigations of larger, iodine-substituted biomolecules such as 5-iodouracil [138] have a link to important applications in medicine, namely radiotherapy, since the very same 'soup' of molecular fragments and emitted electrons can be responsible for the destruction of a cancerous cell, via introducing and irradiating such a radio-sensitizer molecule.

Figure 17. Charge dependence of the total kinetic energy of fragment ions for CH3I and CH2I2 molecules. Reproduced from [136] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageIn the next example, Xe clusters were studied in an FEL-pump near-infrared (NIR)-probe experiment at SACLA, again using ion time-of-flight spectroscopy to detect and analyze the atomic Xe fragments from Coulomb explosion of clusters [137]. In this much larger quantum system, the focus was on the collective phenomena such as the formation, expansion and evaporation of electronic nanoplasma. By probing the transient excited states of the atomic Xe fragments, the experiment was able to capture the ultrafast population (∼12 fs) and depopulation (∼250 fs) of these excited states during nanoplasma formation in Xe clusters. Here, not only photoionization and Auger processes, but also inelastic scattering and interatomic Coulomb decay (ICD) come into play. The nanoplasma formation and MD are the two processes that cannot be treated in isolation, but in a combined description of the system. Such type of experiments have a direct relevance for the use of XFEL pulses in determining the structure of nanosized objects.
Current and future challenges
X-ray FELs have already proven to be excellent tools for the investigation of MD, but are not without technical challenges. The self-amplified spontaneous emission (SASE) that produces FEL radiation at the LCLS and SACLA facilities showcased above, is noisy and presents photon energy jitter that makes it difficult to fine-tune to specific atomic resonances, for example, or to perform electron spectroscopy with high resolution. The most complete information about the photodissociating sample can in principle be gathered by multiparticle coincidence measurements, recording all created charged particles, and their momenta. In reality, this is impossible for all but the smallest systems. However, combining electron and multi-ion detection is very beneficial also for much larger systems. Here, some practical challenges relate to the ability of the detection system to cope with the specific pattern of FEL operation—large number of particles can be produced and must be detected in a short interval followed by long periods of inactivity. On the one hand, this can make successful recording and disentanglement of coincident datasets overwhelmingly difficult; on the other hand, low repetition rates have made the collection times of more complex datasets prohibitively long.
Time-resolved pump–probe experiments that offer the most direct access to MD have their own additional challenges. For example, obtaining full and stable spatial and temporal overlap of the pump and the probe pulses has proven often to be time-consuming and failure-prone processes in the earlier experiments at LCLS and SACLA, partly due to the challenges in developing suitable and accurate calibration and diagnostics tools; a goal often requiring close collaboration between the facility and the scientists.
One of the next scientific challenges is to move beyond the simple showcase molecules and investigate complex photo-induced processes in a broad category of systems, that occur in the sub-fs and attosecond regimes.
Advances in science and technology to meet challenges
A key trend in these complex experiments is that no deeper interpretation is possible without close theoretical support. Theoretical models, novel approaches and practical simulation tools such as XMDYN [130, 133] are being developed at a fast pace. Experiment-wise, a variety of data analysis tools have been harnessed. Particularly in multiparticle detection experiments, even gradual and small improvements in technology can make a crucial difference—for example employing novel 'funnel' type microchannel plate detectors could improve four-ion coincident detection efficiency more than five-fold.
The advances in FEL technology are being made at a very fast pace. Perhaps most crucial for molecular dynamics experiments are the dramatically increased repetition rates, new, highly flexible x-ray pump–x-ray probe creation schemes and improvements on the pulse jitter, such as using self-seeding and shortening the FEL pulse duration to under femtosecond range for accessing even faster dynamics.
Concluding remarks
The first decade of experiments on molecular dynamics using FELs in the soft- and hard x-ray regimes has already yielded a much more detailed insight into photoinduced ultrafast processes than has been possible with conventional light sources.
The next generation x-FELs such as the LCLS-II, the European X-FEL and others that provide high-frequency attosecond x-ray pulses and versatile pump–probe schemes will ensure that the FEL-based science meets the next challenges in the investigations of molecular dynamics.
Acknowledgments
10. Interplay between electron dynamics and nuclear dynamics
Robin Santra
Deutsches Elektronen-Synchrotron DESY, Germany
Universität Hamburg, Germany
Status
When an Nel-electron system is singly ionized, the remaining (Nel − 1)-electron system may or may not be in a non-stationary state. This depends on the degree of coherence present in the (Nel − 1)-electron system. The concept of coherence is defined through the reduced density matrix obtained by tracing the density matrix of the entire Nel-system over the degrees of freedom of the ionized electron. Thus, from the perspective of system-bath models, the (Nel − 1)-electron system is the 'system' and the ionized electron is the 'bath'.
The topic of valence-electron dynamics following ionization has risen in importance because of the development of light sources that enable electron removal on a time scale that is comparable to the natural time scale for valence-hole dynamics. For example, the observation of valence-hole dynamics in Kr atoms was made possible via optical tunnel ionization using a strong quasi-single-cycle optical pulse [14]. These measurements demonstrated the need for a reduced-density-matrix description and revealed a degree of hole coherence of about 60%—limited by the finite duration of the ionizing pulse.
An alternative to optical tunnel ionization is one-photon ionization using an ultrashort XUV pulse from a high-harmonic-generation source or from a free-electron laser. Theory has demonstrated that in ultrafast XUV photoionization of atomic Xe, the maximum degree of coherence in the (Nel − 1)-electron system is limited by the electron–electron-interaction-mediated entanglement between photoelectron and hole [139].
In polyatomic systems, an additional effect appears that limits electronic coherence: the motion of the atomic nuclei. In this situation, not only the photoelectron, but also the nuclei act as a bath for the (Nel − 1)-electron system. In other words, in order to characterize the coherence properties of the hole, the density matrix of the entire system must be traced over the nuclear degrees of freedom as well.
In theoretical work [140, 141], it has been shown that, for a number of representative molecular systems, decoherence of an initially perfectly coherent hole wave packet [a wave packet of the (Nel − 1)-electron system] is typically completed within just a few femtoseconds (see figure 18). The main reason for this effect is the (zero-point) nuclear motion in the ground-electronic state of the target molecule: over the range of molecular geometries sampled by the nuclear motion, the Born–Oppenheimer potential energy surfaces (BO PESs) of the (Nel − 1)-electron eigenstates involved in the electronic wave packet are not perfectly parallel to each other. As a consequence, averaging over these nuclear geometries gives rise to a characteristic time scale for electronic dephasing.
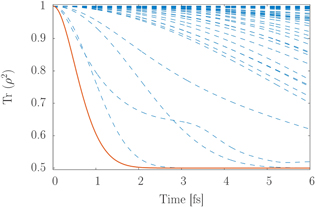
Figure 18. A measure of electronic coherence (the so-called purity) is plotted as a function of time for a hole wave packet in phenylalanine involving two (Nel − 1)-electron eigenstates. The dashed lines show the individual contributions to electronic decoherence from each of the 63 normal modes. The solid line shows the combined effect of all normal modes. A purity of 0.5 corresponds to a completely incoherent superposition of the two electronic states. Reprinted figure with permission from [141]. Copyright 2017, by the American Physical Society.
Download figure:
Standard image High-resolution imageIn regions of nuclear configuration space where BO PESs intersect and so-called conical intersections are formed, the BO approximation breaks down and the question of electronic coherence becomes more subtle. If the electronic wave packet is formed at nuclear geometries sufficiently close to a conical intersection, the electronic phase information is not entirely lost before the molecule passes through the conical intersection. Then, the electronic coherence imprinted in the photoexcitation process can affect the time evolution of the molecular geometry and, hence, offers a potential route towards chemical control [142].
Current and future challenges
Depending on whether the valence-electron removal is triggered by optical tunnel ionization or XUV one-photon absorption, different hurdles must be overcome when attempting a theoretical description of the associated dynamics. In the following, I will focus on XUV photoionization, under the assumption that the sudden approximation is valid. Under these conditions, it is possible to concentrate on the properties of the residual (Nel − 1)-electron system.
Using an XUV photon, in principle any valence-hole state can be accessed. It is known that particularly in the inner-valence regime, strong electron-correlation effects predominate [143]. Therefore, computationally expensive electronic many-body theory is required. So far, such calculations are available primarily near the equilibrium geometry of the target molecule in its ground state. However, in order to predict XUV-induced chemical dynamics, it is necessary to construct BO PESs over a wide range of nuclear geometries.
This is a challenge for the following reason. Typically, XUV photoionization causes molecular fragmentation. At the corresponding nuclear geometries, the Hartree–Fock reference state generally is no longer a good first-order approximation to the electronic ground state of the target molecule. The reason why this is significant is because standard many-body methods such as coupled-cluster theory or Green's functions typically rest on the assumption that the Nel-electron ground state can be approximated using a single Slater determinant, based on a mean-field framework such as ground-state Hartree–Fock.
In addition to the failure of the single-determinant approximation, there is the challenge that a rather large number of (Nel − 1)-electron BO PESs must be computed. Consider a molecule consisting of Nat atoms. At the electronic mean-field level, we expect that the number of initially occupied molecular orbitals—and, therefore, the number of BO PESs reached by electron removal—scales as Nat. Thus, for a molecule containing ten second-row atoms, we would already need to calculate tens of BO PESs.
If one allows for configuration mixing of the one-hole configurations with the two-hole one-particle configurations, the number of potentially relevant BO PESs no longer scales merely as Nat, but, in the worst case, scales as  Thus, in the ten-atom system alluded to above, it may be necessary to compute thousands of BO PESs. Note in this context that standard molecular configuration-interaction codes do not allow the user to compute more than a few electronic eigenenergies.
Thus, in the ten-atom system alluded to above, it may be necessary to compute thousands of BO PESs. Note in this context that standard molecular configuration-interaction codes do not allow the user to compute more than a few electronic eigenenergies.
The high number of (Nel − 1)-electron eigenstates generally required for describing XUV-induced dynamics has another consequence: the energy splittings between neighboring BO PESs tend to be small. This is illustrated in figure 19. Hence, conical intersections are no longer well-isolated entities, but there is a high density of conical intersections as a function of nuclear displacements. In other words, not only must one deal with severe electron-correlation effects, one must also cope with an increased incidence of non-Born–Oppenheimer processes [144, 145].
Figure 19. Time evolution of the 63 one-hole outer-valence BO PESs of photoionized H+(H2O)21. The black curve shows the instantaneous electronic state, starting from the energetically highest outer-valence state. In order to reach the electronic ground state, the system has to move through a large number of conical intersections. Reproduced from [144] with permission from the Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageFurther note that some, or even many, of the BO PESs may be associated with metastable states that can decay by spontaneous electron emission. This challenge is difficult to handle within standard Hermitian quantum mechanics, but may be treated, in principle, using methods developed within the framework of non-Hermitian quantum mechanics [146]. However, such methods are still not routinely available in molecular electronic-structure codes.
Advances in science and technology to meet challenges
The challenges must be addressed using a two-pronged strategy. First, dedicated advances in electronic-structure theory are required. These must take into account strong electron-correlation effects and must provide access to a large number of electronic eigenstates characterized by a high spectral density. Methods based on the one-particle Green's function are a natural choice for the context described here. However, among other things, the limitations imposed by the Gell-Mann and Low theorem must be overcome.
Second, advances are required to handle the nuclear dynamics problem. Mixed quantum–classical methods, which traditionally work in the adiabatic representation, lose their effectiveness when dealing with a quasi-continuum of electronic states. Pure quantum methods that rely on a local, diabatic representation (such as the vibronic-coupling Hamiltonian) are unsuitable for situations where a wide range of nuclear configuration space must be covered and where the density of conical intersections is high.
Concluding remarks
In a contribution to a Roadmap of ultrafast x-ray atomic and molecular physics [147], I discussed challenges for theory in connection with high-intensity x-rays. In the present contribution, I have been focusing on femtosecond dynamics triggered by photoionization of only one valence electron (a process that may be accompanied by the ejection of additional electrons). In spite of many past investigations in this area, there are still important unsolved problems, which offer exciting opportunities for future research.
Acknowledgments
I thank the Hamburg Centre for Ultra fast Imaging (CUI) and SFB 925 for support. This contribution builds on work with Caroline Arnold, Sophia Bazzi, Lorenz Cederbaum, Zheng Li, Mohamed El-Amine Madjet, Stefan Pabst, Nina Rohringer, Oriol Vendrell, and Ralph Welsch.
PHOTOIONIZATION
11. Photoionization of ions
Alfred Müller
Justus-Liebig-Universität Gießen, Germany
Status
Photoionization (PI) is the removal of one (or several) electron(s) from an atom or an ion by a single photon either directly or in a multi-step process involving intermediate autoionizing states. Indirect PI mechanisms include multiple excitation of outer-shell electrons with subsequent electron ejection and the production of an inner-shell vacancy followed by Auger decay(s).
PI and the related spectroscopy are behind many of the ground-breaking advances in modern physics. Besides fundamental atomic-physics insights, studies of PI provide key information for applications in many fields of research such as plasma physics, the physics of planetary atmospheres and astrophysics including research on interstellar and intergalactic media.
Experiments on PI of ions are impeded by the difficulty to provide dense pure targets of ions preferentially at low temperatures to avoid Doppler broadening. Relatively high densities can be provided by laser-produced plasmas or in ion traps. The conceptually cleanest experiments use ion targets in the form of accelerated beams. Advantages of photon–ion interacting-beams arrangements are the relatively straightforward measurement of absolute PI cross sections, the selectivity in providing pure ensembles of mass-over-charge analyzed ions and the low ion temperature that allows for high-precision spectroscopy. This contribution focuses on the photon–ion merged-beams technique [148].
The first interacting-beams PI experiments were conducted with negative ions looking into the release of the least-bound electron for which filtered optical light from a bright projection lamp was sufficient. A boost of PI experiments with negative ions came with the development of optical and UV lasers. PI of positive ions requires higher photon energies and, hence, the first merged-beams experiments with positive ions had to await the development of bright synchrotron radiation sources. A review of work on PI of positive and negative ions until 2005 has been provided by Kjeldsen [149]. Presently, only two experimental facilities using merged beams are regularly producing PI results, MAIA [150] at SOLEIL, and PIPE [151] at PETRA III.
The bulk of previous experiments with ions has yielded total single ionization cross sections for valence-shell PI. Due to limitations in photon energy and photon flux not many measurements on deep inner shells could be reported. The development of new high-brilliance synchrotron radiation sources, particularly SOLEIL and PETRA III, provides possibilities to extend PI experiments to higher photon energies and to processes with smaller cross sections. In addition, spectroscopy of electrons released by PI of ions is now feasible [150].
PETRA III is presently the most brilliant synchrotron radiation source. Small bandwidth photon beams at energies Eγ of 250–3000 eV are available for PIPE experiments while the upper limit of Eγ at MAIA is 1000 eV with much smaller photon flux.
Current and future challenges
Presently, great efforts are devoted to the detailed understanding of multi-electron processes such as multiple ionization by a single photon accompanied and followed by multiple-electron excitations and decays. Partial absolute cross sections for multiple ionization associated with deep inner shells are being determined with the number ν of electrons removed from the target ion by a single photon reaching up to ν = 6 [152].
As an example for multi-electron processes, non-sequential triple-Auger decay in a four-electron interaction was unambiguously observed with C+ ions [153] which provide the minimum number of electrons in the L shell to facilitate the process. The outstanding sensitivity of the PIPE setup with its strong suppression of detector background has made it possible to measure cross sections as small as a few hundred barns. This sensitivity facilitated the observation of the direct triple-Auger decay.
Direct removal of two electrons by a single photon was recently observed with F− ions. Simultaneous ionization of the K and L shells were detected in the triple ionization channel on top of a contribution arising from K-shell ionization with subsequent emission of two electrons. The cross section for direct double ionization was measured over a wide energy range [154].
Such studies on multi-electron processes can be supported by the coincident detection and spectroscopy of electrons and ions subsequent to PI. An example is the investigation of the Auger decay of the 4d → nf (n = 4, 5) resonances in the Xe5+ ion [155].
The theoretical description of multi-electron processes in atoms and ions is challenged by the new experiments (see, e.g. [152]). New approaches have to be developed and tested against measured data. Changing the ion charge state along isoelectronic sequences provides a 'knob' in such experiments by which the relative importance of electron–electron versus electron–nucleus interactions can be tuned for better insight into the fundamental mechanisms.
Resonances are usually the dominant features in PI cross sections. High-precision spectroscopy and accurate determination of resonance energies is essential for the detailed understanding of atomic structures and decay dynamics. Applications, for example in astrophysics need very accurate level energies. Theory can provide such energies with small uncertainties for two-and three-electron systems which can serve as reference standards. An example for a high precision experiment is shown in figure 20 where measured and calculated cross sections for PI of metastable C4+(1s2s3S) ions via 2s2p resonances are compared to one another [156]. The photon energy bandwidth was less than 14 meV and the statistical precision of the energy determination was better than 0.1 meV. Such numbers provide a promising basis for meeting a great challenge for the future, namely, the excitation of U89+(1s22s1/2 → 1s22p1/2) at about 280 eV with sub-meV accuracy which would allow for a critical test of quantum electrodynamics and could eventually lead to new physics beyond the standard model.
Figure 20. High-resolution observation of fine structure within the 2s2p3P resonance term in the absolute PI cross section of metastable C4+(1s2s3S1) ions. The solid line is the result of theory. The 3PJ (J = 0, 1, 2) contributions are shown by the dashed lines. The experiment is shifted by 1.4 meV to match the accurate calculation. Reprinted figure with permission from [156] Copyright 2018 by the American Physical Society.
Download figure:
Standard image High-resolution imageCore-level excitation of atoms and ions has become increasingly more accessible with the new light sources providing the basis for interpreting site-specific excitation of molecules and molecular ions. X-ray absorption spectroscopy of molecular ions is opening new avenues to study isolated radicals which are produced in an ion source, accelerated and made to form a fast beam. Such beams of electrically charged molecular fragments can be mass and charge analyzed and can be transported to the photon–ion interaction region before they decay. Flight times between the production of ions and their arrival at the interaction region are typically 1–10 μs. First experiments with molecular ions including (endohedral) fullerenes have been successfully conducted [148] and point the way to further exploration of this immense field of research. PIPE is equipped for measurements on heavy molecules and nanoparticles with masses up to 100 000 u at 1.5 keV beam energy.
PI of neutral atoms such as C and O is extremely important in astrophysics. Accurate level energies and cross sections are required in order to fully exploit modern satellite-based observations. Targets of neutral C and O are not readily available and call for the application of the merged-beams technique. Intense neutral beams can be made available by photodetachment of negative ions within intense laser fields. Thus the present experiments with ions can be extended to atoms which cannot directly be accessed by other methods.
Advances in science and technology to meet challenges
The most obvious requirement for meeting future challenges is the continuation of light-source development. High brilliance at high energies with very narrow bandwidths together with high stability in position, photon energy and beam handling are the keys to new improved PI experiments.
For extending the range of feasible measurements, efforts in target development are necessary. Several high-performance sources of charged particles are available for installation on the ion-source platform at PIPE. State-of-the-art sources of cluster ions and highly charged atomic ions should be made available for PI experiments. While the existing magnetic spectrometer provides pure beams as far as mass and charge of the ions are concerned also the control and manipulation of the electronic state of the target ions is highly desirable. This will require combinations of radiation fields (lasers), evaporation ovens, charge-exchange gas cells and trapping techniques. Ions and particularly molecular ions can be de-excited and cooled by storage in a suitable ring, preferentially at cryogenic temperatures. This requirement calls for a combination of a synchrotron radiation source with an ion storage ring, both being major pieces of equipment. An alternative is emerging from the laser-based development of portable short wavelength radiation sources which can be integrated in existing heavy-ion storage rings. A proof-of-principle experiment on PI of C+ is already envisaged at CRYRING in Darmstadt [157]. Very high charge states and ultimately even U89+ can be made available for photo-absorption experiments by employing high-energy heavy-ion storage rings.
Advances in detection capabilities are as instrumental for meeting future challenges as is the enhanced performance of particle and light sources. Electron detection in photon–ion experiments should be further improved. Particularly with respect to PI of electrically charged molecules (radicals), clusters and nanoparticles the detector should be capable of separating (fragmentation) products emerging from the photon–ion interaction region under different angles and at different kinetic energies. Suitable combinations of magnetic, electrical, and time-of-flight separation fields together with appropriate detectors have to be devised to reach this goal.
Concluding remarks
Given the numerous applications and their increasing demands for a better understanding of PI processes along with the ongoing developments of light sources, target arrangements, detection devices, and experimental schemes the field will remain very active for decades and provide new fundamental insights as well as information required for the numerous fields of PI applications. Even New Physics beyond the Standard Model may emerge from high precision photo-absorption experiments.
Acknowledgments
The construction of the PIPE setup was supported by substantial funding from the German Federal Ministry for Education and Research (BMBF) within the 'Verbundforschung' funding scheme. I gratefully acknowledge long-standing fruitful collaboration with numerous good colleagues, particularly with S Schippers and R A Phaneuf.
12. Molecular-frame photoelectron angular distributions from static to time resolved
Danielle Dowek1, Robert R Lucchese2 and C William McCurdy2, 3
1Université Paris-Sud, France
2Lawrence Berkeley National Laboratory, United States of America
3University of California, United States of America
Status
Photoionization (PI) of molecules is at the heart of many fundamental processes induced by absorption of VUV to x-ray photons, as well the dominant response to intense laser fields. The molecular-frame photoelectron angular distribution (MFPAD), i.e. the photoelectron emission pattern in the non-spherical potential of the molecule, for each orientation of the molecule relative to the propagation or polarization axis of the ionizing radiation, stands out as the most detailed observable characterizing molecular photoionization stereodynamics. Measuring MFPADs for single photon ionization in the dipole limit is equivalent to resolving the interference pattern of the partial waves from which the electron wavefunction is composed at each energy: it allows for the most complete determination of the complex ionization matrix elements, i.e. their magnitude and phase for each asymptotic partial wave [158]. A number of recent reviews ([159–161] and references therein) give a broad summary of the progress in MFPAD research achieved through 2012, and describe the success of MFPAD studies in probing the electronic dynamics of photoionization and subsequent dissociation, as well as intramolecular dynamics and the relaxation of excited molecular states.
The principal challenge of experimental MFPAD studies is the establishment of the molecular frame. Taking advantage of dissociative photoionization (DPI) in an assembly of randomly oriented molecules, powerful electron–ion coincidence techniques that provide the 3D momentum vector of each charged particle have yielded a rich harvest of results since the mid 1990s (see also section 14). For small molecules the molecular orientation relative to the light polarization axis is inferred from the recoil ion fragment momenta. Then the photoelectron momenta measured in coincidence for each DPI event provide MFPADs for any polarization geometry. The molecular frame (MF) is easily established when the dissociation time is short relative to other motions of the molecule. These methods have strongly benefited from the properties of third generation synchrotron radiation including wavelength tunability up to the x-ray range, spectral resolution, a wide range of polarizations from linear to circular and a repetition rate in the MHz range, which is a good match for coincidence techniques. MFPAD studies have revealed remarkable features of photoionization dynamics [159–161], such as orbital angular momentum properties of the photoelectron temporarily trapped in a shape resonance, quantum interferences between autoionization and direct ionization channels, gerade/ungerade symmetry breaking, circular dichroism in the angular distributions (CDAD) or the multi-electron nature of the ionization process due to the breakdown of Koopmans' theorem. Core–shell photoionization is particularly suited for probing MFPADs in small polyatomics with the use of coincidence techniques since core–shell ionization is followed by rapid electronic relaxation or Auger decay producing dissociation into multiple charged ionic fragments. Their momentum-resolved detection in coincidence with the photoelectron give direct access to the MFPADs, as shown in figure 21 for CF4, or to recoil-frame photoelectron angular distributions when only two fragments are produced. Site and element specific core–shell photoionization processes have provided the basis for the study of fundamental quantum processes including two-source interference, coherence, and core-hole localization. Furthermore, far from the ionization threshold, core–shell ionization MFPADs map the diffraction patterns arising from the superposition of the partial waves emitted from a single center within the molecule and scattered by the molecular potential, therefore reflecting the geometrical structure of the molecule [163]. The sensitivity of MFPADs even to very small geometry changes has been demonstrated through vibrationally resolved measurements. For future applications, it is stressed that measured MFPADs also encode the complete polarization state of the ionizing radiation [164].

Figure 21. MFPAD for carbon K-shell photoionization of CF4 with linearly polarized light from frozen-core Hartree–Fock theory and experiment where the photoelectron was measured in coincidence with two F+ atoms produced after an Auger decay. Reproduced from [162]. © IOP Publishing Ltd. All rights reserved. The four red balls in the central images give the location of the F atoms and the double headed green arrow gives the direction of the polarization. The four-lobe MFPAD pattern indicates strong scattering from the F atoms.
Download figure:
Standard image High-resolution imageAn alternative route to measure MFPADs, required in particular to investigate non-dissociative ionization channels, or ionization from complex molecules, relies on the laser control of the spatial alignment of molecules in the laboratory frame (LF). Different strategies have been developed [159–161], from multiphoton ionization of small molecules through rotationally-resolved states by nanosecond lasers or via resonant electronic states by femtosecond lasers, extended to adiabatic or impulsive strong field alignment relying on short nonionizing laser pulses [165]. The latter methods allow the confinement of the molecular axes about a well-defined direction in the LF, in the presence of the laser field or in field-free conditions e.g. at recurrence times of the rotational wavepacket. Photoionization induced by a second femtosecond pulse (optical laser, high-order harmonic generation (HHG) or free electron laser (FEL)) at a controlled delay therefore provides PADs which converge on the MF distributions when the degree of alignment approaches unity. These PADs are mostly recorded using velocity-map imaging (VMI), where the velocity distribution of the photoelectrons is projected onto the 2D plane of the detector. The progress in molecular alignment and orientation has boosted the investigation of MFPADs induced by intense near-infrared femtosecond laser pulses [166].
MFPAD observables allow a comparison between experiment and theory at the most detailed level, probing the theoretical descriptions of molecular photoionization [159–161]. Such comparisons have been facilitated by the development of the functional forms of MFPADs which capture the range of possible MFPAD angular dependence and allow for filtering out unphysical behavior in measured data [161]. The essential challenge in predicting MFPADs is the calculation of the continuum electronic states, which are not described by the methods of bound-state quantum chemistry. A review of well-established methods that have overcome these difficulties has been given in [161]. If the process is dominated by a single ion target state then relatively simple Hartree–Fock like methods can be used, as seen in figure 21. Otherwise a coupled-channel formalism is needed such as the multichannel frozen-core Hartree–Fock or time-dependent density functional theory. If a more complete treatment of correlation including target response is needed then R-matrix or complex Kohn methods should be used. Almost all calculations have been done within the fixed nuclei approximation for the photoelectron dynamics. Thus even when nuclear motion is included, the continuum electron dynamics are assumed to respond adiabatically to any change in molecular geometry.
Current and future challenges
A number of challenges in MF-resolved photoionization involve time-resolved experiments. A general method for MFPAD determination was demonstrated by measuring LFPADs for XUV photoionization of impulsively aligned N2 molecules, as a function of time during the evolution of the launched rotational wave packet [167].
Nuclear and electronic dynamics from the picosecond to the attosecond time scale
Time-resolved MFPADs (TR-MFPADs) at the femto-to-picosecond time-scale provide a powerful tool for studying relaxation dynamics of molecular excited states using pump–probe techniques, where a short laser pulse prepares a vibronic wavepacket and a time-delayed pulse photoionizes the molecule. Taking advantage of field-free alignment, combined experimental and theoretical studies of the relaxation of the CS2*(C) excited state, chosen as a prototype example of non-adiabatic couplings at conical intersections, spotlighted that TR-MFPADs capture the pure valence-electron dynamics of the evolving wavepacket [168]. Addressing such processes in molecules of increasing complexity is a current challenge. Nowadays, the advent of bright and tunable FELs delivering intense femtosecond XUV to x-ray pulses [169] opens new avenues for MF-resolved studies at the femtosecond timescale, using e.g. XUV-pump XUV-probe schemes. Moreover, the availability of optical lasers at XFEL facilities, combined with the demonstration of femtosecond photoelectron diffraction on laser-aligned molecules [92], paves the way towards time-resolved studies of molecular structure during ultrafast dissociation, with angstrom spatial resolution.
Extending further the pump–probe approach, computations have shown that TR-MFPADs can probe electron dynamics or charge migration initiated by a pump pulse at the attosecond to few femtosecond time-scale, i.e. before the onset of the nuclear motion, and reflect the spatial localization of the electron density [170]. In the model presented in [170], excitation by a few femtosecond UV pump prepares a non-stationary coherent superposition of molecular states while the system is probed by sudden ionization using a delayed attosecond XUV pulse: such a scheme poses another challenge for experiments.
Photoemission time delays in the molecular frame
Most recent developments, calling for the characterization of the ionization dynamics at its relevant attosecond time-scale, focus on the determination of the photoemission time-delays (or Wigner delays), as demonstrated in recent years for atoms [171]. For molecules, the challenge is to measure the spatial dependence of photoionization delays probing the electron wavepacket scattered in the non-spherical molecular potential. Beyond the determination of the magnitude and the relative phases of the ionization matrix elements provided by the MFPADs at fixed photon energies, accessing the Wigner time-delays characterizing XUV ionization of a molecule requires that the energy dependence of those phases is obtained across the explored energy range. This can be achieved by performing angle-resolved RABBITT [172] (reconstruction of attosecond beating by interference of two-photon transitions), where ionization by an attosecond XUV pulse train probed by a coherent IR pulse (ω) leads to the production of sidebands in the photoelectron spectra resulting from the interference of two (XUV2n−1 + IR and XUV2n+1 − IR) paths. A first achievement in MF angularly resolved studies relying on the determination of a molecular stereo Wigner time delay (SWTD), i.e. the relative delay between electrons emitted in the direction of, or opposite to, one end of an asymmetric diatomic molecule, was demonstrated in recent measurements combining a RABBITT scheme and a COLTRIMS reaction microscope set-up [173]. Extracting the spatially resolved Wigner delays for each emission direction (θ, ϕ) in the MF, and well defined orientation of the molecule in the LF, stands as a current challenge.
Photoionization of molecules exposed to strong fields
Within the flourishing developments of attosecond science, coincidence and/or molecular alignment approaches to molecular frame photoemission are being gathered to face the challenge of providing detailed insights into the multiple electronic nature of strong-field ionization of molecules and related ultrafast phenomena, including tunnel [174] and above threshold [175] ionization, electron recollision [176], high-order harmonic generation [160], or laser induced electron diffraction [117].
One-photon synchrotron-based studies
One anticipated development in synchrotron-based MFPAD studies is the highly differential angular distribution of two electrons in the continuum, resulting from one-photon double ionization, where the emitted photoelectrons are strongly correlated (see also section 13). Beyond double ionization of H2, results on one-photon double ionization MFPADs for diatomic and polyatomic molecules are scarce. From a theoretical perspective, the angular distributions in the double continuum are sensitive to subtle effects in initial state correlation and to strong continuum correlation effects when the two electrons have relatively low kinetic energy. The extension of methods developed earlier for atomic systems and H2 to molecules such as H2O requires inclusion of the interaction of the continuum electrons with those left in the ionic core, involving several low lying doubly-charged states. Additionally, many more partial waves are needed to obtain converged scattering results. Such a study will provide unparalleled details of the nature of correlation in the ground state electronic wavefunction of the target.
Another challenge will be the measurement of MFPADs in the tender to hard x-ray range [169] including the study of nondipole effects, or ultrafast relaxation dynamics at femto or sub-femtosecond scale based on core-hole lifetime internal clocks, extending e.g. soft x-ray studies of temporally resolved resonant Auger MFPADs [177] (see also section 14).
Advances in science and technology to meet challenges
Combining progress in electron–ion coincidence 3D momentum spectrometry, velocity map imaging and tomography, as well as expanding techniques for alignment and orientation of complex molecules, with the unprecedented opportunities offered by the development of free electron lasers delivering VUV to x-ray intense femtosecond pulses and the availability of laser-based HHG sources delivering attosecond XUV pulses, is at the core of the technological advances needed to fulfill the challenges posed by the MFPAD control of time-resolved electronic and vibronic wavepacket dynamics in gas phase molecules. The substantial ongoing increase of their repetition rate reaching few tens of kHz up to the 100 kHz range is crucial to meeting future challenges. MFPAD studies should also benefit from the availability of variable polarizations.
Although it is possible to understand multiphoton and strong-field dynamics in terms of time-independent energy eigenfunctions, there is a large class of problems involving short light pulses for which this approach is not adequate and a direct solution of the time-dependent Schrodinger equation is needed. Although there have been a number of theoretical methods developed to treat this problem, there have generally been compromises simplifying either the dynamics of bound electrons or the continuum or both. The combination of an adequate treatment of multichannel and correlation effects with an accurate time-dependent wave packet treatment of the continuum remains a challenge and is an active area of research.
Concluding remarks
The measurement of MFPADs led to a breakthrough in the understanding of molecular photoionization dynamics through spectrally resolved studies. MFPADs are now becoming significant probes of time-resolved electron and nuclear dynamics at the shortest time-scales highlighting links between adjacent fields involving ionization in different regimes of light–matter interactions.
Acknowledgments
The work performed at Lawrence Berkeley National Laboratory was supported by the US. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Contract DE-AC02-05CH11231. DD gratefully acknowledges the collaborative support of the ASPIRE Marie Sklodowska-Curie Innovative Training Network (H2020 ID: 674960).
13. Photo-double ionization of atoms and molecules
Paola Bolognesi and Lorenzo Avaldi
CNR-Istituto di Struttura della Materia, Italy
Status
Double and multiple electron emissions from an atom or a molecule by the absorption of a single photon represent minor processes in the radiation–matter interaction. However, they are of extreme interest, because they are completely determined by electron correlation, which plays a crucial role in several processes and in determining physical and chemical properties in a wide class of materials. Furthermore, a proper treatment of two-particle correlation is a central problem in many-body physics [178, 179]. This has motivated the large experimental and theoretical efforts devoted in the last thirty years to the understanding of the photo-double ionization, PDI, of two electron systems, the He atom and the H2 molecule.
In the case of the He atom a single photon with energy larger than 79 eV, the double ionization potential, produces a bare nucleus and two free electrons. With the polarization axis of the incident radiation defining a natural quantization axis the symmetry of the free electron pair, defined by the dipole selection rules, and the Coulomb interaction determine the differential cross section which describes the correlated emission of the pair. The experimental challenge represented by the low cross section (about 1 kbarn) of the process has been overcome on one hand by the advent of the high intensity third generation synchrotron radiation sources and on the other by the development of set-ups that allow the simultaneous detection of the two electrons with several energies over a large solid angle or one/two electrons in coincidence with the recoil ion. The combined experimental and theoretical efforts have lead to a clear identification of three basic mechanisms of PDI. In two of them the photon couples with the system formed by one electron and the nucleus and the second one is emitted either in a knock-out or a shake-off process. The relevance of the former (latter) decreases (increases) with the incident energy. The energy sharing between the two ejected electrons evolves from a flat to a U-shape distribution from threshold to higher energy (figure 22). At the same time the correlated angular distribution changes from a 'butterfly' shapes, determined by the 1P0 symmetry of the wavefunction and the Coulomb interaction in the continuum, to a pure dipole/isotropic shape for the fast/slow electron, respectively. In these mechanisms, the nucleus actively participates and gains a large recoil momentum. A third predicted mechanism, which involves the photon absorption by two electrons spatially close and the nucleus acting as spectator, has been observed only recently [180]. The main feature of the process is the emission of the two electrons with the same energy and back-to-back configuration, a geometry strictly forbidden in a dipole transition but allowed in quadrupole transitions, which contribute to about 0.1% of the total double photoionization cross section.
Figure 22. He atom, (a) energy distribution of one electron at a few energies, E, above threshold. In the top panel the signature of the mechanism corresponding to the absorption of the photon by two-electrons; (d) angular distribution of the electron pair in equal energy sharing condition at E = 20 eV and fully linearly polarized radiation resulting from the contributions due to the Coulomb interaction (c) and dipole photo-absorption angular term (b). The arrows indicate the direction of one electron.
Download figure:
Standard image High-resolution imageIn the case of the H2 molecule, the other two electron system, a few factors make the understanding of PDI even more challenging. The ground state electronic configuration is determined by the two-center nuclear potential, the dissociative final state makes the double ionisation threshold not unique and results in a Coulomb explosion of the two protons. Thus, a kinematically complete description of the break-up process calls for the detection of at least one of the protons. Such an approach allows the reconstruction of the molecular geometry when the PDI event occurred, i.e. to measure the correlated angular distribution of the electron pair in the molecular frame. The results of the few realised experiments [181, 182] and their interpretation by theories based on fully numerical approaches show that the angular distribution of the pair is strongly affected by the molecular orientation as well as by the internuclear distance, i.e. by the electron–ion interaction.
Current and future challenges
In the case of He the dynamic of PDI is fully determined by the final state effects. Nevertheless, an effect of the target electronic structure, due to radial extent and oscillations of the target orbital of the singly charged ion, has been predicted and observed in the angular correlation pattern of the PDI of Mg to Mg2+ (3s−2) [183]. It has been shown that the angular distribution bears the information for electron pairs 'originated' in different regions of the charge density. This has been the first prove that PDI experiments can access initial state effects. The possibility to investigate electron correlation in the molecular target wavefunction and therefore to distinguish among molecular wavefunctions on the base of the different treatment of electron–electron correlation was the motivation for the proposal of PDI experiments in the eighties of the last century.
In the case of molecules however the correlated emission of electron pairs from a multi-center Coulomb potential is a severe challenge for theory as well as for experiments. In the case of diatomic molecules different from H2 as well as several polyatomic molecules the lowest dication states are metastable with barriers to dissociation. This prevents complete experiments and only experiments on randomly oriented samples can be done [184]. Exploratory studies with momentum imaging techniques have been performed on higher dissociative states of H2O and interpreted via classical trajectories on computed potential energy surfaces [185]. These exploratory experiments have shown the richness of the process in more complex systems and are expected to stimulate new activity.
Advances in science and technology to meet challenges
In many-electron systems, the effective electron–electron interaction is not known a priori, but builds up during the photoexcitation process in a dynamic behavior determined by the active surrounding of the emitting atom/molecule. In these cases, an interesting scenario for the emission of an electron pair is that the photon excites one electron that senses its environment for accessible scattering channels (elastic, plasmonic, phononic, etc). By a proper choice of the energies and angles of the electron pair, PDI enables us to zoom into those channels where electron–electron interaction is operational. An example is the electron–electron interaction mediated by charge density fluctuations in a confined geometry, which has been studied in the case of C60 [186] (figure 23). The dominant pathway identified for the process is the following: a valence electron absorbs the photon and rescatters inelastically from multipolar collective modes (plasmons) that mediate the coherent emission of a second electron. These pioneering results show that the measurement of energy and angle resolved electron pair opens the opportunity to explore different regimes of electron correlations, including Coulombic scattering, local field effects and dynamical screening.
Figure 23. Scheme of a set-up for the coincidence detection of the two photoelectrons and coincidence yields for the PDI of C60 versus photon energy at different kinetic energies of the photoelectrons. The full lines are the predictions of the theory that accounts for collective excitations.
Download figure:
Standard image High-resolution imageAnother aspect only partially explored is the dynamics of the electron correlation in the time domain. While the dynamics of the interaction of the ejected electron with the residual ion in single ionization has been investigated with attosecond pulses, only one attempt exists for double ionization [187]. In that work two weak XUV and IR pulses have been used to double ionize the Xe atom via both sequential and non-sequential processes. In equal energy sharing conditions the non-sequential process dominates and the observed quantum interference, due to two paths to the same final dication state, indicates that the two-electron wave packet remains coherent during the measurement. The signature of the electron pair dynamics is encoded in the interference signal variable with the delay between the XUV and IR pulses. The results and their interpretation by lowest order many-body perturbation-theory have provided insights in the different mechanisms (sequential/non-sequential, shake off/knock-out) of PDI. The actual repetition rate of ultrafast lasers hampers these studies, but further development in the sources and more efficient coincidence techniques may allow full energy and angle resolution.
Concluding remarks
Electron correlation plays a crucial role in determining physical and chemical properties in a wide class of materials that exhibit fascinating properties (e.g. high temperature superconductivity, colossal magneto resistence, metal insulator or ferromagnetic anti ferromagnetic phase transitions, self assembly and quantum size effects as well as in biology). PDI, with its unique property to be fully dependent on electron correlations, has provided and will provide the most suited tool to unveil their different facets. The advances in imaging techniques will allow the extension of the range of many-electron systems where kinematically complete experiments are achievable, while high repetition rate and high intensity FEL and HHG sources will fully disclose the investigation of electron correlation in the time domain.
Acknowledgments
The support of the FOE-Eurofel project to the PDI activity at Elettra is acknowledged.
14. Photo-induced reaction dynamics probed by COLTRIMS
Till Jahnke, Markus S Schöffler and Reinhard Dörner
Goethe University, Germany
Status
The experimental toolbox for investigating light–matter interaction is well equipped nowadays. Ultrashort, ultrastrong laser pulses, free electron laser light and synchrotron radiation provide light of different characteristic features—with each radiation source covering unique aspects to drive quantum dynamics in small quantum systems in a fully controlled way. The dynamic response of atoms, molecules and clusters to such irradiation is extremely rich and often leads to many reaction products, such as several electrons and ionic fragments. COLTRIMS reaction microscopes [188] allow the measurement of the momentum vectors of all these products in coincidence independent of their initial emission direction.
The first experiments on photon interactions pioneering this technique dealt with atomic double ionization and Compton scattering (see [188]) investigating electron–electron correlation. Soon after, these instruments were used to examine photoionization dynamics of small molecules [189, 190]: after molecular photoionization, the resulting molecular ions often fragment rapidly along steeply repulsive potential energy curves. In these cases the molecules do not rotate before fragmentation, and thus the emission directions of the ionic fragments coincide with the spatial orientation of molecular bonds at the instant of photoabsorption (see figure 24). This opened a new era of molecular photoionization studies, as such experiments accessed the photoelectron and Auger electron angular distributions in the molecular frame of reference. Such experiments elucidated, for example proton migration in small carbohydrates [192] and pioneering work by Davies et al [193] demonstrated the temporal evolution of such photoelectron emission patterns in NO2 employing a laser pump–probe approach. The unprecedented detail and completeness of these studies unveiled exciting new physical effects, which are often hidden to non-coincident spectroscopy techniques [194]. Photoelectron waves emitted from a well localized K-shell have been used to illuminate molecules from within [195]. This photoelectron diffraction is an extremely sensitive probe of molecular geometry. Even small asymmetries between otherwise equal bonds in CO2 lead to observable asymmetries in the electron angular distributions [196] and the angular distributions are sensitive to the vibrational motion of the molecule. Molecular frame angular distributions obtained upon sulphur 1s-ionization of CS2 demonstrated that the effect of corehole localization can depend explicitly on the reaction dynamics [197]. In weakly bound systems particularly rich nuclear dynamics takes place during interatomic Coulombic decay (ICD). ICD is a non-local electronic decay mechanism with decay times in the regime of several 10 femtoseconds to picoseconds, i.e. on the same time scale as typical nuclear motion. Imaging studies inferred the mean decay width from these nuclear dynamics [198] and showed the vibrational motion of the decaying state [199].

Figure 24. Upper panel: the molecular geometry (CHBrClF) can be inferred from measuring the emission directions of the ions after a Coulomb explosion triggered by K-ionization and multiple Auger decay ([191] John Wiley & Sons. © 2016 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim). Lower panel: scheme of a COLTRIMS Reaction Microscope.
Download figure:
Standard image High-resolution imageCurrent and future challenges
One of the current frontiers of this field is to extend the studies of photon driven reactions to the time domain. Accessing the time domain is traditionally done by employing pump–probe approaches. The advent of higher repetition rate lasers and free electron lasers bears the promise that elaborate multi-coincidence studies with COLTRIMS can be performed, but the technical challenges are still daunting. Up to today, for example, only a few true ion-electron coincidence imaging experiments performed at a free electron laser have been reported despite a decade of efforts [200]. Furthermore, while ultrashort light pulses are obviously a key tool to investigate dynamics taking place on the shortest time scales, they have an intrinsic energy uncertainty. While such (coherent) broadband features may even generate dynamical behavior, they can in many cases prohibit the preparation of distinct initial states or impede triggering definite processes.
The pump–probe approach is the most mature but by far not the only technique to access the atomic or molecular time domain. Recently, new schemes have been developed which fully exploit the power of coincidence detection. These do not require a probe step, which is often disruptive or at least perturbing to the process under investigation. Instead, internal clocks provided by the ionization process or the molecule itself are exploited. One such ultrafast intrinsic clock, for example, is nuclear dynamics occurring on a known repulsive potential energy curve of an excited molecule. The prime example here is ultrafast dissociation of molecules, where narrow bandwidth excitation initiates a rapid dissociation and the electronic decay dynamics can be inferred from the kinetic energies of the final state ions and electrons. According studies [177] have depicted the temporal evolution of a molecular orbital during the dissociation process (see figure 25). A second example utilizes a naturally occurring streaking field [199]. If a (comparably) fast secondary electron is emitted shortly after a low energy photoelectron both electrons will interact in the continuum and the photoelectron is typically decelerated due to post collision interaction (PCI). As the strength of this interaction depends on the time between the emission of the two electrons, the decay time is encoded in the energy loss of the primary photoelectron. Possible decay processes, which may drive this internal clock also include the Auger decay. Accordingly, time scales of a few femtoseconds become accessible by using this scheme termed 'PCI streaking'. Such internal electronic or nuclear motion clocks can be exploited using the COLTRIMS coincidence detection. Here one of the detected particles, e.g. a photoelectron in case of PCI streaking or a proton when examining ultrafast dissociation, is used to decipher the decay time on a single event basis. The temporal evolution of properties of other particles detected in coincidence can then be investigated. The PCI streaking approach has been employed, for example, to study ICD in real time in helium dimers, showing the contraction and vibration of the dimer during the decay [199].
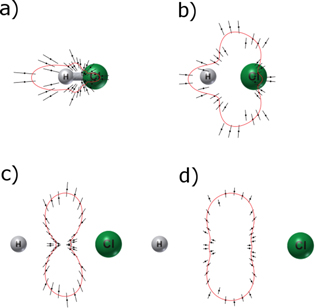
Figure 25. Auger electron angular distribution in the molecular frame as the HCl molecule undergoes the transition from (a) molecule to (d) separated atoms. Reprinted figure with permission from [177], Copyright 2016 by the American Physical Society.
Download figure:
Standard image High-resolution imageA further challenge besides approaching the time domain is exploring the role of quantum entanglement in photon driven processes. The ground state of the atom or molecule under investigation is a single quantum state consisting of many interacting particles. In the gas phase photo break-up of an atom or molecule is a fully coherent process occurring without coupling to any environment. Thus, analogous to parametric down conversion, where entangled photon pairs are created by splitting one photon into two, it is to be expected that the photofragments emitted from one joint initial state can share more than purely classical correlations and form multi-particle Bell-states. These quantum features become accessible only by coincident detection of all the reaction products. Despite the fundamental nature of this many-particle entanglement, so far only few cases demonstrated it directly in an experiment. For example, the entanglement of two electrons emitted from molecular hydrogen has been demonstrated by proving that Cohen/Fano double slit interference does not occur on the individual electron but is observable in the momentum distribution of the electron pair. Already ten years ago, the formation of a Bell-state consisting of the photoelectron and the Auger electron emitted upon K-shell ionization of N2 has been shown. This unexpected finding finally resolved the controversial debate whether K-shell ionization of equivalent sites of a homonuclear molecule leads to a localized or delocalized hole. It was shown that the question is quantum mechanically ill posed and the localization properties are defined by the quantum entanglement between all emission products of the photoreaction [194]. For larger molecules and more multifaceted decay channels, however, the issue of quantum entanglement is much more complex and calls for in-depth theoretical work along with more refined coincidence experiments. As of today there is not even a clear quantitative measure of entanglement, which is well adapted to the observables occurring in studies of molecules and to continuous variables like emission angles and momenta in the continuum.
Advances in science and technology to meet challenges
COLTRIMS coincidence experiments have been driven by major technological advances in the past. They rely on projecting charged particles onto position and time-sensitive detectors (see figure 24). Thus, one of the important drivers is detector technology. As low energy charged particles are to be detected, there is currently no alternative to multichannel plates in combination with some sort of position readout. The decisive parameters are detection efficiency (which is more and more crucial as the number of coincidently detected particles increases), time resolution, position resolution and multiple hit capability. Much progress in terms of efficiency has been made by so-called funneled MCPs or ones with special coating and by implementing imaging spectrometers that have a larger transmission. In terms of time resolution, however, 4th generation synchrotron radiation sources counteract the efforts increasing the detectors time resolution, by the increased length of the photon pulses. The second domain of COLTRIMS where progress can be expected (and has partially already started) are next generation imaging spectrometers. Today, the most basic analyzers rely on homogenous electric field regions and parallel magnetic fields. Great potential lies in more sophisticated electron/ion optics schemes. The goal is to spread out particles in time and to focus different starting position in the interaction region to the same time-of-flight and onto the same impact position on the detector.
The tremendous increase of photon flux at synchrotrons and (X)FELs has already opened the route to extend gas phase coincidence studies to much higher photon energies. These were for long inaccessible because of the steeply decreasing cross section. With today's high photon fluxes coincidence experiment become possible for processes with cross section below 10−24 cm2. Among the opportunities anticipated in high photon energy regime is the possibility to approach the Born limit of photoionization. In this limit photoionization becomes a prime tool for imaging of electronic wavefunctions, because once the ejected electron can be described by a plane wave, photoionization directly maps the Fourier transform of the bound state onto the detector [201]. Compton scattering is another process, which becomes accessible at high energies. Coincidence experiments on Compton scattering in the gas phase are mostly virgin territory. They promise a rich scientific harvest as Compton scattering gives access to the bound orbital, as well. For the future it will become possible to pair the photon detectors at XFELs, which are used today for photon diffraction with a COLTRIMS. This will allow imaging of the Compton scattered photons in coincidence with molecular fragments and secondary electrons. If this is used in a pump–probe experiment it will finally yield molecular movies of the time evolution of both, the electronic wavefunctions and simultaneously the nuclear motion.
A revolutionary new era of studies on fundamental atomic and molecular quantum processes will emerge once 'orbital angular momentum' (OAM) photon beams (or 'vortex beams', 'twisted light') become available. By manipulating the wave front of a photon field a phase modulation is created, which equals that of a photon carrying orbital angular momenta (i.e. in addition to the spin of 1 ħ that a 'normal' photon has). Such light beams are routinely generated in the visible employing table-top laser sources. The properties of 4th generation synchrotron sources, as they are in the first design stage nowadays, will allow for the first time the routine generation of these novel photon beams at high photon energies. A prerequisite for such photon beams is a high transverse coherence, which will be available at future diffraction-limited radiation sources. The photon–matter interaction using OAM photons is a completely untouched area of fundamental research. Possible vortex photon beams allow, for example, addressing completely different states than beams consisting of 'normal' photons.
Concluding remarks
COLTRIMS coincidence experiments provide the most detailed and complete account of photon driven dynamics in the gas phase. They are the eyes into the many-body quantum world, which deliver pictures of unseen detail and completeness. The information content provided by these experiments is so rich that already today hypothesis-driven data-mining is as important as the experiment itself. It is the imagination during that process and the ingenuity of questions asked to the data which often drives the progress.
Acknowledgments
We are indebted to Horst Schmidt Böcking, Lew Cocke, Mike Prior, Joachim Ullrich, Ottmar Jagutzki, Achim Czasch, Marc Simon, Lenz Cederbaum and Kyioshi Ueda for the decades of providing ideas, knowledge, (experimental) expertise and support in general.
15. Circular dichroism in photoemission of chiral molecules: from static to time-resolved
Laurent Nahon1, Yann Mairesse2 and Olga Smirnova3
1Synchrotron SOLEIL, France
2CNRS-CELIA Talence, France
3Max Born Institute & Technical University Berlin, Germany
Status
Holding a sea snail in your hand during your beach vacations you may wonder about the beauty and the function of countless chiral objects, including the snail and the hand. Chiral objects cannot be superimposed onto their mirror-images; what is the purpose of this asymmetry in Nature? Our ability to shake hands is one of them. Chiral molecules, including elementary bricks of life such as amino acids and nucleic acids sugars form chiral complexes, a 'molecular handshake', when embedded into a chiral medium. Thanks to this chiral recognition we can perceive odors and tastes and metabolize drugs. Discriminating enantiomers and understanding chiral interactions are major challenges in pharmacology, food and fragrance industry, chemistry, life sciences and physics. We spotlight here a recent disruptive step: the 'electric dipole revolution' in chiral discrimination.
The chiral response originating from magnetic light–matter interactions is generally markedly weak. It can be boosted by increasing the light frequency—e.g. in x-ray absorption circular dichroism [202]—or the light intensity—in chiral high-order harmonic generation [203]. Both methods provide dichroisms in the few % range, surpassing standard absorption circular dichroism by orders of magnitude and enabling ultrafast time-resolved experiments.
A powerful alternative to increase chiral signals is to find observables producing enantio-sensitive signals in the electric-dipole approximation: this is the 'electric dipole revolution'. This revolution broke into chiral discrimination when several extremely efficient methods emerged, exciting and probing chiral dynamics in bound electronic, vibrational (photoexcitation circular dichroism PXCD) [204] and rotational states (enantio-sensitive microwave spectroscopy ESMS) [205] of molecules. Its herald, photoelectron circular dichroism (PECD), was predicted by Richie [206], quantified by Powis [207], and then observed by Bowering et al [208]. The revolutionary methods—PXCD and its detection via photoionization (PXECD), ESMS and PECD—exploit the same physical mechanism [209], exciting vectorial observables such as induced polarization (PXCD, ESMS) or photoelectron current (PECD, PXECD), and detecting them using a chiral reference frame fixed by the experimental set-up. The 'electric dipole revolution' deposed the magnetic field and substituted it by a chiral experimental arrangement: the directions fixed by the two detectors together with the plane fixed by the electric field constitute a chiral reference frame needed to measure vectorial observables and distinguish enantiomers (Instead of circularly polarized light, one can also define the plane using two orthogonally polarized electric fields, with different frequencies (ESMS)). Several 10's of % [210] difference in signals from the two detectors in PECD highlights the extreme efficiency of dipole-based techniques (see figure 26).

Figure 26. Principle of a PECD experiment. A gaseous sample of randomly oriented chiral molecules is ionized by left or right circularly polarized light, producing a forward/backward asymmetric electron angular distribution.
Download figure:
Standard image High-resolution imageThe combination of synchrotron-based studies and tremendous advances in modeling one-photon ionization of chiral molecules from valence and core states has underscored the basic features of PECD: its initial-orbital dependence, its complex dependence on photoelectron energy reflecting the scattering dynamics in the asymmetric molecular potential and therefore its sensitivity to static and dynamical molecular structures [211] such as isomers, conformers, clustering and vibrational motion [212]. The advent of table-top laser-based PECD [213, 214] exploiting resonant multiphoton ionization coupled sometimes to ion momentum imaging in a coincident scheme enabled accurate determination of enantiomeric excess [215], time-resolving ultra fast chiral molecular dynamics [204, 216, 217] and revealed the universal nature of PECD from weak to strong field ionization regimes [218].
Current and future challenges
The electric dipole revolution introduces highly efficient methods but also poses new challenges. We leave the rich and exciting field of ESMS outside the scope of this paper.
Enantio-sensitive spectroscopy in complex systems: biomolecules, nanomaterials, liquids
Many chiral systems, and especially biomolecules, are floppy i.e. can be found in several conformations. While the PECD signal for each conformer may be different and even have an opposite sign, isolating individual signals is challenging. For example, a hard-to-asses distribution of conformers in molecular beams obscures interpretation of measurements. Designing new schemes for efficient selection or tagging of conformers is thus an acute challenge. One route is to select conformers using electrostatic selectors. Alternatively, individual conformers can be tagged spectroscopically using high resolution in a ns-REMPI scheme.
So far, most PECD studies focused on easy-to-handle systems (terpenes and oxirane derivatives). Bringing biomolecules such as amino acids in the gas phase as intact species is a challenge. A successful example is detecting PECD in alanine [220], suggesting PECD as a possible origin of the homochirality of life operative in interstellar medium [210]. Can we use PECD to study how elementary bricks of life form larger biopolymers such as peptides? Could supramolecular chirality, such as alpha-helix, be studied by PECD?
Chiral clusters and nanomaterials are a playground for exploring self-organization in chiral systems. The evolution of PECD from monomers to clusters to nanoparticles (NPs) could monitor the competition between order and disorder, e.g. through the decrease of PECD due to inelastic electron scattering versus its enhancement due to micro-crystallization. Functionalized nanoparticles, e.g. metallic NPs capped with chiral species shells, could also be investigated by PECD, which should be sensitive to possible chiral induction from the ligand-shell to the interface, or even to the metallic NP substrate. These individual contributions might be disentangled thanks to the different electron binding energies.
Finally, solvation processes in chiral systems from nanosolvation in a molecular beam towards full solvation in a liquid jet could be efficiently probed by PECD, with direct relevance to in vivo conditions.
Developing ultrafast imaging of chiral dynamics and learning to 'read' the images
Ultra fast imaging of chiral dynamics has begun. Time-dependent PECD [216], PXECD [204], chiral high harmonic generation (cHHGm) [203], phase-resolved PECD giving access to the forward/backward asymmetry in the photoionization delays [221], could open a view into asymmetric chemical reactions, the coupling between chiral electronic dynamics and torsion modes capable of changing molecular handedness, the role of decoherence in stabilizing transient chirality, etc. Bringing the 'electric dipole revolution' to high harmonic generation (cHHGd) [222] and other nonlinear optical spectroscopies will broaden the scope of accessible dynamics. The extension of the first time-resolved measurements (see figure 27) to more complex systems is an ultimate goal. As in any nonlinear spectroscopy, the challenge is to correlate measured signals with specific types of dynamics. The short-term goal is to perform such experiments on smaller, well characterized systems, to determine the sensitivity of every observable to different classes of dynamics. The excitation of vibrational wavepackets by stimulated impulsive Raman scattering could reveal the importance of different molecular sites, associated with different vibrational modes, in the probe signal (PECD, cHHG or phase-resolved PECD). Pump–probe measurements of photodissociation dynamics leading to achiral species could provide a dynamical picture of the death of chirality. PECD will be a natural observable of ultrafast isomerization and conformational changes, whose measurement is challenging. Increasing the excitation photon energy to the XUV and soft-x-ray range will trigger multi-electron relaxation dynamics driven by electronic correlation and would as well provide site and chemical-specificity. Time-resolved measurements could reveal the transfer of the forward/backward asymmetry through correlation. Pre-alignment of the molecular samples by laser pulses—challenging for large molecules with low symmetry—could enhance the asymmetries, offering new information [223].
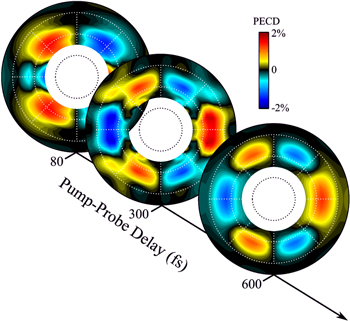
Figure 27. Time-resolved PECD measurement in fenchone, photoexcited by absorption of two linearly polarized photons of 3.1 eV energy and probed by ionization by two circularly polarized 1.55 eV photons. Reproduced from [217] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution image'Reading' ultrafast images requires theory at all levels of complexity: from simple models [218, 219] to sophisticated simulations [224, 225]. These are challenging. A quantitative description of photoionization may require inclusion of two-hole excitations, fundamentally difficult in the computationally efficient density functional theory while computationally demanding for biomolecules in ab initio methods. Compounding the problem is the coupling between electronic and nuclear motions, evolving on the highly multidimensional potential surfaces. Interpreting these dynamics will necessitate answering fundamental questions: what determines the sign of PECD? How does it depend on the photoionization mechanism: one-photon, REMPI, or strong-field [215], and on the localization of the initial orbital versus the chiral center(s)? What is the role of the photoionization propensity rules, different in different photoionization regimes?
Adapting the 'electric dipole revolution' to practical applications
Both PECD and cHHG [222] can be used to perform accurate determination of enantiomeric excess. In PECD accuracies below 1% can be reached, now with laser-based setups [215]. Coincidence electron–ion detection enables analysis of multicomponent samples, with high potential for industrial applications (http://massspecpecd.com). Challenges include reducing measurement times from several hours to minutes or seconds to follow the evolution of chemical samples in real time, and new methods for disentangling the contribution of multiple species with or without (in the case of isomers) coincidence detection.
Advances in science and technology to meet the challenges
Compact high-power fiber-laser systems are revolutionizing ultrafast science by enabling femtosecond and attosecond pulses to be generated with unprecedented repetition rates and photon fluxes, from the visible to the XUV. A key challenge is generating XUV light with circular polarization. One route is offered by free electron lasers, but their repetition rate should be increased to allow time-resolved coincidence imaging. Another possibility is to drive high-harmonic generation with tailored light fields or to use resonances, which allow one to tune the harmonic ellipticity. From the photoexcitation point of view, broadband ultrashort UV sources will also have to be developed.
Advances are needed to bring fragile biomolecules as neutrals in the gas phase, e.g. by aerosol thermodesorption or laser-desorption, ensuring compatibility with imaging techniques and high repetition rate light sources. Size-selected large clusters and large biopolymers could be produced as ions by electrospray techniques, where high densities should be achieved while coupling with photoelectron spectrometers, ideally in coincidence with ion detection.
Coincidence electron–ion imaging spectrometers will benefit from the repetition rate boost of fiber lasers, but the electronics and detection schemes may have to be improved to admit highest count rates.
Modeling ultrafast chiral response requires the description of coupled electronic–nuclear dynamics together with photoionization. The potential energy surfaces governing the dynamics could be computed 'on-the-fly', as demanded by nuclear trajectories spawned into this multidimensional space [225]. Algorithms winnowing out trajectories to speed up computation [226] would be a game changer. Coherent electronic-nuclear wavepackets are characteristic of PXCD [204], with signals sensitive to their coherence. Here, fully quantum description of electrons and nuclei may benefit from looking at the problem as that of an open quantum system, sorting degrees of freedom into 'system' and 'bath', and treating the bath accordingly.
Concluding remarks
Photoionization processes in chiral systems involve new chiroptical observables, sensitive to molecular structures and ideal to template molecular dynamics. A joint development of theoretical and experimental methods will allow the extension and understanding of chiral interactions from model chiral molecules up to much more complex chiral systems as encountered in biology and nanosciences.
Acknowledgments
David Auyso, Samuel Beaulieu, Valérie Blanchet, Antoine Comby, Baptiste Fabre, Gustavo Garcia, Andres Ordonez, Bernard Pons, Ivan Powis, Maurice Tia are warmly acknowledged.
COMPLEX SYSTEMS
16. Fragmentation of gas-phase biomolecules
Thomas Schlathölter
University of Groningen, The Netherlands
Status
Almost 20 years ago, a large number of groups from the ICPEAC community embarked on a journey destined to a fundamental understanding of the molecular mechanisms underlying radiation therapy. In particular, interactions of low-energy electrons [227], keV ions [228], highly charged ions (HCI) [229] and photons [230] with gas-phase DNA building blocks or their model systems were studied. It was soon established that nucleobases are much more resilient towards particle-collisions than deoxyribose molecules. Energetic fragments were observed particularly for HCI impact and these ions were found to potentially induce severe secondary DNA damage.
From an experimental perspective, the appeal of studying molecules such as nucleobases and deoxyribose was the straightforwardness of preparing a sufficiently dense gas-phase target by means of evaporation at modest temperatures. From a theoretical point of view, the level of complexity, i.e. the number of constituent atoms in a nucleobase was sufficiently small to allow high-level quantum-mechanical treatment.

Figure 28. Sketch of a future crossed-beam type experiment.
Download figure:
Standard image High-resolution imageThe studies on DNA building blocks delivered a wealth of important data such as cross sections (which are input in track structure calculations) and greatly improved our understanding of dynamics in strongly perturbed polyatomic molecules. However, a major fundamental question persisted: are collision-induced dynamics in small gas-phase DNA building blocks representative for the molecular processes occurring in living cells, where radiation interacts with entire double stranded DNA strands that have complex tertiary and quaternary structures defined by DNA interactions with water and proteins?
To answer this question while at the same time sticking to the strategy of evaporating DNA building blocks, the Caen–Groningen collaboration studied keV HCI collisions with clusters of nucleobases [231]. In these clusters, nucleobases form a hydrogen-bound network. As a consequence of the intermolecular hydrogen-bonds, a nucleobase in a cluster exhibits fragmentation channels absent in a single gas-phase molecule, proving the critical importance of a chemical environment for the molecular response.
For the nucleoside thymidine, Maclot et al [232] have compared fragmentation by HCI collisions with state-selective photoelectron-photoion coincidence data. This data served as a basis to precisely determine the excitation energy distribution of thymidine induced by HCI collisions.
A limitation of preparing molecular targets by evaporation is the thermal stability of the molecules. It is impossible to evaporate intact DNA strands in useful amounts. Even for DNA building blocks such as deoxyribose, evaporation can lead to coexistence of structurally conformers, which can respond to collisions in very different ways.
A much more gentle alternative technique was explored by the Aarhus group, who used electrospray ionization (ESI) to produce beams of complex biomolecular ions and studied their collisions with neutral atoms. In collaboration with the Stockholm and Caen groups, they were able to study collision-induced dissociation of the nanosolvated intact nucleotide adenosine monophosphate (AMP) anion [233]. The isolated AMP anion was found to break preferably between deoxyribose and nucleobase. With increasing number of attached water molecules m, fragmentation is quenched and AMP is fully protected when m > 13.
Current and future challenges
ESI has become a key technique for preparation of biomolecular targets for collision studies, as it allows for bringing biomolecular systems of virtually unlimited complexity into the gas phase. Our group in Groningen and the SOLEIL group in parallel tackled the problem of low target density by collecting the electrosprayed ions in a radiofrequency (RF) ion trap which can be interfaced with synchrotron or keV/MeV ion beamlines.
Pioneering studies on photon and keV ion induced ionization and fragmentation of the gas-phase protonated oligonucleotide (dGCAT + H)3+ confirmed the important role of glycosidic bond cleavage leading to base loss and also the fragility of the deoxyribose unit. However, the actual fragmentation patterns are entirely different from what is observed in isolated DNA building blocks, stressing the relevance of studying such large systems [234].
With the establishment of ESI and related methods, the size of the system became virtually unlimited as with ESI even an entire virus can be brought into the gas-phase. At present, the influence of DNA tertiary structure on radiation damage is explored. Milosavljevic et al were able to determine ionization energies of various gas-phase protonated proteins as a function of protonation state and could link these data to map structural transitions from folded to unfolded states [235]. Applications of the method include proteomics, biomolecular radiation damage and drug-receptor interactions but also astrochemical problems or studies on magnetic molecules.
The combination of ESI with ion mobility spectrometry, a technique sensitive to the molecular geometric cross section, would even allow for production of conformationally pure targets. It is clear that for instance the application of this experimental strategy to biopolymers (e.g. polynucleotides and polypeptides) will open up a virtually unlimited library of tailored target systems, to investigate charge migration, proton migration, isomerization or conformational changes in general, but also specific problems from the field of biomolecular radiation damage. Not only sequence and length of the biopolymers can be arbitrarily chosen, but the polymers can be chemically modified, for instance by incorporating halogenated nucleobases where the halogen atom can be selectively core excited/ionized.
A next breakthrough step will be towards real crossed-beam experiments (a first study on keV ion collisions with a protonated peptide have been published very recently [236]). For collisions in RF ion traps, many photoproduct properties such as momenta and correlations are lost and information on emitted electrons is fully unavailable. However, with sufficienly intense continuous ESI beams or, or with bunched beams extracted from RF traps, it will still be possible to generate 'free' targets (a schematic for a possible future crossed beam experiment is shown in figure 28). Only then, will it be possible to still use state-of-the-art experimental techniques for biomolecular ion manipulation but then in combination with the entire arsenal of molecular physics techniques and in particular advanced coincidence spectroscopies (PEPIPICO-schemes, velocity map imaging, reaction microscopes etc).
The appeal of gas-phase experiments is not only the possibility of investigating collision induced dynamics in the absence of a disturbing chemical environment but also the possibility to study for instance nanosolvation. Up to now, only very few groups succeeded in attaching water molecules to gas-phase biomolecular systems. An experimental technique that allows for controlled addition of an arbitrary number of H2O molecules is in high demand.
Last but not least, the step towards real time-resolved measurements has to be done. We need to find answers to the following questions: how does energy deposited in a complex biomolecular system develop in space and time? How—and how fast—does this electronic excitation couple to nuclear degrees of freedom and ultimately to conformational changes and/or fragmentation? How can we use this knowledge to solve problems of technological and societal relevance? The pioneering work of Calegari et al [15] has shown the feasibility of tracking attosecond charge migration in the amino acid phenylalanine, a relatively simple biomolecular system. For investigating ultrafast processes in much larger biomolecular systems with sufficient detail, the availability of 'free' targets extracted for instance from ESI sources (discussed above) is also a requirement
It is clear that the current experimental progress will need corresponding advances on the theory side. Complex biomolecular systems are far beyond reach for ab initio quantum calculations and must be tackled by molecular dynamics type approaches. The combination of a quantum treatment of the initial excitation/ionization and a molecular dynamics treatment of the subsequent dynamics (multiscale modelling) is an ongoing challenge for theory.
Advances in science and technology to meet challenges
The field is currently benefitting from a large number of technological developments, a few of which are particularly interesting. Electrostatic ion traps and storage rings are ideal tools for studies on dilute targets of extremely heavy molecular systems that are delivered by ESI sources. The double electrostatic storage ring DESIREE [237] allows for investigation of meV ion–ion interactions.
For RF guiding, trapping and manipulation of biomolecular ions, more and more printed-circuit-board (PCB) devices such as ion funnels, RF guides and traveling-wave ion-mobility spectrometers are being developed PCB-based devices for production of complex biomolecular ions and their manipulation and analysis are compact, versatile and cost-effective and will allow for easy interfacing with existing experimental setup.
Last but not least, we are currently witnessing enormous developments in the field of ultrafast pulses of energetic photons. With the advent of x-ray free electron laser facilities and attosecond pulse lasers new tools for biomolecular fragmentation studies are currently becoming available and even direct diffraction imaging of biomolecular processes are now feasible.
Concluding remarks
Fragmentation studies on gas-phase biomolecules have long focused on small biomolecules such as amino acids and nucleobases. These systems were considered extremely complex from a physics point of view and overly simplified for application of the obtained results in medicine, biology and material sciences. The field has now reached a turning point, where experimental developments already allow for investigation of very large gas-phase biomolecular systems such as DNA complexes or proteins of direct biological relevance. The experimental challenge for the near future is time-resolved experiments on ultrafast electronic and structural dynamics in gas-phase DNA and polypeptides. From a theoretical point of view, it will be most challenging to implement the initial excitation/ionization in a quantum mechanically correct way and to follow the subsequent dynamics of the large biomolecular systems by molecular dynamics type approaches.
17. Fullerene spectroscopy and dynamics
Eleanor E B Campbell
University of Edinburgh, United Kingdom
Status
Since the discovery of C60 by Kroto, Smalley and co-workers in 1985, the molecule has been shown to be a fruitful and interesting model system for exploring the properties of complex molecules. Due to its high symmetry and ease of handling, C60 has often been the molecule of choice for the atomic and molecular physics community to explore molecular systems with many degrees of freedom. Fullerene studies have shown the existence of a range of phenomena and motivated development of theoretical tools that have subsequently been applied to other complex molecules and clusters.
Early studies of ns laser photo-excitation/ionization showed behavior that can be considered to be highly statistical in nature. This involved thermionic photoelectron emission, metastable C2 emission from excited cations and radiative decay. The unusual properties of C60 and other fullerenes, i.e. their relatively low ionization energies and high energetic thresholds for fragmentation, made it possible to observe all of these decay processes on the timescale of conventional mass spectrometers. These studies have been well covered in a number of review articles and statistical models have been applied to explain the experimental results (see e.g. [238, 239] and references therein). A statistical interpretation in terms of efficient energy equilibration followed by thermionic electron emission explained the early photoionization studies, involving multiphoton absorption of pulses in the visible and UV regime. This has been validated by more recent electron imaging experiments at the IR free electron laser FELICE where the vibrational degrees of freedom were directly excited [239]. Single-photon ionization experiments in the VUV, using synchrotron radiation, showed excitation of a plasmon resonance in C60 at ca. 20 eV and interesting oscillations in the HOMO/HOMO-1 photoionization cross sections that could be related to the electron density on the carbon cage [239]. Time-dependent DFT calculations can reproduce the main features of the ionization dynamics [240]. The influence of encapsulated metal atoms on the single-photoionization cross sections has also been investigated with evidence for significant redistribution of metal electron oscillator strength [241]. Photoelectron imaging studies have provided surprising evidence for the occurrence of low energy thermoelectronic emission (thermal emission from a hot electron bath prior to coupling with vibrational degrees of freedom) following single-photon VUV absorption in the 27–65 eV energy range [242].
A similar thermoelectronic emission was observed in multiphoton excitation experiments using femtosecond laser pulses. Three different regimes can be identified for multiphoton ionization of C60 [243], figure 29: above threshold ionization for pulse durations, τ < 30 fs [239]; thermoelectronic emission from hot electrons for 50 fs < τ < ps, both regimes characterized by electron emission from vibrationally cold parent ions and the production of multiply-charged parent species; and thermionic electron emission accompanied by extensive fragmentation and no multiple ionization, for timescales longer than the electronic-vibrational coupling timescale (a few hundred femtoseconds). Photoionization studies on empty and endohedral fullerenes using ultrashort x-ray pulses, accompanied by MD simulations, show a high degree of multiple ionization accompanied by statistical fragmentation, similar to results obtained for intense fs laser pulses in the UV and visible spectral region [244]. Attosecond studies that can probe the photoemission delay are providing detailed insight into the role of the plasmon resonance on the electron dynamics [239].

Figure 29. Comparison of photoelectron kinetic energies and time-of-flight mass spectra for excitation with laser pulses of different pulse duration (produced by stretching the 25 fs pulse) but approximately the same fluence, demonstrating the different ionization mechanisms for excitation on different timescales. Upper: above threshold ionization, Middle: thermoelectronic emission, Lower: thermionic emission, inset ( signal) shows the occurrence of delayed (>μs) thermionic emission from the parent molecule. Reprinted figure with permission from [243], Copyright 2000 by the American Physical Society.
signal) shows the occurrence of delayed (>μs) thermionic emission from the parent molecule. Reprinted figure with permission from [243], Copyright 2000 by the American Physical Society.
Download figure:
Standard image High-resolution imageThe efficient coupling between electronic and vibrational degrees of freedom that leads to a rapid equilibration of excitation energy has the consequence that it is extremely difficult to apply conventional multiphoton ionization spectroscopy techniques to obtain high resolution spectroscopic information about the properties of excited electronic states of gas phase fullerenes. Only limited resonant two-photon ionization data on cold gas phase molecules exists and although single-photon absorption measurements in rare gas matrices or He droplets have been performed, the influence of the matrix is uncertain [245]. Angle-resolved photoelectron spectroscopy using fs laser excitation, that relies on the efficient energy coupling and does not require vibrationally cold molecules, has been used to obtain information about diffuse low-lying Rydberg ('Super-Atom Molecular Orbital') states of fullerenes and endohedral fullerenes [246]. The high resolution absorption and emission spectroscopy of fullerenes in the UV-IR has become highly topical with the discovery of relatively large amounts of C60 and  in space. The confirmation of
in space. The confirmation of  as the first unambiguously identified source of diffuse interstellar bands was recently made possible by high resolution He-tagging spectroscopy studies of cryogenically cooled He-
as the first unambiguously identified source of diffuse interstellar bands was recently made possible by high resolution He-tagging spectroscopy studies of cryogenically cooled He- [245].
[245].
Current and future challenges
A major current challenge is to produce sufficiently dense molecular beams of isolated fullerenes with well defined levels of internal excitation energy (and specifically vibrationally and rotationally cold molecules). This is important for determining the absorption spectra in the UV to near-IR range for comparison with diffuse interstellar bands, for investigating the excitation mechanisms leading to IR emission and for determining reaction rate constants of relevance for astrochemistry. There are currently no high resolution electronic spectra of neutral fullerenes that have sufficient accuracy to provide a meaningful comparison with astronomical observations. Not only C60 is relevant here, also higher fullerenes, hydrogenated fullerenes and possibly some endohedral fullerenes are expected to be important constituents of the interstellar medium and play a role in astrochemistry [247]. There is also the important challenge of developing accurate, predictive theories for such large systems that can provide aid to the spectroscopists in identifying the most likely candidates for detection in space and also for providing input for astrochemical models.
Sources of gas phase molecules with well-defined (and controllable) internal energy content will also be valuable for exploring and understanding the complex dynamics induced by excitation with intense, ultrashort laser pulses in experiments that are beginning to become possible (see e.g. section 9). This would enable the role of laser-induced nuclear dynamics to be more clearly decoupled from that of thermally excited vibrational modes of large, complex molecules and provide a more accurate comparison with detailed theoretical calculations. Endohedral fullerenes can be a particularly fruitful category for studying the effects of confinement on the spectral properties of atoms/ions or small molecules and the timescales for energy transfer and equilibration.
Fullerene experiments are often justified on the basis of using them as model systems to understand more complex molecules of e.g. biological significance. This may be appropriate but advances in the ability to produce gas phase targets of such molecules combined with complex multi-coincidence measurement techniques may soon negate that argument. Instead one should not be afraid to justify experiments on the basis of fundamental curiosity-driven science. As in the past, this is likely to lead to important new insights that may be even more far-reaching.
Advances in science and technology to meet challenges
In the author's opinion, the challenge of understanding the formation and role of fullerenes in space is the most exciting area that can be expected to develop in the coming years. This requires the availability of well-characterised molecular targets (both charged and neutral) and the application of new experimental facilities that are beginning to become available. In particular, the cryogenic storage rings at Stockholm University (DESIREE), MPI Heidelberg (CSR) and RIKEN (RICE) as well as other facilities to be built or upgraded to cryogenic operation in Leuven and Lyon, will enable experiments under well-controlled extreme conditions that are relevant for astrochemical conditions. In combination with laser excitation and photon detection, many novel experiments can be envisaged that will provide new information of direct relevance for exploring and modelling fullerene astrochemistry. Laboratory advances will be matched by advances in observational possibilities. The James Webb Space Telescope, due to be launched in 2021, will provide unprecedented sensitivity, spatial and spectral resolution in the near to mid-IR part of the spectrum and will provide a wealth of data to provide clues to the formation of fullerenes in space and the possible role of other fullerene species. The laboratory and observational advances will also need to be accompanied by advances in the efficiency and accuracy of predictive theory and advanced data mining techniques to extract the relevant signals from the vast amounts of observational data that are to be expected.
Concluding remarks
Fullerenes have been convenient and extremely fruitful model systems for understanding the fundamental electron dynamics of complex molecules over the past 25 years. With the current advances in extreme light facilities and detection techniques this role is likely to continue into the foreseeable future. In addition, the more conventional absorption/emission spectroscopy and reactions of fullerene species under conditions relevant for astrochemistry is likely to be an important area of research in coming years.
Acknowledgments
The author gratefully acknowledges the contributions of many collaborators and students over the past 30 years.
18. Clusters interacting with attosecond and FEL pulses
Jan M Rost
Max-Planck Institute for the Physics of Complex Systems, Germany
Status
Clusters consist typically of atoms or small molecules with a density of more than 1022 cm−3, i.e. close to solids. Yet, clusters can be handled like individual atoms or molecules in experiments since they are available in beams following supersonic expansion from a gas jet. This property has rendered them ideal targets for probing time-gated high-density interaction of light and matter. In other words, clusters absorb or scatter a large number of photons within the fraction of the (short) duration of the light pulse. This leads inevitably to serious radiation damage, and in most cases, Coulomb-explosion, whereby electrons and ions are ejected with partially high velocities. Therefore, fast transport of ever new targets via the cluster beam through the overlap region with the laser pulses is a valuable and actually unique property of clusters among high-density targets to achieve good statistics in experiments.
The first spectacular result with FEL light at FLASH in Hamburg (at that time still called 'TESLA test facility') was the unexpected large number of photons absorbed by Xenon clusters, eventually explained by inverse Bremsstrahlung through ions, higher charged in a transient nanoplasma than concluded from the spectrum of the (recombined) ions [248]. Nanoplasma formation is the result of the pertinent two ionization mechanisms in clusters, namely outer ionization for electrons leaving the cluster, and inner ionization for electrons which remain trapped by the large positive background charge of the cluster and form a transient plasma.
Apart from rare gas clusters, special cluster forms such as fullerenes (see section 9) and helium droplets [249] have become popular targets. The former are extremely simplified models for large (bio-) molecules, while the latter, in particular if doped, exhibit intriguing dynamics and attain superfluid properties if large enough, giving rise to vortex lines which were made visible in an XFEL experiment [250]. Both of them exhibit collective behavior of the transient nanoplasma [251] whose formation is a general signature of impulsive excitation of clusters [137]. Of pertinent interest are also composite clusters. These can be core–shell systems, e.g. with a heavier rare gas species (xenon) embedded into an argon or helium hull. Protonated systems, e.g. clusters formed with methane molecules, get rid of the absorbed high-frequency photon energy by ejecting protons reducing the plasma temperature [252].
Clusters also build a bridge to nanoparticles and much larger structures such as viruses [253].
The probably most serious disadvantage of cluster sources is their loose definition as a target since they are not mass selected but are generated in the supersonic beam with a statistical (log-normal) size distribution about a mean size of N constituents. In addition, traditional intense pulse experiments suffer from a statistical (typically Gaussian) intensity distribution about the laser focus since one does not know where the target, still much smaller than the focus volume, was hit.
Both problems have been overcome simultaneously in recent XFEL experiments by imaging the cluster along with recording traditional parameters such as ions and electrons and eventually fragments [254]. This permits the determination of cluster size and light intensity of interaction events in individual laser shots 'single-shot experiments' a posteriori. One surprise in such experiments was the finding that clusters often are not 'clean' targets but occur as twin systems, with a smaller and a larger cluster forming one system, see figure 30.
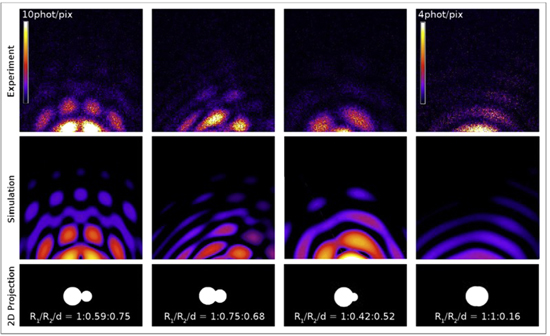
Figure 30. Snapshots of xenon twin-cluster morphology: typical experimental patterns (first row) are in good agreement with simulated ones (second row). The third row shows 2D projections with ratios of bigger radius to smaller radius to the distance d between the cluster centers (third row), all in nm. Taken with 20 fs pulses delivering about 1012 photons of 92 eV energy. Reproduced from [254]. © IOP Publishing Ltd. CC-BY 3.0.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Clearly, imaging objects on the nanoscale or even smaller is an extremely powerful technique which is only at its infancy. Quests go on for higher spatial and temporal resolution. Unconventional proposals such as holographic projection with the help of a cluster as a reference particle in the target (in this case a virus) have already been realized [253]. Also, attention has been given recently to the recombination dynamics in clusters, that is, to the long-term fate of inner-ionized electrons [255]. Recombination dynamics is also an important feature on the way to understand the transition to equilibrium.
In general, XFEL or attosecond pulse experiments follow a time-dependent perspective in a pump–probe scheme, often with a combination of an infrared and a high-frequency pulse or of two high-frequency pulses, slightly detuned in frequency, such that the first and the second pulse can be distinguished. For small systems such experiments extract the same information as time-independent measurements as long as the coherence or full information of the system is preserved (in this case time-resolved and energy resolved observables are roughly speaking related by Fourier transform).
Conversely, unique information can be retrieved by time-resolved experiments if the target exhibits transient dynamical behavior whose signature is lost once particles have arrived at the detector, where they provide the information for measurement without time-resolution. One example is the transient creation of highly charged ions which are responsible for the large photo absorption observed in the early FEL experiments with Xe clusters [248] as discussed above. Other proposed experiments include the ultrafast but transient nanoplasma response to an attosecond pulse excitation [255, 256] or more systematically the monitoring of the non-equilibrium dynamics of complex systems during the excitation and in the early times of relaxation, respectively, dissipation, of the excitation energy.
Such questions parallel the interest of condensed matter physics in the fundamental question, to which extent, how and on which time scale, finite quantum systems relax to equilibrium.
In the context of ultrashort pulses and in particular for clusters an interesting question is also if relaxation (to equilibrium) is always governed by an almost homogenous (and slow) dissipation of energy into many channels, respectively, degrees of freedom. Can it happen that dissipation of electronic energy happens suddenly and localized, e.g. through a specific transfer of energy to particular ions? This would correspond to true charge migration, eventually on ultrafast time scales, while electron dynamics following short pulse excitation without irreversible relaxation is more a coherent beating of electron density of all eigenstates in the energy interval covered by the energy width of a short pulse.
In the violent excitation of clusters so far addressed the energy intake is so large that the systems behave to a large extent classically. Moreover, rare gas and metallic clusters become very similar in producing a nanoplasma. However, much more gentle activation of non-equilibrium dynamics would preserve initially coherence and would permit us to investigate the genuine quantum effects of dephasing and decoherence with ultrashort pulses in clusters representing complex but finite quantum systems. First steps towards this goal are described in [137].
Advances in science and technology to meet challenges
Advances to get a handle on non-equilibrium dynamics and decoherence rely on progress in the theoretical description to propose experimentally accessible observables and their interpretation. Since we deal with truly time-dependent and excited complex systems, where many degrees of freedom are activated through the light pulse, this is already a challenge. What makes the problem even more difficult is the absence of the thermodynamic limit or other small parameter regimes in finite systems even if it becomes computationally feasible to describe such dynamics one needs to develop relevant simplifications and approximations for two reasons: first, to really gain insight which always is linked to meaningful simplification and therefore generalization, and secondly, to have a basis for meaningful coarse-grained formulations for even larger, also biologically relevant systems, approaching the microscale. This makes clusters also for the future fascinating but challenging objects.
Experimentally, the control over the cluster parameters has to and will be steadily improved. Sometimes progress is almost disruptive, as through imaging, e.g. which suddenly has opened the possibility to gain information about cluster size and incident light intensity in an unexpected fashion [253, 254, 258]. A better characterization of single XFEL pulses and better statistics through high repetition rates is also desirable. With seeded beams, already available at ELECTRA in Triesete for XUV photons, and, e.g. LCLS II geared towards high pulse rates, these problems are addressed with next generation concepts. Moreover, novel analysis tools aided by machine learning certainly promise a positive impact [259].
Concluding remarks
Investigation of structure and properties of clusters, and in particular metal clusters, began some 35 years ago often by nuclear physicists. Ever since then clusters have served as paradigmatic systems to develop and explore experimental as well as theoretical techniques to answer pressing questions arising also in other, even more complex systems. Today, clusters do not only bridge the gap towards smaller systems such as molecules but also towards larger systems at the nano- and biologically relevant micrometer scale. Ultrashort intense light pulses have added another chapter to this success story enabling a time-dependent perspective. In the future this combination is expected to provide unique insight into non-equilibrium dynamics of finite systems.
Acknowledgments
It is a pleasure to thank Ulf Saalmann for a continuing research journey through the world of atoms and clusters interacting with intense pulses. I am also grateful to all students who accompanied us on this journey exploring with us uncharted territory.
NEW LIGHT SOURCES FOR NEW CHALLENGES
19. X-ray free-electron laser facilities
Michael Meyer
European XFEL GmbH, Germany
Status
Free-electron lasers (FELs) operating in the short-wavelength regime, i.e. at photon energies up to several tens of keV, have strongly advanced and revolutionized many research areas related to photon–matter interaction. Built up from long linear accelerators for ramping up the energy of electron bunches to values of several GeVs and long undulator sections to enable self-amplified stimulated emission (SASE), these large scale facilities are characterized by a dramatic increase (factor of 108) in average and peak brightness compared to other sources, such as synchrotron radiation facilities (see figure 31). Highly coherent x-ray pulses with duration in the femtosecond regime and pulse energies up to several mJ can be produced. Intensities up to 1018 W cm−2 are available for scientific applications in dedicated experimental areas. These unique performances open up numerous novel research opportunities. Nonlinear multi-photon processes can now be driven at short wavelengths, i.e. favoring the interaction with core electrons; time-resolved studies of ultra fast processes are enabled with simultaneous site-specific monitoring; and single-particle imaging of clusters, nanoparticles and biomolecules can be realized due to the high number (exceeding 1014) of photons in a single x-ray pulse.
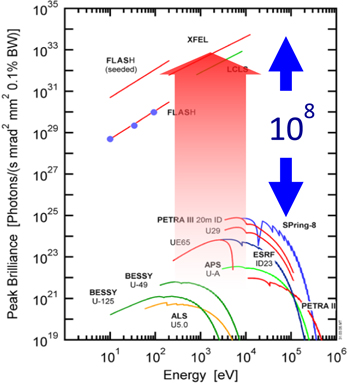
Figure 31. Comparison of peak brilliance for different FEL and synchrotron radiation sources. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature Nature Photonics [260] 2007.
Download figure:
Standard image High-resolution imageFirst user operation of a short-wavelength FEL started in 2005 with FLASH at DESY in Hamburg, Germany [260] with lasing in the VUV–XUV regime. The SCSS test facility at Spring-8 in Japan and FERMI at ELETTRA in Italy followed in the softer photon energy range. The first x-ray FEL, the LCLS at SLAC in the USA [261], went online in 2009 followed by SACLA in Japan, PAL in Korea, European XFEL in Germany and SwissFEL in Switzerland (for more information about the individual sources see e.g. https://lightsources.org). Many further FEL projects are presently under consideration all over the world demonstrating the strong impact of x-ray FELs on the present research as well as the large expectations for future research enabled by these powerful facilities.
Beside continuous improvement of the general FEL characteristics, several new features have been developed during the last years. Stable wavelength operation and wavelength tuning are achieved by seeding techniques (e.g. [262]) based on external sources or on self-seeding schemes. Operation with circularly polarized radiation and variable linear polarization [262] has enabled investigations of dichroic phenomena and two-color lasing at different x-ray energies has been successfully demonstrated at different facilities (e.g. [263]). Experiments combining x-ray pulses with a synchronized optical laser are standard at all facilities and combination with THz radiation produced by the same electron beam allows almost jitter-free operation [264]. Alternatively, various split-and-delay schemes were developed, which divide optically the FEL pulses and use both parts for x-ray pump–x-ray probe type of investigations. Finally, stability and precision on the attosecond level was realized even for the relative spectral phase between two coherent FEL pulses and used for the demonstration of coherent control in atomic photoionization [265].
Current and future challenges
After the first proof-of-principle experiments demonstrating the outstanding FEL performances, many challenges have still to be met to fulfill the requirements for more sophisticated applications. Since the generation of the FEL pulses is a highly nonlinear process, variations in pulse energy, wavelength as well as in the spectral and temporal width are observed between the individual pulses. Especially the SASE process causes rather non-uniform pulse shapes, which are characterized by a large number of spikes in the spectral and temporal distribution. Stabilization and control of all parameters in combination with their precise online diagnostic for each single pulse is essential to realize reproducible experimental results and to provide a meaningful basis for the comparison with theoretical models.
Dedicated optical components of highest quality are used for the transport of the x-ray beam to the experimental areas and for providing a micrometer focus to the interaction region with the sample. Pointing stability of the focused beam and preservation of the almost perfect wavefront of the FEL beam has to be assured with the same micrometer precision after transport over distances of several hundreds of meters from the undulator to the experiment. Maintaining these conditions even under the huge head load produced by the irradiation with the intense x-ray pulses is extremely challenging, especially for facilities providing a high repetition rate, such as the European XFEL with 27 000 pulses per second [266].
The generation of ultrashort x-ray pulses has proven to be particular important for many, not only time-resolved studies, but also imaging and nonlinear applications. Few-femtosecond pulses are generated by dedicated bunch compression schemes and single spike lasing can lead even to sub-femtosecond durations. More promising techniques, such as x-ray laser-enhanced attosecond pulse generation (XLEAP), are currently under development and tested at LCLS. As illustration for a possible outcome of XLEAP, the expected performances are given for the SASE3 soft x-ray undulator at the European XFEL [267], providing pulses of 300–500 as width with still remarkable intensities of several hundreds of μJ (see table 1).
Table 1. Photon beam parameters for the SASE3 soft x-ray undulator at the European XFEL when operating in the XLEAP mode [269].
| Photon energy (eV) | 600 | 870 | 1000 | 1500 | 2300 |
|---|---|---|---|---|---|
| Undulator cells | 6 | 7 | 6/7 | 8 | 8 |
| Duration (as, FWHM) | 500 | 500 | 400/550 | 300 | 300 |
| Peak power (GW) | 600 | 500 | 100/650 | 800 | 400 |
| Pulse energy (μJ) | 350 | 300 | 45/350 | 250 | 100 |
Use of these extremely short x-ray pulses for time-resolved studies in combination with an external optical laser generates strong demands on the temporal stability and reproducibility of the x-ray pulses. Especially for long pulse trains at high repetition rate FELs sophisticated feedback systems were developed providing synchronization of better than 30 fs and promising sub-10 fs in the future [268]. For lower repetition rates, the pulse-to-pulse characterization by THz streaking techniques can be implemented and recent experiments have demonstrated attosecond resolution when additionally angle-resolved electron spectroscopy is applied [269]. Alternative approaches are based on seeding techniques, as demonstrated for example at FERMI [263], using the same optical laser for the seeding process and for the experiments.
Advances in science and technology to meet challenges
Although the technology to generate FEL radiation in the short wavelength regime is relatively new, it is based on a long-standing and highly developed technology of electron beam manipulation in long linear accelerators. Therefore, tremendous advances were achieved during the last years in order to provide for example ultrashort pulses with sub-femtosecond durations via dedicated electron bunch compression schemes, wavelength-stabilized pulses via various seeding schemes, circularly polarized pulses and multi-color lasing as well as the generation of repetition rates in the kHz regime due to the application of super-conducting technology. In addition, techniques which are already well established for table-top lasers are successfully transferred to the x-ray regime such as phase control, chirped-pulse amplification (CPA) and THz streaking techniques.
For the optical transport of the intense radiation, super-polished mirrors have been produced providing shape errors of about 2 nm and corresponding slope errors in the order of 50 nrad for mirrors of 80–100 cm length. The precision to fabricate such mirrors is to a large extend determined by the limitation in measuring the quality of polishing with an adequate high resolution.
The efficient use of higher repetition rates in order to enhance the data acquisition performances and counting statistics is directly related to advances in detector technology, enabling for example recording of several hundreds of high-resolution diffraction patterns during one second (e.g. [266]). Coincidence techniques will strongly benefit from the higher number of x-ray pulses supported by the fact that enormous technical expertise was already acquired during operation at synchrotron radiation sources.
Concluding remarks
Science at x-ray free-electron lasers has become a very active and promising research field and many new facilities are presently under development. In addition, all operating facilities have initiated large R&D programs to further improve the performances of the x-ray pulses and to provide them routinely during user experiments. It is clear that this short review can only represent a temporal snapshot of these operations and the selected examples can only illustrate the broad range of activities, but do not claim completeness. There is still a vast potential for further improvements and developments, which can and will be fully exploited in the future by the fruitful collaboration between experts in accelerator science, x-ray optics and x-ray science.
Acknowledgments
Many stimulating discussions with the colleagues at the European XFEL and with scientists in various collaborations are acknowledged.
20. Where does ELI-NP stand now?—Two arms of 10 PW and 20 MeV gamma beam systems
Kazuo A Tanaka
Extreme Light Infrastructure: Nuclear Physics, Romania
Status
ELI-NP has been selected in 2006 by the European Strategy Forum on Research Infrastructures (ESFRI) as making ultra-intense laser fields with intensities reaching up to 1022–1023 W cm−2 and high brilliance gamma-ray beam with 20 MeV photon energy possible for nuclear physics. The uniqueness of the facility is to have these laser and gamma beams as stand-alone systems or even to have unusual combinations of both laser and gamma beams together for some specific experiments. Since the construction fund has been approved in 2012, the three ELI pillars are being built in the Czech Republic, Hungary and Romania. The Romanian pillar is ELI-Nuclear Physics (ELI-NP). The new facility is designed to serve a broad national, European and International science community. Its mission covers scientific research at the frontier of knowledge involving two domains. The first one is high power laser system (HPLS: laser power <10 PW)-driven experiments related to nuclear physics, strong-field quantum electrodynamics and associated vacuum effects [270]. The laser pulse compression has been based on the chirped pulse amplification (CPA) technique for obtaining 25 fs laser pulses and is to which the Nobel Physics prize has been awarded in 2018 [271]. The second is based on a Compton backscattering high-brilliance and intense low-energy gamma beam system (GBS: gamma beam photon energy <19.5 MeV), a marriage of laser and accelerator technology which will allow us to investigate nuclear structure and reactions as well as nuclear astrophysics with unprecedented resolution and accuracy. In addition to fundamental themes, a large number of applications with significant societal impact are being developed such as medical applications and radio isotope production. The ELI-NP has been located in Măgurele south of Bucharest, Romania. The project is implemented by 'Horia Hulubei' National Institute for Physics and Nuclear Engineering (IFIN-HH). The project started in January 2013. The new facility will be fully operational by the end of 2020.

Figure 32. High power laser system (HPLS) of 10 PW 2 arms. The central laser wavelength is 820 nm with the laser bandwidth of 10 nm. The pulse width is <25 fs with 250 J laser energy output. The focused laser intensity may reach 1023 W cm−2. The final laser beam size is 50 × 50 cm2. HPLS sits on the vibration-dumped floor in the 70 × 40 m2 clean room. 100 TW or 1 PW laser outputs can be extracted for separate experiments simultaneously with 10 PW output.
Download figure:
Standard image High-resolution image
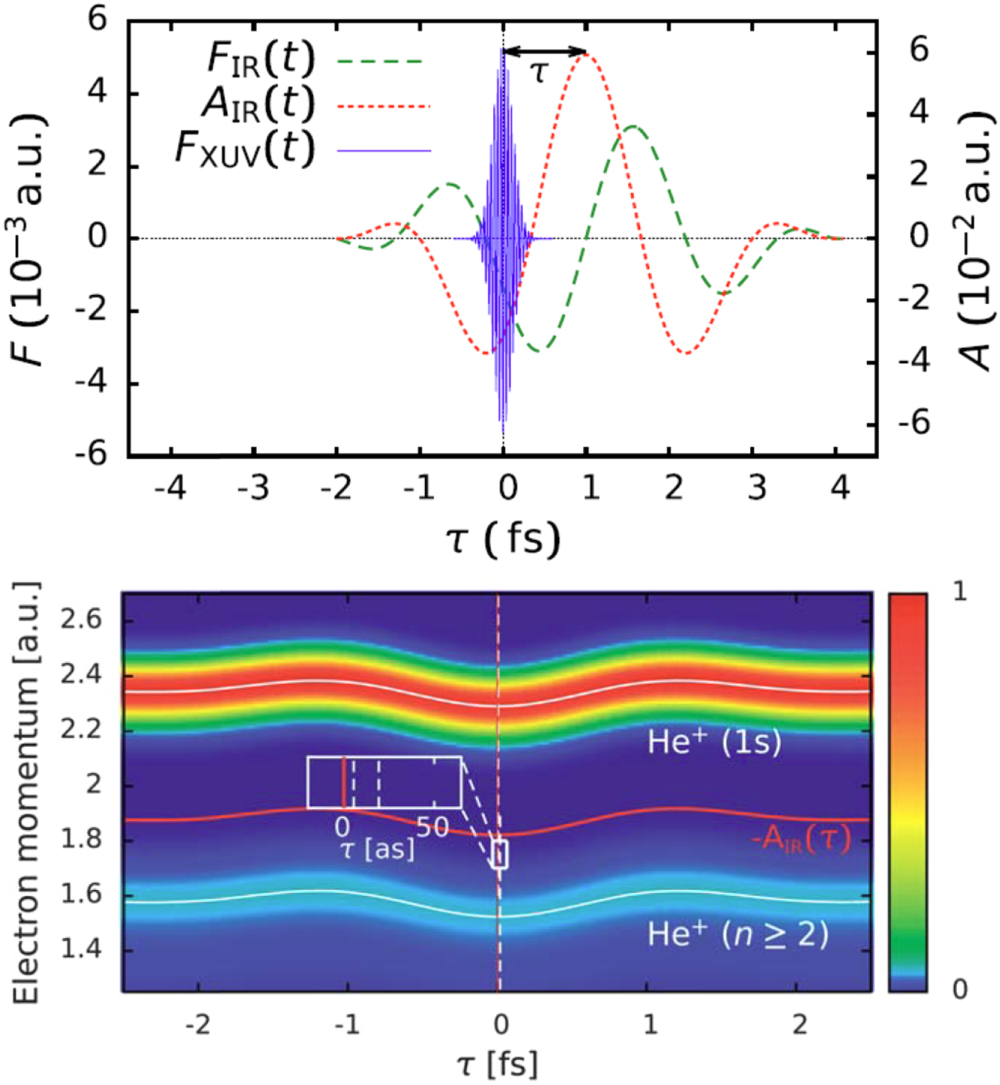{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 33. Main experimental areas for two beams of 10 PW and one beam of PW laser systems. The pipes deliver the laser pulse in vacuum to the experimental chambers. One on the left is for gas jet target that is used for electrons accelerated by 10 PW laser pulse via a 30 m focal length mirror. Accelerated relativistic electrons collide with another 10 PW laser beam for QED experiments. The right chamber is used for the laser solid target interaction experiments as well as the nuclear fission–fusion.
Download figure:
Standard image High-resolution image{kind=link}
Current and future challenges
The HPLS is being installed on time by Thales French and Thales Romania, as shown in figure 32. A fairly good pulse contrast is expected to have a 1013 ratio between the main laser pulse and the pre-pulse levels. As of June 2018, 3 PW laser output was demonstrated with a very good Strehl ratio 0.88. This contrast ratio is a key factor to conduct experiments for producing high energy (>200 MeV) protons. The central laser wavelength is 820 nm with a laser bandwidth of 10 nm. The pulse width is <25 fs with 250 J laser energy output. The focused laser intensity may reach 1023 W cm−2. The final laser beam size is 50 × 50 cm2. HPLS sits on the vibration-dumped floor in the 70 × 40 m2 clean room.
The GBS is able to provide gamma-ray beams with continuously tunable energy in the range from 200 keV to 19.5 MeV with a number photons of 109 s−1. Production of quasi-monochromatic gamma beams is based on the inverse Compton scattering (ICS) process of laser light pulses off relativistic electron bunches. To generate high-brilliance beams, the solution adopted in the case of ELI-NP was to minimize the interaction volume and maximize the number of photons and electrons within the interaction volume by using low-emittance electron beams and high-brilliance laser pulses. The resulting GBS is based on a high-quality electron–photon collider with luminosity at the level of about 2 × 1035 cm−2 s−1, that is almost one order of magnitude better than what was achieved at the LHC in CERN. The Gamma Beam System at ELI-NP will become the state-of-the-art system in the world delivering beams with photon spectral densities and bandwidth orders of magnitude better than what is today available. Eight independent experimental areas will be constructed where users can prepare their experiments independent of the on-going ones. The experimental chambers have typically the volume of 24 m3 with a 10−6 mbar vacuum.
Advances in science and technology to meet challenges
High power laser system experiment: one typical challenge of the possible experiments is the strong QED in the laser electron collisions where 10 PW laser pulse will be focused onto the relativistic electron flux created via the wake field acceleration mechanism through the gas jet target [272], at the experimental chamber shown in figure 33. The effect is measured with the ratio of the laser electric field in the electron rest frame: ERF and the Schwinger field: ES 1016 V cm−1 at which the vacuum breaks into electron–positron pairs,
From this equation (1), it is advantageous to achieve high electron energy, namely γ for large η. We expect to have more than GeV electron energy through wake field acceleration while the focused laser intensity is of the order of 1022 W cm−2. A large flux of gamma rays will be created via the inverse Compton scattering process, but the production mechanism should be affected with the nonlinear quantum effect and will be rather different from the classical one.
Gamma beam system experiment: the NRF technique is a unique way of investigating low-lying dipole excitations in atomic nuclei and provides a specific research niche for the ELI-NP facility. Gamma-ray photons are scattered off bound nuclear states. In particular, the pencil-size bright gamma beams at ELI-NP will provide access to minimum weight targets and will open the actinide region for NRF studies. NRF experiments at present-day facilities require large-mass targets, of the order of grams, while at ELI-NP milligram targets will be used. Thus, the availability frontier will widen further with the start of the facility. Other studies, which will benefit from the improvement of the availability frontier, are studies of p-process nuclei, which are available in small quantities. At a longer term, parity-doublet studies [273] would be of a high impact, addressing both the sensitivity and precision frontiers. An important part of the NRF program, which will be possible at the high energy beam line, are high-resolution studies of the E1, M1 or E2 strength in the region of the soft collective modes, below the neutron evaporation threshold. Day-one experiments related to photonuclear reactions will also be directed towards the studies of the photodisintegration of 7Li with the ELISSA array, related to the Big-Bang nucleosynthesis, high-precision studies of photo-neutron cross sections. The motivation for studying the 7Li(γ, t)4He reaction serves for not only a final check on one of the reactions that could solve the so-called 'cosmological Li problem' but also to resolve a recent disagreement between theoretical models and the experimental data.
Concluding remarks
The current status of ELI-NP facility is reported here. The 2 beam HPLS of 10 PW outputs is under construction on time. Recent performance test demonstrated 3 PW output with an indication of very good focusability. The components of the GBS have been delivered to the ELI-NP building. One of the highlights of the GBS is to deliver the highest number of photons after the collimator. Various detectors for measuring gamma rays, neutrons etc have been produced and tested. Early phase Day 1 experiments will start some time in 2020 including electron acceleration of up to GeV leading to nonlinear quantum QED exeperiments with HPLS. NRF and photonuclear reaction experiments with 7Li will be planned using GBS.
Acknowledgments
Work has been supported by the Extreme Light Infrastructure Nuclear Physics (ELI-NP) Phase II, a project co-financed by the Romanian Government and the European Union through the European Regional Development Fund and the Competitiveness Operational Programme (1/07.07.2016, COP, ID 1334).