

Abstract
The electronic structures of the 10 K layered yttrium carbide halide superconductor Y2C2I2 is characterized by bands of low dispersion and narrow peak-valley features in the electronic density of states at the Fermi level. In order to investigate to what extent the superconducting properties can be modified by external pressure we have studied the pressure dependence of the superconducting critical temperature and the crystal structure of Y2C2I2 to pressures of 7.4 GPa. Up to ~2.5 GPa we observe an increase of Tc from 10 K to about 12 K. A structural phase transition from a 1s to a 3s stacking variant occurs at about 2.5 GPa above which Tc rapidly decreases to a value of ~7.5 K at 7.5 GPa. Density functional calculations corroborate the structural phase transition to occur at a critical cell volume of ~270 Å3 corresponding to a pressure of ~2.4 GPa, in good agreement with the experimental findings. The pressure dependence of Tc and inter-atomic distances and angles are discussed with respect to the results of density functional calculations of the electronic and crystal structure.
Export citation and abstract BibTeX RIS
Introduction
Superconductivity in binary yttrium carbides has long history starting with the early discovery of superconductivity with a Tc of ~4 K in the body-centered tetragonal CaC2-type yttrium dicarbide, YC2 [1], followed by the subsequent remarkable increase of Tc to ~17 K found in the binary and quasi-binary Y-Th sesquicarbides adopting the body centered cubic Pu2C3 structure [2, 3]. The crystal structures of the rare earth dicarbides and sesquicarbides feature C–C dumbbell units embedded in a metal atom environment. Research on rare earth sesquicarbides was revived at the beginning of this millennium when Tc's up to 18 K could reliably be established in high-pressure synthesized Y2C3 [4, 5]. Especially the rare earth sesquicarbides evolved to an interesting research ground for studying multi or nodal gap effects, the effects of a non-centrosymmmetric crystal structure and strong electron-phonon coupling on the superconducting properties [6–9]. By investigating the electronic structure and electron-phonon coupling in Y2C3 using density functional calculations Singh and Mazin found that the Fermi level falls in a broad manifold of states with mixed character derived from Y d and antibonding states associated with the C–C dumbbell units in the structure [10]. Their calculation of the electron-phonon coupling revealed that phonon modes associated with C–C dumbbell bond stretching have large matrix elements, but their contribution is reduced due to the high frequencies of the bond stretching modes. Most of the electron phonon coupling arises from modes associated to Y vibrations [10]. This scenario is in stark contrast to the electronic structure of YC2 where the electronic density of states near the Fermi level is largely featureless reflecting the reduced electron count in the rare earth dicarbides [11].
Reducing the dimensionality and forming 2D structures has been proven to be a fertile strategy to identify new superconductors with elevated Tc's [12, 13]. Following this concept we prepared Y and La carbide halides with composition M2C2X2 (M = Y, La; X = Cl, Br, I) which showed superconducting transition temperatures up to 10 K [14–16]. The compounds M2C2X2 crystallize with layered crystal structures (see figure 1). They contain close-packed metal atom double layers which can be regarded as a 2D section of the dicarbide structure. The C–C dumbbell units are located in the octahedral voids of the metal atom double layers. These are sandwiched by layers of halogen X atom layers to form X–M–C2–M–X slabs as elementary building units which connect via van der Waals forces to stacks along the crystallographic c axis, analogous to the crystal structures e.g. of the heavy transition metal nitride halides, HfNCl or ZrNCl [17, 19].

Figure 1. Projections along [0 1 0] of the crystal structures of 1s-Y2C2I2 and 3s-Y2C2I2 (high pressure phase). Carbon atoms are represented by the small (black), yttrium atoms are displayed as larger (gray) and iodine atoms by large (dark gray) spheres. The red shaded area highlights the change of the iodine coordination by transforming from the 1s to the 3s structure type. Distances between the iodine atoms are given. Stacking sequences are indicated and unit cells outlined.
Download figure:
Standard image High-resolution imageThe electronic structures of the Y2C2X2 (X = Br, I) compounds has been investigated by first-principles calculations within the full-potential linearized augmented plane wave method based on density functional theory (DFT) using the experimentally determined crystals as input [18]. Similar to the results found for Y2C3 the Fermi level falls in a regime characterized by bands of low dispersion and corresponding peaks in the electronic density of states [18].
1s- and 3s stacking sequences have been observed for crystal structures of the M2C2X2 compounds differing in the sequential arrangement of the metal atom and halogen double layers (see figure 1). The 1s ↔ 3s transition re-arranges the van der Waals contacts in the halogen double layers but leaves the topology of the Y2C2 double layers unchanged [20–22].
By choosing appropriate Cl-Br-I mixtures in the quasiternary phases Y2C2(X,X')2 (X, X' = Br, I) Tc could be raised to 11.2 K e.g. for the ratio Br:I ≈ 1:3 indicating considerable sensitivity to chemical pressure (see table 1). Interestingly, with decreasing unit cell volumes of the phases starting from 1s-Y2C2I2 with Tc ~ 10 K the superconducting critical temperatures pass through a maximum with Tc ~ 12 K and drop to 5 K as observed for 3s-Y2C2Br2 [15]. Intercalation of Na atoms as electron donors in-between the bromine double layers in 3s-Y2C2Br2 raised Tc from 5 K to almost 6 K pointing to the importance of the interlayer distance as well as to the relevance of the electron count [23].
Table 1. Crystallographic parameters and superconducting critical temperatures Tc of the Y and La carbide halides with composition M2C2X2 (M = Y, La; X = Cl, Br, I).
| Compound | Tc (K) | a (Å) | b (Å) | c (Å) | β (deg) | Ref. |
|---|---|---|---|---|---|---|
| 3s-Y2C2Br2 | 5.0 | 6.958(1) | 3.767(1) | 9.932(1) | 99.97(1) | [14] |
| 1s-Y2C2Cl2 | 2.3 | 6.830(2) | 3.712(1) | 9.332(2) | 95.01(3) | [15] |
| 1s-Y2C2I2 | 9.85 | 7.217(2) | 3.879(1) | 10.435(2) | 93.55(3) | [15] |
| 1s-Y2C2I2 | 9.97 | 7.217(2) | 3.879(1) | 10.435(2) | 93.55(3) | [16] |
| 1s-La2C2I2 | 1.6 K | 7.644(3) | 4.138(1) | 10.776(3) | 93.10(4) | [22] |
| 3s-La2C2I2 | 1.7 | 7.624(2) | 4.141(1) | 10.903(3) | 100.74(4) | [22] |
| 1s-Y2C2Br0.5I1.5 | 11.2 | 7.154(2) | 3.851(1) | 10.388(1) | 93.92(4) | [15] |
Note: All compounds crystallize in the monoclinic spacegroup C2/m (no. 12) with two formula units per unit cell, and all atoms are located on the Wyckoff position 4i.
High pressure experiments can provide essential information on the lattice and electronic properties of superconductors. Whereas application of pressure often leads to a decrease of Tc, predominantly due to lattice hardening, experiments on unconventional superconductors in many cases revealed substantial increases of the critical temperatures with pressure.
First experiments applying external pressure up to 1 GPa showed the Tc's of the 1s-type phases Y2C2I2 and La2C2Br2 (Tc ~ 7 K) to increase with pressure whereas 3s-Y2C2Br2 (Tc ~ 5 K), and mixed (Br/I) 3s-type phases exhibited a decrease of their Tc's with pressure [24].
Applying higher pressures up to ~2.5 GPa Tc of 1s-Y2C2I2 increased and leveled off at 12 K combined with a rather anisotropic decrease of the lattice constants under pressure [25, 26]. The relative compression of the c lattice parameter was found to be about twice as large as those found for a and b. With pressure the distance between the carbon atoms in the C–C dumbbell located in the octahedral voids in the Y metal atom double layers was observed to show a slight decrease up to 1.77 GPa. Indication for a change of the stacking sequence from 1s to 3s and a steep and a recovery of the C–C distance towards 3 GPa was found [26].
Here we report the results of a follow-up investigation of the structural and the superconducting properties of Y2C2I2 extending the pressure range up to ~7.4 GPa. To track the interatomic bond distances especially of the light carbon atoms our measurements of Tc under pressure are complemented by performing neutron powder diffraction (NPD) time-of-flight experiments at room temperature under pressure. The NPD measurements reveal the details of the structural phase transition from the 1s- to the 3s-type stacking variant occurring at ~2.5 GPa. The 1s ↔ 3s phase transition coincides with the maximum Tc. In addition, we have performed ab initio density functional calculations of the crystal structure which confirm the 1s → 3s structural phase transition to occur at 2.4 GPa and predict structural details.
Experiment
Sample preparation
Polycrystalline samples of Y2C2I2 were prepared from Y metal, YI3 and glassy carbon powder at 1050° as described in detail elsewhere [20, 21]. YI3 had been synthesized before from Y metal and I2 at 950 °C. The glassy carbon powder was treated at 950 °C under high vacuum, <10−5 mbar. In order to compensate for minute losses of carbon powder a slight excess, ~3% was added intentionally. It has been found that a carbon deficiency causes a significant decrease of the superconducting transition temperature from 10 K down to 7 K and a substantial broadening of the superconducting transition, and thus a carbon deficiency can be monitored by investigating the superconducting properties of the samples [15]. Phase purity of the samples was verified by powder x-ray diffraction measurements collected on STOE x-ray powder diffractometers using CuKα1 and MoKα1 radiation (see figure 3) proving the samples to be of the 1s-type.

Figure 2. Experimentally observed dependence of the superconducting critical temperatures of various Y2C2(X,X')2 (X, X' = Br, I) phases versus the volume of their unit cells (see [15]).
Download figure:
Standard image High-resolution image
Figure 3. (Red) Circles represent the x-ray powder diffraction pattern collected with MoKα1 radiation of the sample of Y2C2I2 used for the PEARL NPD experiment. The (black) solid line results from the profile refinement of the pattern using the FULLPROF program package assuming contributions from 1s-Y2C2I2 only. The vertical (green) bars mark the angles of the Bragg reflections used to simulate the refined pattern. The (blue) solid line underneath gives the difference between measured and calculated diffraction patterns.
Download figure:
Standard image High-resolution imageAttempts of multiphase profile refinements of the x-ray diffraction patterns put an upper limit of ~2% for the 3s stacking variant in the pristine samples. Due to their moisture sensitivity all starting materials and the samples were handled under dried argon atmosphere or in vacuum.
Magnetic susceptibility measurements
Before carrying out the neutron experiments the magnetic susceptibilities of the samples used for the NPD experiments were measured with a SQUID magnetometer (MPMS, Quantum Design) between 2 K and 12 K in an external field of 1 mT. After the NPD measurements had been performed the pressure was released and magnetic susceptibilities of the samples were measured at the same condition to check the change for the superconducting transition temperature. The measurements of Tc were repeated after the samples had been annealed at 1050 °C for 24 h in a sealed tantalum capsule under argon atmosphere to release strain. Magnetic susceptibilities under pressure were measured in the SQUID magnetometer using a home-built Cu–Be pressure cell (8 mm outer diameter) similar to that described in detail by Tateiwa et al however with the non-magnetic composite ceramic anvils replaced by CVD diamond anvils with 0.6 mm culets [27, 28]. A sample hole of 0.2 mm diameter was drilled into the hardened Cu-Be gaskets which had been pre-indented to a thickness of ~0.15 mm. The pressures were determined in situ by measuring the superconducting critical temperature of tiny high purity (99.9999%) Pb pieces placed next to the sample [29]. Silicon oil was used as the pressure transmitting medium.
NPD measurements
NPD measurements at room temperature were carried out using the dedicated high pressure time-of-flight diffractometer PEARL installed at the ISIS Facility, Rutherford Appleton Laboratory. Additional measurements with lower resolution were carried out on the time-of-flight diffractometer POLARIS. Diffraction patterns were collected under various pressures using a Paris–Edinburgh type pressure cell (Pmax ~ 7.5 GPa, sample volume ~ 100 mm3). A deuterated methanol/ethanol mixture (MeOD/EtOD) and Fluorinert were used as pressure transmitting media for the PEARL and the POLARIS experiment, respectively. Before use both liquids were carefully dried and degassed to avoid chemical reaction with the samples. The pressures were determined from the lattice parameters of NaCl added as an internal standard to the samples [30]. Since the samples are air- and moisture sensitive, the neutron diffraction patterns were collected in an evacuated sample chamber in order to avoid deterioration of the sample and background scattering from air. Multiphase profile refinements of the diffraction patterns were carried out using the GSAS + EXPGUI program package [31, 32]. In addition to the patterns of the sample the diffraction patterns of the pressure cell anvil materials, WC (spacegroup P-6m2) and Ni (spacegroup Fm-3m), and of NaCl (spacegroup, Fm-3m) were included in the profile refinements.
Experimental results
Pressure dependence of the superconducting critical temperature
The superconducting critical temperatures versus pressure of Y2C2I2 had been determined before up to pressures of ~2 GPa [22]. At small pressures Tc grew linearly with a rate of about 1.4 K GPa−1 and at 2.5 GPa Tc leveled off at a value of ~11.8 K. The current measurements using a diamond anvil pressure cell extended the pressure range up to ~5 GPa. Our new data are displayed in comparison with those published previously in figure 4. They confirm the increase below ~2.5 GPa and identify a maximum Tc of 11.8 K between 2 and 3 GPa followed by a rapid, almost linear decrease. At 5 GPa Tc reached a value of 9 K.

Figure 4. Tc versus pressure of Y2C2I2. The present data are represented by circles, data marked by squares were taken from [22], Tc of the PEARL sample recovered after the pressure had been released is represented by the triangle. Dashed and dotted lines are guides to the eye.
Download figure:
Standard image High-resolution imagePressure dependence of the crystal structure and structural phase transformation
Figure 5 displays selected NPD patterns of Y2C2I2 collected at room temperature and pressures up to 7.4 GPa. Apart from the characteristic decrease of the cell parameters (see figures 6 and S-1 in the supplemental) (stacks.iop.org/JPhysCM/28/375703/mmedia) diffraction patterns collected for pressures between 2.5 and 3.5 GPa, in addition to the Bragg reflections from the 1s-Y2C2I2 phase, exhibit additional Bragg reflections. These can be attributed to Y2C2I2 with the 3s-stacking sequence. Above ~3.5 GPa we observed exclusively reflections from the 3s-Y2C2I2 phase. Structural parameters for both phases were derived from the patterns measured at different pressures by performing Rietveld profile refinements including reflections from 1s- and 3s-type of Y2C2I2 and reflections from the pressure cell material, the gasket and from the NaCl manometer. The results are compiled in the supplemental information6.

Figure 5. Time-of-flight NPD patterns of Y2C2I2 (circles) collected at room temperature at the instrument PEARL at the indicated pressures. The blue solid curves represent the results of the Rietveld profile refinement using the GSAS program package assuming contributions from 1s-Y2C2I2, 3s-Y2C2I2, scattering from the pressure cell and the gasket, and from NaCl used as an internal manometer. The vertical bars (black: 1s-Y2C2I2; red: 3s-Y2C2I2; blue: WC; green: Ni) indicate the d-spacing values for the Bragg reflections used to simulate the refined pattern. The green solid line underneath gives the difference between measured and calculated diffraction patterns.
Download figure:
Standard image High-resolution image
Figure 6. Lattice parameter c of Y2C2I2 versus pressure, PEARL and POLARIS data, are indicated. Blue and black symbols have been used to mark the data points resulting from the refinement of the PEARL and POLARIS patterns assuming the 1s and 3s structure type, respectively. Solid red lines are guides to the eye.
Download figure:
Standard image High-resolution imageAs can be expected from the layered character of the crystal structure the compressibility of Y2C2I2 is anisotropic [26] with c decreasing about twice as much as the in-plane lattice parameters a and b (see figure 6). With the transition from 1s to 3s stacking the decrease reduces markedly indicating a stiffening of the lattice and an increase of the bulk modulus. At the phase transition c exhibits a jump whereas a and b pass continuously through the phase transition, however with a reduced slope in the 3s phase (see figure S-1 in the supplemental information)6.
The pressure versus unit cell volume data of 1s-Y2C2I2 and 3s-Y2C2I2 (figure 7) were fitted to the Birch–Murnaghan equation-of-state (EOS) given by [33]
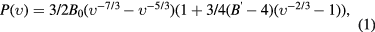
where  with V0 being the cell volume at zero pressure. B0 is the bulk modulus and B' is the derivative of the bulk modulus with respect to pressure. The results of the fits to the experimental data are compiled in table 2 and compared to the theoretical findings (see below).
with V0 being the cell volume at zero pressure. B0 is the bulk modulus and B' is the derivative of the bulk modulus with respect to pressure. The results of the fits to the experimental data are compiled in table 2 and compared to the theoretical findings (see below).

Figure 7. Measured pressure versus volume relationship of the 1s-Y2C2I2 and 3s-Y2C2I2 stacking variants (filled and empty circles) and fits (solid red line) of the Birch-Murnaghan equation of state (equation (1)) to the data. The derived bulk moduli and zero-pressure volumes are compiled in table 2.
Download figure:
Standard image High-resolution imageTable 2. Unit cell volume at zero pressure, V0, bulk modulus, B0, and derivative of the bulk modulus with respect to pressure, B', of 1s-Y2C2I2 and 3s-Y2C2I2 as derived by fitting the Birch–Murnaghan EOS (equation (1)) to the experimental and theoretical pressure versus volume data.
| V0 (Å)3 | B0 | B' (1/GPa) | ||
|---|---|---|---|---|
| 1s-type | Experiment | 291.8(2) | 32.5(5) | 5.4 (fixed) |
| Theory | 288.7(6) | 33.6(1.0) | 5.4(1) | |
| 3s-type | Experiment | 287.0(8) | 36.2(1.4) | 5.4 (fixed) |
| Theory | 287.6(6) | 34(1) | 5.4(1) | |
Note: When fitting the 3s-Y2C2I2 experimental data B' was fixed to the value obtained from the theoretical data.
Experimental and theoretical bulk moduli, B0, and zero-pressure volume V0 are in good agreement and consistent with the experimental values reported earlier [26]. The theoretical bulk modulus of the 3s-type is slightly larger than that of the 1s-type indicating a moderate lattice hardening.
Structurally the 1s–3s transformation appears to be reversible. After completely releasing pressure the 1s-type phase of Y2C2I2 is fully recovered, as seen by NPD. However, a reduction of the superconducting critical temperature to ~7 K (see figure 8) was observed for the pressure released sample. The initial superconducting transition temperature of 10 K can be regained by annealing the sample to 1050 °C for one day indicating that the reduction of Tc for the pressure released sample is likely due to frozen-in strain left by the phase transformation.

Figure 8. Magnetic susceptibilities of the sample of Y2C2I2 measured before the PEARL diffraction experiment (a), measured after pressure release (b) and after pressure release and additional annealing for one day at 1050 °C (c).
Download figure:
Standard image High-resolution imageThe profile refinement of the NPD patterns also allowed to extract the atom positional parameters. The PEARL data for selected pressure values and the carbon and yttrium positional parameters x and z are compiled in tables S-1 and S-2 and shown in figures S-2 and S-3 in the supplemental information. The 3s-type data above 3 GPa describe a new structural variant for Y2C2I2.
The atom coordinates of the high-pressure phase 3s-Y2C2I2 are close to those found e.g. for Gd2C2Br2, Y2C2Br2, or La2C2I2 [15, 20–22]. As a consequence of the different stacking sequence in 3s-Y2C2I2 the iodine van der Waals contacts are slightly different from those in the 1s-type causing a small reduction of the inter-plane distances between neighboring iodine double layers. Figure 1 displays a comparison of the crystal structure of 1s-Y2C2I2 and of 3s-Y2C2I2 highlighting the characteristic I–Y–C2–Y–I building blocks and the different stacking sequences of the 1s- and the 3s structure-type. Figure 9 displays the pressure dependence of the C–C distance as refined from the high resolution PEARL data. The C–C distance shows an increase up to ~2.5 GPa and a decrease above in the 3s-Y2C2I2, however with a reduced magnitude of the slope.

Figure 9. (a) Distance of the carbon atoms in the C–C dumbbell in the 1s and 3s structure of Y2C2I2 versus pressure as calculated from the neutron diffraction data under pressure (tables S-1 and S-2 in the supplemental information). The atom positional parameters of the carbon and the yttrium atoms and the monoclinic angle β used for the calculations have been interpolated according to the blue solid lines in figures S-2–S-4 in the supplemental information, respectively. The additional data point at zero pressure (upright triangle) has been taken from [41]. (b) Angle enclosed by the blue (dashed) line connecting the (red) yttrium atom and the line connecting the carbon atoms in a C–C dumbbell (cmp. inset). The (red) dashed lines represent the results of the present density functional calculations. The vertical black dashed line marks the critical pressure for the transition from the 1s to the 3s structure type.
Download figure:
Standard image High-resolution imageThis finding is opposite to results of a preceding first NPD investigation under pressure [26]. Therein, underestimating experimental errors arising from the Rietveld profile analysis of diffraction patterns collected at a medium resolution powder diffractometer, we had concluded the C–C interatomic distance in Y2C2I2 to decrease with pressures up to 1.8 GPa and to increase back to the zero-pressure value at 3 GPa.
Electronic structure calculation
The superconducting properties and especially the development of Tc with pressure are very sensitively determined by the structural phase transition near 3 GPa evidenced by the NPD results. In order to support these findings we have performed ab initio total-energy calculations using DFT calculations [34]. As has been reviewed in detail in [35], phase stability, electronic and dynamical properties of materials under high pressure can be well described by DFT-based total-energy calculations employing the plane-wave method and the pseudopotential theory encoded in the Vienna ab initio simulation package (VASP) [36–40].
We used the projector-augmented wave scheme (PAW) [39] implemented in this package to take the full nodal character of the all-electron charge density in the core region into account. In order to achieve sufficient precision and highly converged results and an accurate description of the electronic properties the basis set included plane waves up to an energy cutoff of 520 eV. The description of the exchange-correlation energy was performed within the generalized gradient approximation (GGA) with the PBEsol prescription [42]. In view of the metallic character of Y2C2I2 we employed a dense sampling of special k-points (700 k-points) in the Brillouin Zone integration in order to achieve very well converged energies and forces. At each selected volume, the crystal structures were fully relaxed to their equilibrium configuration through the calculation of the forces and the stress tensor. In the equilibrium configuration, the forces on the atoms are less than 0.004 eV Å−1 and the deviation of the stress tensor from a diagonal hydrostatic form is less than 0.1 GPa. The pressure versus cell volume relations for the 1s- and the 3s phase were fitted to the Birch–Murnaghan EOS (equation (1)). The parameters (see table 2) agree well with the experimental findings.
The 1s → 3s transition essentially involves a re-arrangement of the halogen van der Waals contacts with only little energy change involved. Accordingly the calculated enthalpies (per two formula units) displayed in figure 10 are almost identical for the low and the high pressure phase.

Figure 10. Enthalpies (per two formula units) of the (blue square) 1s- and the (red circle) 3s phase. The solid lines are fits of a 3rd order polynomial to the data.
Download figure:
Standard image High-resolution imageOnly the difference between the enthalpy of the 3s and the enthalpy of the 1s phase reveals a tiny (~100 meV) disparity of the enthalpies of the two phases (figure 11). The enthalpy difference disappears at a transition pressure of 2.4(1) GPa corresponding to a unit cell volume of 270 Å3, in good agreement with the experimental observation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11. Enthalpy difference of the 1s- and the 3s phase of Y2C2I2. The 3s phase is stable above 2.4 GPa, corresponding to a calculated unit cell volume of 270 Å3 (see figure 7).
Download figure:
Standard image High-resolution image{kind=link}
Figure 9 also shows a comparison of the calculated C–C distance and the angle enclosed by the C–C dumbbell and the apical yttrium atoms. The calculated C–C distance remains constant throughout the covered pressure range whereas the Y ··· C–C angle exhibits a decrease with increasing pressure. In order to enable a meaningful comparison of the experimental and theoretical findings as a function of pressure we interpolated the experimental data with smooth polynomial like approximants and used these to calculate the experimental distances and the angles versus pressure. Except in the transition regime around 2.5 GPa the experimental data for the Y ··· C–C angle follow the trend indicated by the DFT. In contrast the C–C distance deviates from the DFT prediction.
Discussion and summary
Similar to observations for several unconventional superconductors [43], a dome-shaped pressure dependence is found for Y2C2I2 with a maximum Tc of ~12 K next to the 1s–3s structural phase transition. The linear increase of Tc at low pressures and the leveling off at ~12 K towards ~2 GPa has been described before and attributed to a pressure effect on the electronic band structure which leads to a marked pressure dependence of the Hopfield parameter, N(EF)· , where N(EF) is the electronic density at the Fermi energy and
, where N(EF) is the electronic density at the Fermi energy and  is the average squared electronic matrix element. Electronic structure calculations had revealed maxima in the electronic density of states (DOS) near the Fermi level and a dip at the Fermi energy ('pseudogap') [19]. The peaks in the DOS result from bands of low dispersion near the Fermi level. Application of pressure closes the pseudogap and increases the electronic density of states by about 35% [26]. Gap-filling has largely been completed at pressures above ~3 GPa, and for higher pressure values Tc exhibits a linear decrease with a value of ~ −0.8 K GPa−1, very similar to the decrease found for mixed Br–I phases Y2C2(BrI) [24]. A reduction of Tc with pressure has to be attributed to the dominance of the pressure induced 'lattice-hardening' as also indicated by the larger bulk modulus leading to a reduction of the electron-phonon coupling constant.
is the average squared electronic matrix element. Electronic structure calculations had revealed maxima in the electronic density of states (DOS) near the Fermi level and a dip at the Fermi energy ('pseudogap') [19]. The peaks in the DOS result from bands of low dispersion near the Fermi level. Application of pressure closes the pseudogap and increases the electronic density of states by about 35% [26]. Gap-filling has largely been completed at pressures above ~3 GPa, and for higher pressure values Tc exhibits a linear decrease with a value of ~ −0.8 K GPa−1, very similar to the decrease found for mixed Br–I phases Y2C2(BrI) [24]. A reduction of Tc with pressure has to be attributed to the dominance of the pressure induced 'lattice-hardening' as also indicated by the larger bulk modulus leading to a reduction of the electron-phonon coupling constant.
Based on a formal ionic description of Y2C2I2, Y in the oxidation state +3 provides two electrons to fill halogen p levels and π* antibonding levels of the C–C dumbbell and to constitute a C–C double bond [44]. The overlap of the π* states with energetically nearby Y d states leads to delocalization and to the metallic character of Y2C2I2. Depopulation of the π* antibonding states effects a shortening of the C–C bond length as compared to typical C–C distances found e.g. in molecular compounds. Figure 9(a) displays the pressure dependence of the C–C distance. It shows an increase up to ~2.5 GPa and a constant value or a moderate decrease above, however with a significantly reduced magnitude of the slope.
Although limited by the rather large experimental errors, the experimentally determined C–C distances appear to increase with pressure in the 1s phase. The finding that application of pressure up to ~3 GPa leads to an increase of the C–C distance i.e. increasing population of antibonding C2 π* states in the first moment appears to be counterintuitive. However, it can be understood as a consequence of the volume reduction and the concomitant up-shift of the Fermi energy, effectively filling the π* levels. In fact, in the 3s-type Y2C2Br2 the low dispersive π* bands fall ~0.1 eV below the Fermi energy and the geometry relaxed DFT calculations for 3s-Y2C2Br2 indicated an increase of the C–C by ~4% [19].
The beginning of the negative pressure dependence of Tc, the reversal of the pressure behavior of the C–C distances is initiated by a structural phase transition changing the layer stacking from the 1s to the 3s variant. The slight non-monotonous jump near 2.5 GPa seen especially in the c lattice parameters (see figure 6) points to a first order character of the structural phase transition. The hysteresis found in Tc of the pressure released sample (figure 8) and the coexistence of both phases in the pressure regime around 2.5 GPa support this notion. Interestingly all other M2C2X2 systems with a negative pressure coefficient of Tc already at low pressures also crystallize with the 3s stacking variant or are very close to a 1s–3s transition [24, 45]. Whereas the 1s → 3s structural transition leaves the topology of the Y2C2 double layers unchanged it essentially re-arranges the van der Waals contacts in the iodine double layers [20, 21]. In fact, our DFT calculations also corroborate this transition with enthalpy differences between the high-pressure and the low pressure phase of the order of 100 meV (see figure 11). The calculations show the 3s-type to be more stable below a cell volume of ~270 Å3 corresponding to a pressure of ~2.4 GPa, in good agreement with the experimental observation.
As has already been discussed before (see figure 2) the 1s → 3s structural transition can also be initiated by chemical pressure induced by varying the halogen constituent and using mixtures of different halogen elements. The critical pressure is close to ~270 Å3 and a maximum Tc of ~12 K is similarly observed. The latter, however, does not coincide with the 1s → 3s structural transition as has been found in the present investigation. We tentatively attribute this discrepancy to locally varying pressure components brought about by the random arrangement of the mixed halogen constituents with locally differing ionic radii.
A closer inspection of the yttrium metal atom distances within the double layers reveals that the 1s → 3s phase transition also effects a slight distortion of the yttrium metal atom neighborhood for the C–C dumbbell with partly increased Y–Y distances. This elongation and a slight tilting of the C–C dumbbell axis away from the direct connection to the apical yttrium atoms (nearest neighbor yttrium atoms of the C atoms) by ~4 deg allows to accommodate the elongated C–C dumbbell in the metal atom double layer consistent with the results of the electronic structure calculation. Therefore the structural phase transition likewise involves van der Waals energy components in the iodine double layers and also electronic contributions from the slight re-arrangement of the yttrium atoms in the metal atom layers modifying also their interaction with the carbon atoms
In summary, we have studied the pressure dependence of the superconducting critical temperature of the layered yttrium carbide halide superconductor Y2C2I2 up to pressure of ~7.4 GPa. We observe an increase of Tc with pressure below ~2.5 GPa and a decrease above. Using NPD under pressure we found that the maximum Tc coincides with a structural phase transition changing the crystal structure from a 1s stacking variant to the 3s-type which leads to a re-arrangement of the van der Waals contacts in the iodine interlayers separating the Y2C2 double layers but also minute changes of the electronic interaction of the C and the Y atoms. Our experimental observations are in good agreement with the results of electronic structure calculations.
Acknowledgments
AM wants to acknowledge the financial support from Spanish MINECO under projects MAT2013-46649-C4-1/3-P and MAT2015-71070-REDC. We thank K Syassen for a careful reading of the manuscript and helpful discussions.
Footnotes
- 6
See supplemental material at (stacks.iop.org/JPhysCM/28/375703/mmedia) for the structural parameters.