

Abstract
In this letter, we demonstrate for the first time that cobalt phosphide nanowires (CoP NWs) exhibit remarkable catalytic activity toward electrochemical detection of hydrogen peroxide (H2O2). As an enzymeless H2O2 sensor, such CoP NWs show a fast amperometric response within 5 s and a low detection limit of 0.48 μM. In addition, this nonenzymatic sensor displays good selectivity, long-term stability and excellent reproducibility.
Export citation and abstract BibTeX RIS
1. Introduction
Hydrogen peroxide (H2O2) has been generally applied to environmental protection, clinical studies, food, pharmaceutical, chemistry and many other fields [1–3]. Specially, H2O2 is also a common reactive oxygen species [4] and the presence of excess H2O2 in cells may lead to oxidative stress that can induce cancer, Alzheimer's disease and Parkinson's disease [5–7]. Therefore, it is highly necessary to sensitively and selectively detect H2O2 under physiological conditions. Many techniques have been developed for H2O2 detection, such as chemiluminescence [8], fluorescence [9, 10], colorimetry [11] and electrochemical methods [12, 13], etc. Among them, the electrochemical technique has received considerable attention with its merits of rapid response, high sensitivity, good selectivity and simple apparatus [14, 15].
Enzymes such as horseradish peroxidase [16], cytochrome [17] and myoglobin [18] are efficient for electroreduction of H2O2 and thus used widely for electrochemical H2O2 sensing. Unfortunately, the enzyme-based sensors suffer from several drawbacks associated with complex fabrication procedures, environmental instability, low reproducibility and high cost, limiting their large-scale applications [19–21]. To overcome these obstacles, it is highly desirable to develop non-enzymatic electrochemical H2O2 sensors.
In recent years, noble metal materials have been intensively utilized in the construction of electrochemical H2O2 sensors [22, 23]. Given the deficiency and high cost of the precious noble metals, the large-scale practical utilization of noble metal-based sensors for H2O2 detection has been severely impeded. In contrast, transition metal-based nanomaterials have received increasing interest in electrochemical H2O2 sensors [24, 25], but suffer from limited electrical conductivity. It is well known that transition metal phosphides (TMPs) are an important class of compounds with metalloid characteristics and good electrical conductivity [26], but their use as an electrocatalyst for enzymeless H2O2 detection has not been reported before.
In this letter, we report on our recent finding that cobalt phosphide nanowires (CoP NWs) behave as an efficient noble metal-free catalyst for electrochemical reduction of H2O2. This nonenzymatic H2O2 sensor exhibits a fast amperometric response time of less than 5 s, a linear range of 0.001–12 mM and a low detection limit of 0.48 μM at a signal-to-noise ratio of 3. It also shows favorable selectivity due to its high anti-interference property and excellent resistance to chloride poisoning.
2. Materials and methods
2.1. Chemicals
All reagents were used as received. The water used throughout all experiments was purified through a Millipore system. NaH2PO4, Na2HPO4 · 7H2O, NaH2PO2, HCl, CoSO4 · 7H2O and urea were purchased from Beijing Chemical Corp. Glycerol, H2O2, glucose, lactose, fructose, ascorbic acid (AA), urea, ethanol, uric acid (UA) and NaCl were bought from Beijing Chemical Corporation. The 0.1 M phosphate buffer solution (PBS) was prepared by mixing 81 ml of 0.1 M Na2HPO4 · 7H2O and 19 ml of 0.1 M NaH2PO4 and was employed as the base electrolyte in electrochemical experiments. A fresh solution of H2O2 was prepared daily.
2.2. Preparation of Co(CO3)0.5(OH) · 0.11H2O nanowires and CoP NWs
Co(CO3)0.5(OH) · 0.11H2O nanowires were obtained via a hydrothermal reaction. Typically, 0.56 g CoSO4 · 7H2O and 0.1 g urea were completely dissolved in 40 ml solution which contained 33 ml deionized water and 7 ml glycerol under magnetic stirring. The above solution was transferred into a 50 ml Teflon–lined stainless autoclave and the autoclave was maintained at 170 °C for 20 h in an electric oven. After the autoclave cooled down slowly at room temperature, the precipitate was collected and washed with water and ethanol several times by centrifugation and dried at 60 °C.
To prepare CoP NWs, 200 mg Co(CO3)0.5(OH) · 0.11H2O nanowires were placed in the hot center of a tube furnace, and an alumina boat containing 500 mg NaH2PO2 was placed at the farthest upstream position. Subsequently, the two alumina boats were heated at 300 °C for 2 h with a heating speed of 0.5 °C min−1 in Ar atmosphere, and then naturally cooled to room temperature under Ar.
2.3. Fabrication of H2O2 biosensors based on CoP NWs
Prior to modification, the glassy carbon electrode (GCE, diameter 3 mm) was respectively polished with 1, 0.3, and 0.05 μm alumina slurry, and cleaned by brief ultrasonication, then cleaned electrode was dried under nitrogen flow. 2 mg CoP NWs were dispersed in 1 ml of aqueous ethanol solution (1:1). The CoP NW-modified GCE (CoP NWs/GCE) was prepared by casting 10 μl of CoP NWs suspension (2 mg ml−1) on a GCE surface and dried in air as working electrode.
2.4. Characterizations
Powder x-ray diffraction (XRD) data were acquired on a RigakuD/MAX 2550 diffractometer with Cu Kα radiation (λ = 1.5418 Å). Scanning electron microscopy (SEM) measurements were carried out on a XL30 ESEM FEG scanning electron microscope at an accelerating voltage of 20 kV. Transmission electron microscopy (TEM) measurements were performed on a HITACHI H-8100 electron microscopy (Hitachi, Tokyo, Japan) with an accelerating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) analysis was performed on an ESCALABMK II x-ray photoelectron spectrometer using Mg as the excitation source.
2.5. Electrochemical measurements
Electrochemical measurements were performed with a CHI 660D electrochemical analyzer (CH Instruments, Inc., Shanghai) in a standard three-electrode system, using CoP NWs/GCE as the working electrode, a Ag/AgCl electrode as the reference electrode, and a platinum wire as the counter electrode. 0.1 M PBS (pH = 7.4) was used as the base electrolyte. Cyclic voltammetry (CV) curves were recorded from 0.2 to –0.8 V. The current−time curve was conducted at –0.5 V in 5 ml 0.1 M PBS solution under stirring. The modified electrode was washed with deionized water and then dried in nitrogen before each measurement.
3. Results and discussion
CoP NWs were derived from its Co(CO3)0.5(OH) · 0.11H2O precursor via low-temperature phosphidation reaction. Figures 1(a) and (b) show the x-ray diffraction (XRD) patterns of the precursor and phosphided product, respectively. As shown in figure 1(a), the diffraction peaks of the precursor are in accordance with the standard diffraction pattern of Co(CO3)0.5(OH) · 0.11H2O (JCPDS No. 48−0083). The XRD pattern for the phosphided product (figure 1(b)) can be well indexed to the (011), (111), (112), (211), (103) and (020) planes of CoP (JCPDS No. 65–1474), confirming the successful conversion of Co(CO3)0.5(OH) · 0.11H2O into CoP. The scanning electron microscopy (SEM) image shows the precursor materials consist of nanowires that are several micrometers in length, as shown in figure 1(c). Also note that phosphided product still preserves the 1D morphology (figure 1(d)). The energy-dispersive x-ray (EDX) spectrum (figure 1(e)) verifies a 1:1.1 atomic ratio for Co:P, consistent within experimental error with the expected 1:1 stoichiometry of CoP. The high resolution transmission electron microscopy (HRTEM) image taken from one single CoP nanowire (figure 1(f)) reveals lattice fringes with an interplanar distances of 0.26 nm, corresponding to the (211) plane of CoP. Figure S1(a) displays the x-ray photoelectron spectroscopy (XPS) spectra in the Co(2p) and P(2p) regions for CoP NWs. As shown in figure S1(a), The Co 2p XPS spectrum shows a doublet containing Co 2p3/2 and Co 2p1/2 at 779.1 and 793.9 eV [27]. The peaks at 785.6 and 803.2 eV are attributed to two shake-up satellites [28]. The binding energies (BEs) of P 2p3/2 and P 2p1/2 appear at 129.5 and 130.4 eV (figure S1(b)) [29, 30]. The peaks at 781.9 and 133.8 eV are assigned to oxidized Co and P species resulting from superficial oxidation of CoP [31]. The Co 2p BEs of 779.1 and 793.9 eV are positively shifted from that of Co metal (778.1−778.2 eV), and the P 2p3/2 BE of 129.5 eV is negatively shifted from that of elemental P (130.2 eV) [32]. According to the XPS analysis, the Co in CoP has a partial positive charge (δ+) while the P has a partial negative charge (δ−), implying transfer of electron density from Co to P, which is in good agreement with previous report [33].
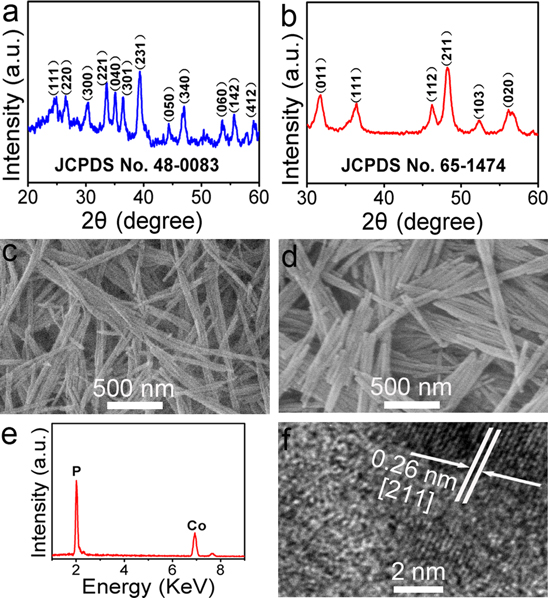
Figure 1. XRD patterns of (a) o(CO3)0.5(OH) · 0.11H2O precursor and (b) CoP NWs. (c) SEM image of Co(CO3)0.5(OH) · 0.11H2O precursor. (d) SEM image and (e) EDX spectrum of CoP NWs. (f) HRTEM image of one single CoP nanowire.
Download figure:
Standard image High-resolution imageTo investigate the electrocatalytic activity of the CoP NWs toward H2O2 reduction, we designed a non-enzymatic H2O2 sensor by depositing the CoP NWs on a bare GCE surface. Figure 2(a) shows the CV curves of bare GCE and CoP NWs/GCE in saturated 0.1 M PBS at pH 7.4 with and without 1 mM H2O2, respectively. As observed, the bare GCE exhibits negligible response to H2O2 reduction with pretty low reduction current. In contrast, the CoP NWs/GCE exhibits a remarkable catalytic current peak about 141 μA in intensity at −0.53 V. It is also important to note that the CoP NWs/GCE exhibits no electrochemical response in the absence of H2O2. All the above observations indicate that the CoP NWs/GCE exhibits a notable catalytic performance for H2O2 reduction. Furthermore, the effect of scan rate on the CoP NWs/GCE was also examined. As shown in figure 2(b), the peak currents increase accordingly with the scan rates in the range of 10–200 mV s−1. The peak currents increase linearly with the square root of the scan rate, demonstrating a diffusion-controlled process of H2O2 reduction on the CoP NWs/GCE electrode.
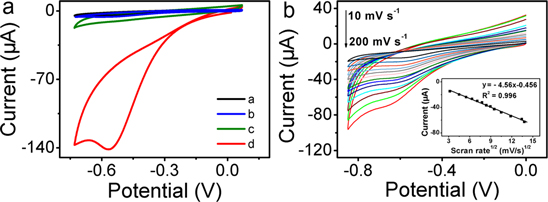
Figure 2. (a) CV curves of bare GCE (curve a) and CoP NWs/GCE (curve c) in 0.1 M PBS at pH 7.4. CV curves of bare GCE (curve b) and CoP NWs/GCE (curve d) in 0.1 M PBS with the presence of 1 mM H2O2 (scan rate: 50 mV s−1). (b) CV curves for CoP NWs/GCE in 0.05 mM H2O2 at scan rates from 20 to 200 mV s–1. Inset: corresponding plot of current versus the square root of scan rate.
Download figure:
Standard image High-resolution imageFigure 3(a) shows the amperometric responses of the CoP NWs/GCE upon successive addition of various concentrations of H2O2 into the stirring 0.1 M PBS (pH 7.4) at –0.5 V. It can be observed that the CoP NWs/GCE exhibits quick response to the change of H2O2 concentration and achieves the maximum steady-state current within 5 s. The inset of figure 3(a) displays the amperometric responses to low H2O2 concentration. Figure 3(b) shows the calibration curve of the sensor. The linear detection range is from 0.001–12 mM. As shown in figure S2, CoP NWs/GCE displays a multi-linear range for the concentration of H2O2 from 0.01 mM–0.1 mM, 0.1 mM−0.2 mM, 0.2 mM−1 mM and 1 mM−12 mM with a detection sensitivity of 3463, 7029, 375 and 26.8 μA cm−2 mM–1, respectively. The existence of more than one linear range on nonenzymatic metallic devices has been observed for H2O2 detection [34, 35]. The detection limit is calculated to be 0.48 μM at a signal-to-noise ratio of 3, which is lower than most of the previous reported nanomaterials based biosensors, as shown in table S1.
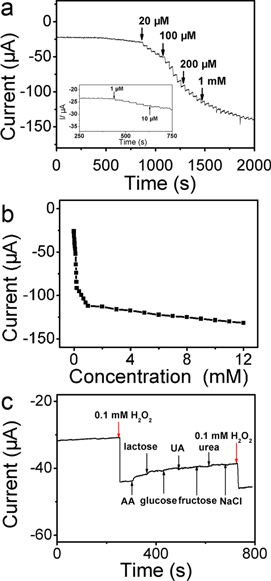
Figure 3. (a) Typical steady-state responses of the CoP NWs/GCE to successive injection of H2O2 into the stirred saturated 0.1 M PBS at pH 7.4 (applied potential: –0.5 V). (b) The corresponding calibration plot for the CoP NWs/GCE. (c) Effects of 10 mM ascorbic acid (AA), lactose, glucose, uric acid (UA), fructose, urea and NaCl on the amperometric responses of CoP NWs/GCE with 0.1 mM H2O2.
Download figure:
Standard image High-resolution imageOne important influence factor for electrochemical analysis application is the interference from electrochemical signals. The influence of some potentially interfering reagents, such as ascorbic acid (AA), lactose, glucose, uric acid (UA), fructose and urea on the detection of H2O2 were introduced one by one during the H2O2 sensing process. In addition, chloride may poison the electrode [36–38]. The influence from NaCl was also investigated to evaluate the interference. As shown in figure 3(c), a quick and obvious current response is presented after the addition of 0.1 mM H2O2 at −0.5 V into 0.1 M PBS under stirring, whereas no further significant current change is observed with the continuous addition of 10 mM ascorbic acid (AA), lactose, glucose, uric acid (UA), fructose, urea and NaCl. Nevertheless, the current increases again with another addition of 0.1 mM H2O2. These results indicate both electro-active species and chloride ions show no interference on the response of this electrode toward H2O2 detection, which proves a good selectivity and an excellent resistance to chloride poisoning of this fabricated CoP NWs/GCE.
The long-term stability of the CoP NWs electrode was evaluated by testing the current response at −0.5 V to 0.1 mM H2O2 every week. As shown in figure 4(a), the CoP NWs/GCE retains 90.18% of its initial response after four weeks of storage at room temperature. Moreover, a relative standard deviation of 5.59% was obtained for the responses at potential of −0.5 V to 0.1 mM H2O2 recorded on five independently fabricated CoP NWs modified electrodes (figure 4(b)), indicating good reproducibility.

{kind=link}
{kind=link}
{kind=link}
Figure 4. (a) Long-term stability of CoP NWs/GCE toward 0.1 mM H2O2. (b) Current responses of five-paralleled CoP NWs/GCE toward 0.1 mM H2O2.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
In summary, CoP NWs have been demonstrated as an efficient catalyst toward electrochemical reduction of H2O2. As a non-enzymatic H2O2 sensor, it shows a fast amperometric response time of less than 5 s, a linear range of 0.001−12 mM and a low limit of detection of 0.48 μM at a signal-to-noise ratio of 3 with favorable selectivity. Our present study is important because it opens new opportunities in exploring the use of TMPs nanostructures, a big family of water-splitting catalysts [32, 33, 39–44], for electrochemical detection of small molecules.