
Abstract
Adsorption of noble gases on metal surfaces is determined by weak interactions. We applied two versions of the nonlocal van der Waals density functional (vdW-DF) to compute adsorption energies of Ar, Kr, and Xe on Pt(111), Pd(111), Cu(111), and Cu(110) metal surfaces. We compared our results with data obtained using other density functional approaches, including the semiempirical vdW-corrected DFT-D2. The vdW-DF results show considerable improvements in the description of adsorption energies and equilibrium distances over other DFT based methods, giving good agreement with experiments. We also calculated perpendicular vibrational energies for noble gases on the metal surfaces using vdW-DF data and found excellent agreement with available experimental results. Our vdW-DF calculations show that adsorption of noble gases on low-coordination sites is energetically favored over high-coordination sites, but only by a few meV. Analysis of the two-dimensional potential energy surface shows that the high-coordination sites are local maxima on the two-dimensional potential energy surface and therefore unlikely to be observed in experiments; this provides an explanation of the experimental observations. The DFT-D2 approach with the standard parameterization was found to overestimate the dispersion interactions, and to give the wrong adsorption site preference for four of the nine systems we studied.
Export citation and abstract BibTeX RIS
1. Introduction
Van der Waals (vdW) interactions play a very important role in the adsorption of noble gases on surfaces and mesoporous materials. Accurate determination of the interaction energy, equilibrium distance, and site preference are crucial to understanding the adsorption. Rare gas adsorption on metal surfaces has been studied extensively for many years using theoretical and experimental approaches [1–3]. It was initially expected that noble gases would adsorb preferentially on high-coordination sites on transition metal surfaces, because high-coordination sites maximize van der Waals interaction with the surface. However, the preponderance of experimental data now indicate that a variety of noble gases only adsorb on low-coordination sites of transition metal surfaces, both for close-packed [4–9] and open surfaces [10].
While experiments have identified the unexpected low-coordination site binding preference, they have not been able to identify the reason for this preference. Atomistic modeling can be used to probe the details of adsorption energetics. However, it is critical to use a level of theory in the modeling that correctly accounts for the essential physics of the interactions. The adsorption of noble gases on metals has been modeled using several different methods, including classical potential models [1, 11, 12], kinetic Monte Carlo [13, 14], and density functional theory (DFT) [15–20]. In principle, the structural and energetic properties of these systems could be computed from ab initio methods. However, highly accurate quantum mechanical methods, such as coupled cluster approaches, are out of the question for periodic systems, whereas second-order Møller–Plesset perturbation theory, which have been extended to periodic systems, will fail for metallic systems. While DFT is generally accepted as the method of choice for studying periodic systems, it is well known that the use of DFT to study noble gas adsorption on metal surfaces is problematic, because the interaction is very weak and dominated by vdW (dispersion) attraction as noted above. Standard DFT methods do not properly account for dispersion interactions since the exchange–correlation (XC) functionals in DFT are not exact. The dispersion interactions are primarily determined by electron correlation where the electron density overlap is small. However, the local density approximation (LDA), which is known to suffer from overbinding, often gives interaction energies for weakly bound systems that are approximately correct [21–23]. In particular, for noble gases adsorption, LDA energies are only fortuitously correct, giving reasonable energies as a result of a cancelation of errors between the exchange and correlation potentials [24]. Despite its shortcomings, LDA has been used to study noble gas adsorption on metals.
Müller [15] used LDA to study Xe adsorption on Pt(111), where the surface was approximated with a cluster model. The atop site was found to be energetically preferred over the hollow site by 30 meV and the Xe–Pt bond distance was found to be 3.0 Å. Da Silva et al [16] studied Xe adsorption on Mg(0001), Al(111), Ti(0001), Cu(111), Pt(111), and Pd(111) using LDA. They found that Xe prefers the low-coordination atop site over the hollow site in all cases by ∼ 1–51 meV . Other noble gases (He, Ne, Ar, and Kr) [25] preferred atop site adsorption as computed from the LDA functional. This study showed that the Perdew–Wang 91 (PW91) [26] and Perdew–Burke–Ernzerhof (PBE) [27] generalized gradient approximation (GGA) functionals likewise predict that the atop site is preferred over the hollow site for these same systems, with the exception of Ar and Ne on the Pd(111) surface, where the fcc site has lower energy. In all of these studies, GGA calculated adsorption energies (in absolute value) are greatly underestimated and LDA gives equilibrium distances that are too short compared to experimental results.
Extending the applicability of DFT to dispersion-dominated systems has attracted a great deal of interest in recent years. A number of methods have been developed for condensed phase systems, including the van der Waals density functionals (vdW-DF) introduced by Lundqvist et al, [28–30], the DFT-D methods of Grimme and co-workers [31, 32], the dispersion-corrected atom-centered pseudopotential methods of Rothlisberger et al [33], and Tkatchenko–Scheffler DFT+vdW schemes [34, 35]. The performance of different vdW-corrected DFT methods was recently reviewed by Tkatchenko et al [36]. Among these methods, the vdW-DF approach is perhaps the most promising because it introduces a new density functional (depending only on the electron density) within the spirit of a first-principles approach, where the dispersion interactions are recovered using a nonlocal correlation functional. Unfortunately, the vdW-DF functional is not exact and it violates some important limits [37]. Nevertheless, its utility has been established for different systems where vdW interactions are important. For example, vdW-DF has given good results for layered systems like graphite, benzene dimers, DNA base pairs, metal organic frameworks (MOFs), and carbon nanotubes [38]. This functional has also been applied to investigate the interaction between molecules and different metal surfaces [39–43]. For example, Toyoda et al [39] investigated pentacene adsorption on Cu(111), Ag(111), and Au(111). Atodiresei et al [40] studied C6H6, C5NH5, and C4N2H4 on the Cu(110) surface. Recent calculations using the vdW-DF functional for benzene on Au(111) [43], benzene on Cu(111) [44], and H2 on Cu(111) [45] all show good agreement between calculations and experimental data. In all of these studies, the inclusion of vdW corrections was shown to be important for the binding of molecules on the metal surfaces.
Recently, we applied vdW-DF to study the adsorption of Xe on metal surfaces including Pt(111), Pd(111), Cu(111), and Cu(110) [19]. We found that the hollow sites are local maxima rather than minima on the two-dimensional potential energy surface, and therefore adsorption at high-coordination hollow sites should not be observed in experiments, as is indeed the case. We showed that including nonlocal vdW interactions is crucial for obtaining results in quantitative agreement with experiments for adsorption energies, equilibrium distances, and vibrational energies. In the present paper we extend our previous study in two ways: (1) we include the noble gases Ar and Kr and show that our previous findings are quite general in applying to other noble gases; (2) we compare our vdW-DF calculations with results from DFT-D2, where dispersion energies are corrected using Grimme's DFT-D2 scheme. The comparison between vdW-DF2 and DFT-D2 shows that DFT-D2, at least with the recommended parameterization, qualitatively fails in describing these systems.
2. Theoretical methods and computational details
Several different functionals, including the local LDA/Perdew–Zunger [46], the semilocal GGA/PBE, DFT-D2/PBE [31], and the nonlocal vdW-DF1 and vdW-DF2 functionals are employed in this study. The vdW-DF functional calculations were carried out non-self-consistently using an in-house code that implements the fast Fourier transformation evaluation technique introduced by Román-Pérez and Soler [29]. This method has NlogN scaling with system size and is more efficient than the original direct summation method having an N2 scaling [28]. The charge densities needed for the non-self-consistent vdW-DF calculations were computed self-consistently using the PBE functional [27] in a pseudopotential planewave formulation (projector augmented wave method) [47] as implemented in the Vienna Ab initio simulation package (VASP) [48–51].
The correlation energy in the vdW-DF functionals is given as the sum of a local term computed using LDA (ELDA,c) and a nonlocal term (Enl,c) calculated as:
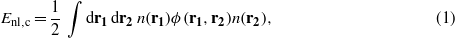
where ϕ is the vdW-DF kernel and n(r) is the charge density. There is more than one choice for the exchange functional to use with the vdW-DF functional [52, 28, 53]. In our investigations, we used the recommended choices for the exchange functional namely, revPBE for vdW-DF1, and a modified form of PW86 for vdW-DF2. Note that in this study, all of the vdW-DF calculations were performed using the PBE charge density, unless otherwise noted.
Using a non-self-consistent calculation with the PBE density, the total vdW-DF energy is written as:
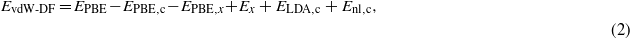
where EPBE,EPBE,c, and EPBE,x represent the PBE total energy, PBE correlation energy, and PBE exchange energy, respectively. Ex is the exchange energy appropriate for the vdW-DF functional used. To check on whether self-consistency in vdW-DF is important for our systems, we performed some of the calculations using the LDA density. In this case, the total vdW-DF/LDA energy is given by:
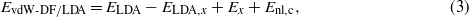
where ELDA and ELDA,x are the total LDA energy and LDA exchange energy, respectively.
We used a supercell approach to study the adsorption of noble gases on (111) surfaces in the  ° structure, where the gas was adsorbed on one side of the metal surface only. The noble gas monolayer adsorption on Pd(111) and Pt(111) were modeled using a repeated slab of seven atomic layers, while Cu(111) and Cu(110) surfaces were modeled using eight atomic layers. In order to simulate 1/4 monolayer (ML) adsorption on Pt(111), a supercell with 2 × 2 unit cells and four atomic layers was used. Interaction between the periodic images along the stacking direction were mitigated by using a large vacuum spacing of 20 Å, in conjunction with dipole corrections [54]. As a check, we carried out calculations with adsorption on both sides of the slab for some systems and found equivalent results to gas adsorption on one side only. Isolated atoms were studied using a cubic 15×15×15 Å3 supercell. We employed a Monkhorst–Pack [55] (8 × 8 × 1) grid to sample the Brillouin zone for surfaces, and used a planewave energy cutoff of 600 eV in all of our calculations. We carefully checked the convergence with respect to the k-point mesh, vacuum thickness, and planewave energy cutoff, and we found that the above parameters were sufficient to ensure convergence of the adsorption energies to within ∼ 1 meV .
° structure, where the gas was adsorbed on one side of the metal surface only. The noble gas monolayer adsorption on Pd(111) and Pt(111) were modeled using a repeated slab of seven atomic layers, while Cu(111) and Cu(110) surfaces were modeled using eight atomic layers. In order to simulate 1/4 monolayer (ML) adsorption on Pt(111), a supercell with 2 × 2 unit cells and four atomic layers was used. Interaction between the periodic images along the stacking direction were mitigated by using a large vacuum spacing of 20 Å, in conjunction with dipole corrections [54]. As a check, we carried out calculations with adsorption on both sides of the slab for some systems and found equivalent results to gas adsorption on one side only. Isolated atoms were studied using a cubic 15×15×15 Å3 supercell. We employed a Monkhorst–Pack [55] (8 × 8 × 1) grid to sample the Brillouin zone for surfaces, and used a planewave energy cutoff of 600 eV in all of our calculations. We carefully checked the convergence with respect to the k-point mesh, vacuum thickness, and planewave energy cutoff, and we found that the above parameters were sufficient to ensure convergence of the adsorption energies to within ∼ 1 meV .
The (111) surface unit cell is shown in figure 1, with black, gray, and light gray spheres represent the first (atop), second (hcp), and third (fcc) metal layers, respectively. The metal atoms of the slab were fixed in their relaxed bulk positions, in agreement with previous theoretical investigations [25] that showed that relaxation effects of the topmost surface layer for the Xe/metal system do not play a significant role in the interaction mechanism between Xe adatoms and metal surfaces. Seyller et al [5] experimentally observed that the substrate structure is essentially unrelaxed with respect to the bulk for Xe/Pt(111) system. The PBE calculated lattice constants are 3.95, 3.97, and 3.63 Å for Pd, Pt, and Cu, respectively, which are close to the experimental values of 3.89, 3.92, and 3.61 Å [56]. It has been reported that the lattice constant for Pd computed from the vdW-DF2 functional is larger than that computed from the PBE functional by 0.14 Å [57]. Our calculations show that using the larger lattice constant does not significantly change the adsorption energies. For example, the adsorption energy of Xe/Pd(111) using the larger lattice constant is decreased by only about 3 meV, which is 1% of the total adsorption energy.
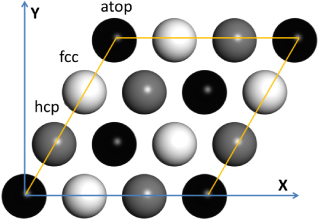
Figure 1. Top-view of the (111) surface unit cell, where black, gray, and light gray balls represent atop, hcp, and fcc sites, respectively, from the first, second, and third metal layers.
Download figure:
Standard image3. Results and discussion
3.1. Comparison of different density functionals
Xenon monolayer adsorption on Pd(111) and Pt(111) surfaces was examined first to gauge the performance of the different XC functionals. Figure 2 shows the adsorption potential energies of the Xe monolayer on Pd(111) and Pt(111) at the atop site as a function of the perpendicular distance between Xe and top-layer metal. The adsorption energy is given by
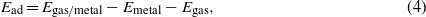
where Egas/metal,Emetal, and Egas represent the total energy of (noble gas+metal surface), bare metal surface, and isolated noble gas atom, respectively. Calculated and available experimental data are presented in table 1. The LDA value of Ead for Xe with atop site adsorption on Pd(111) is −396 meV and the equilibrium distance between the Xe monolayer and the Pd surface is d = 2.9 Å. Our Ead value compares favorably with previously published LDA results of −453 [16] and −435 meV [25]. Calculations with the PBE functional give Ead =− 53 meV , in good agreement with literature values of −76 [16] and −55.3 meV [25]. Our calculated equilibrium distance is d = 3.6 Å for the PBE functional. The adsorption energy reported from experiments is −320 meV [58], while three experimental distances were reported: 3.61 Å [7], 3.5 [59], and 3.07 [7]. Thus, LDA overestimates the adsorption energy in absolute value and underestimates the equilibrium distance. PBE, on the other hand, severely underestimates the magnitude of the adsorption energy but yields an equilibrium distance that is in better agreement with experiments.

Figure 2. Potential energy curves for (a) Xe/Pd(111) and (b) Xe/Pt(111) at the atop site as computed from LDA (up triangles), PBE (down triangles), vdW-DF1 (squares), vdW-DF2 (circles), and DFT-D2 (stars).
Download figure:
Standard imageThe DFT-D2 method has been shown to be efficient and accurate for many weakly bound systems. However, this method is known to be problematic for metallic systems [32]. Additionally, screening can mitigate vdW interactions in bulk metals, and the DFT-D2 cannot properly account for this effect, at least with the default C6 and vdW radius parameters [60]. As a consequence, it is expected that DFT-D2 would overestimate vdW interactions for adsorbed species on metal surfaces [61]. Indeed, the DFT-D2 calculated adsorption energy for Xe/Pd(111) is −420 meV, even larger in magnitude than the LDA result, and the equilibrium distance of 3.0 Å is smaller than the PBE value and comparable to the LDA value.
Our calculations with the vdW-DF2 (vdW-DF1) functional give an adsorption energy of −309 (−330) meV and an equilibrium distance of 3.7 (3.8) Å for Xe/Pd(111). The vdW-DF1 binding energy is larger in magnitude than the vdW-DF2 value by 21 meV, which is consistent with the observation of Lee et al [30] that vdW-DF1 overestimates the vdW attraction. The adsorption energies from both vdW-DF1 and vdW-DF2 are in excellent agreement with experimental data. The predicted equilibrium distance from vdW-DF2 is in slightly better agreement with experiment than vdW-DF1, which is also expected [30]. We note from figure 2 that the major difference between vdW-DF1 and vdW-DF2 appears at longer distances, rather than near the potential minimum. The vdW-DF2 functional gives a shorter-ranged potential; this is consistent with the previous reports that C6 coefficients obtained using the vdW-DF2 functional are underestimated compared to those obtained using vdW-DF1 [37]. Also, the large-r behavior of the adsorption potential curves in LDA and GGA show the typical exponential decay, in contrast to the much slower decay rate of the vdW-DF potential.
The Xe/Pt(111) system was also studied using the above functionals and results are compared with the Xe/Pd(111) system in figure 2. The experimentally measured adsorption energy ranges from −320 to −270 meV, while two different adsorption distances are reported: 4.2 and 3.4 Å. These data are given in table 1. The LDA calculated adsorption energy of −305 meV for Xe/Pt(111) agrees with experiments and is much smaller than the value of −396 meV for Xe/Pd(111). The computed equilibrium distance of 3.2 Å for Xe/Pt(111) is smaller than the smallest reported experimental value, but larger than that for Xe/Pd(111) by 0.3 Å. Similarly, the PBE functional also gives smaller adsorption energy and larger equilibrium distance for Xe/Pt(111) compared to Xe/Pd(111) (see table 1). Surprisingly, DFT-D2 gives a much larger adsorption energy of −605 meV for Xe/Pt(111) compared with −420 meV for Xe/Pd(111). The DFT-D2 value is about a factor of two larger in magnitude than the experimental data for Xe/Pt(111). This large deviation, together with incorrect ordering of adsorption site preference for several systems that will be discussed later, indicates that the DFT-D2 method, at least with recommended parameterization, is not suitable for investigating noble gas adsorption on metals. Note that the improved pairwise schemes of Tkatchenko and Scheffler [34], where the C6 coefficients depend on the bonding environment, may provide a more accurate description of the adsorption energies. The adsorption energy of −305 (−329) meV from vdW-DF2 (vdW-DF1) calculations for Xe/Pt(111), is only slightly smaller than that for Xe/Pd(111), while the equilibrium distance for Xe/Pt(111) is slightly larger than Xe/Pd(111), as expected (see table 1).
Table 1.
The equilibrium distance (d, in Å), adsorption energy(Ead, in meV), and vibrational energy (hν, in meV) for atop site adsorption of noble gas atoms on several metal surfaces obtained with LDA, PBE, vdW-DF1, vdW-DF2, and DFT-D2. The values in brackets represent the difference in adsorption energies between the fcc and atop sites:  . Positive values in brackets indicate that the atop site has lower Ead. The experimental data are from [4, 5, 7, 8, 58, 59, 62–66].
. Positive values in brackets indicate that the atop site has lower Ead. The experimental data are from [4, 5, 7, 8, 58, 59, 62–66].
| LDA | PBE | vdW-DF1 | vdW-DF2 | DFT-D2 | Exp. | ||
|---|---|---|---|---|---|---|---|
| Xe/Pd(111) | d | 2.9 | 3.6 | 3.8 | 3.7 | 3.0 | 3.61, 3.5, 3.07 |
| Ead | −396 [36] | −52 [6] | −330 [3] | −309 [4] | −420 [3] | −320 | |
| hν | 6.5 | 2.1 | 2.7 | 3.4 | 4.8 | ||
| Kr/Pd(111) | d | 2.9 | 3.9 | 3.8 | 3.6 | 3.3 | |
| Ead | −234 [23] | −43 [1] | −235 [1] | −200 [4] | −260 [−28] | ||
| hν | 6.4 | 1.5 | 3.2 | 4.1 | 4.9 | ||
| Ar/Pd(111) | d | 3.0 | 4.0 | 3.7 | 3.6 | 3.2 | |
| Ead | −154 [17] | −17 [0] | −203 [0] | −171 [3] | −181 [22] | ||
| hν | 7.9 | 1.9 | 4.4 | 5.4 | 8.1 | ||
| Xe/Pt(111) | d | 3.2 | 4.0 | 4.0 | 3.8 | 3.1 | 4.2, 3.4 |
| Ead | −305 [39] | −36 [2] | −329 [2] | −305 [5] | −607 [8] | −320 to −270 | |
| hν | 5.2 | 1.4 | 2.8 | 3.6 | 6.5 | 3.5, 3.7 | |
| Kr/Pt(111) | d | 3.2 | 4.2 | 3.9 | 3.8 | 3.3 | |
| Ead | −185 [21] | −24 [0] | −238 [1] | −197 [3] | −390 [−39] | −161 | |
| hν | 5.6 | 1.3 | 3.3 | 4.1 | 4.9 | 3.9 | |
| Ar/Pt(111) | d | 3.2 | 4.2 | 3.8 | 3.6 | 3.4 | |
| Ead | −123 [12] | −15 [0] | −205 [0] | −171 [2] | −248 [27] | ||
| hν | 7.2 | 1.8 | 4.6 | 5.4 | 8.2 | 4.9 | |
| Xe/Cu(111) | d | 3.3 | 4.4 | 4.1 | 4.0 | 3.4 | 3.6 |
| Ead | −255 [7] | −16 [1] | −283 [1] | −270 [1] | −283 [−15] | −190 | |
| hν | 3.8 | 1.1 | 2.4 | 2.9 | 4.4 | 2.6 | |
| Xe/Pt(111) 1/4 ML | d | 3.0 | 3.8 | 3.9 | 3.7 | 3.2 | |
| Ead | −301 [51] | −32 [5] | −247 [4] | −240 [7] | −656 [33] | ||
| hν | 5.4 | 1.8 | 2.9 | 3.8 | 3.2 | ||
| Xe/Cu(110) | d | 2.9 | 3.8 | 3.8 | 3.6 | 3.2 | |
| Ead | −257 [56] | −31 [6] | −176 [3] | −163 [5] | −205 [−48] | ||
| hν | 5.6 | 1.3 | 2.3 | 2.9 | 3.5 |
We also performed vdW-DF2 calculations using charge densities obtained from a self-consistent LDA calculations to compare with PBE charge density based vdW-DF2 results. The adsorption energies computed from vdW-DF2/LDA are −360 and −359 meV for Pd(111) and Pt(111), respectively. The equilibrium distances computed from vdW-DF2/LDA are 3.8 and 3.9 Å, for Pd(111) and Pt(111), respectively. The difference between the vdW-DF2 (where PBE density was used) and vdW-DF2/LDA adsorption energies for Xe/Pd(111) is −51 meV, while the difference in equilibrium distance is about 0.1 Å, with similar differences observed for the Xe/Pt system. This is in reasonably good agreement given the large differences between the PBE and LDA results on these systems. Hence, as was observed before [28], we conclude that self-consistency does not have a profound effect on the vdW-DF results.
3.2. Comparison between low-coordination and high-coordination sites adsorption
We here examine the differences in adsorption energies between low- and high-coordination sites for various surfaces as computed from different functionals. Since the fcc and hcp sites for the (111) surfaces have been found to be energetically very similar [16, 19], we consider only fcc sites in the following, with the understanding that our results are equally applicable to hcp sites. We see from table 1 that adsorption at low-coordination sites is energetically preferred for all systems studied with various functionals, except DFT-D2. It is striking that LDA, PBE, and both vdW-DF functionals give consistent site preference results for all systems studied. Because the DFT-D2 functional gives results that do not agree with the other functionals, we will discuss the DFT-D2 results after those from the other functionals. In table 1, we show in brackets the relative adsorption energies of noble gas atoms at the atop and fcc sites (or row and trough sites for Cu(110)), where positive values indicate atop site preference. For Xe/Pd(111), the PBE and LDA calculations predict the atop site is preferred over the fcc site by 6 and 36 meV, respectively. Our LDA value is in good agreement with two different published LDA calculations by Da Silva et al, who reported preferences of 51 meV [16], and 44 meV [25] for atop versus fcc adsorption. In comparison, vdW-DF1 and vdW-DF2 calculations show a preference of only 3 and 4 meV in this case. The results for adsorption site preferences for Xe/Pt(111) are very similar to those for Xe/Pd(111). Hence, there is an order of magnitude difference in the site preference energies between the LDA and either the PBE or vdW-DF potential energies. As shown in table 1, the adsorption energy for the Xe monolayer on the Cu(111) surface is smaller than on Pd(111) and Pt(111) surfaces, based on the calculations with the different XC functionals. The adsorption energies from vdW-DF2 and LDA are −270 and −255 meV, respectively, both larger than the experimental value of −190 meV. The calculated equilibrium distance of 4.0 Å is larger than experimental value of 3.6 Å, while LDA value of 3.2 Å again underestimates the equilibrium distance. The energy differences between atop and hollow sites are found to be smaller for Xe/Cu(111), i.e., 7 and 1 meV, for LDA and vdW-DF2, respectively.
In addition to noble gas monolayer adsorption results, we also calculated 1/4 ML adsorption of Xe on Pt(111). With dilute Xe coverage, the adsorption energy is reduced to −240 meV based on vdW-DF2, smaller than monolayer adsorption energy by 65 meV, due to the reduced Xe–Xe interaction for low coverage adsorption. We note that the energy difference between atop and fcc site adsorption increases from 5 to 7 meV in going from monolayer to 1/4 ML adsorption. Ellis et al [67] reported a value of 9.6 meV for the corrugation of the potential for low Xe coverage on Pt(111), which is very close to our calculated value of 7 meV.
We extended our study to the non-close-packed Cu(110) surface. We modeled the experimental Cu(110)–(12 × 2)–14Xe structure [10] using a simple model with 1 Xe atom on a 1 × 1 Cu(110) unit cell (4 Cu atom in each layer). The vdW-DF2 calculated adsorption energy is −163 meV on the atop site of the top of the Cu rows. This is 5 meV lower in energy than adsorption on the four-fold hollow site in the trough. Our calculations showing that the low-coordination row top sites are energetically favored over the high-coordination trough sites is in agreement with experimental observations [10].
We now turn our attention to the results of the DFT-D2 calculations. It is striking that the DFT-D2 calculations predict that adsorption at high-coordination sites is energetically more favorable than at low-coordination sites for four of the nine systems studied, i.e., Kr/Pd(111), Kr/Pt(111), Xe/Cu(111), and Xe/Cu(110). This result is in contradiction with calculations from the other functionals and available experiments. Analysis of the damped atom-pairwise dispersion correction shows that this term dominates the energy difference between low- and high-coordination sites, and consequently determines the site preference. As shown in figure 3(a), the dispersion contribution to the difference in energy between high- and low-coordination site adsorption favors the high-coordination sites only for Kr/Pd(111), Kr/Pt(111), Xe/Cu(111), and Xe/Cu(110). Therefore, it is the dispersion correction that dominates the site preference in the DFT-D2 calculations. This result can be clearly seen from the scatter plot of the vdW contributions to the adsorption energy difference between high- and low-coordination sites plotted as a function of the difference in the DFT-D2 adsorption energies, shown in figure 3(b). In contrast, the differences in the dispersion energies from vdW-DF (Enl,c) for high- and low-coordination sites are at least an order of magnitude smaller than for the DFT-D2 calculations, as can be seen by comparing the bar graph and points in figure 3(a). This confirms our previous finding that it is the balance between electrostatic energy and kinetic energy that determines the site preference for the vdW-DF functional [19].

Figure 3. (a) The difference in the vdW energies between high- and low-coordination sites, E(vdW, high) − E(vdW, low), as computed from DFT-D2 (bars) and vdW-DF2 (filled circles). Note the different scales on the left and right axes. (b) The difference in the vdW energies plotted as a function of the difference in the adsorption energies for the DFT-D2 functional.
Download figure:
Standard imageOur calculations show that the trend in the vdW-DF2 adsorption energies for Ar, Kr, and Xe follows the same trend as the atomic polarizabilities, which are 1.64, 2.49, and 4.02 Å3, respectively [64]. This trend can be seen from figure 4 for adsorption of these gases on both Pd(111) and Pt(111). Moreover, we see from table 1 that the atop site adsorption energy on Pd(111) decreases from −309 (Xe) to −200 (Kr) to −171 (Ar) meV based on vdW-DF2 functional. The trend is very similar for the Pt(111) surface. In general, the energy difference between atop and fcc sites decreases going from Xe to Kr to Ar, based on LDA, PBE, vdW-DF1, and vdW-DF2 functionals. For example, vdW-DF2 gives energy differences of 5, 3, and 2 meV for Xe, Kr, and Ar on Pt(111), respectively; LDA gives larger energy differences of 39, 21, and 12 meV for Xe, Kr, and Ar on Pt(111), respectively, but with the same trend. This is expected, because the magnitude of adsorption energy decreases from Xe to Kr to Ar. However, the DFT-D2 functional does not follow the trend of the other functionals. As shown in table 1, the energy differences computed from DFT-D2 are 3, −28, and 22 meV for Xe, Kr, and Ar adsorption on Pd(111), respectively. The energy differences for the Pt(111) surface computed from DFT-D2 follow a similar pattern with 8, −39, and 27 meV for Xe, Kr, and Ar, respectively. These anomalous results from DFT-D2 can be attributed to the inaccuracy of the vdW energy term in DFT-D2 as discussed above.

Figure 4. Potential energy curves for Ar (squares), Kr (circles), and Xe (triangles) adsorption on (a) Pd(111) and (b) Pt(111) at both atop (filled) and fcc (open) sites obtained from vdW-DF2 calculations.
Download figure:
Standard imageWe have computed occupation probabilities of atop and hollow sites (fcc and hcp) for Xe/Pd(111) assuming a Boltzmann distribution at 60 and 90 K—temperatures at which the  ° structure was reported to be stable [58, 59, 7]. We assumed a degeneracy for atop and hollow sites of 1 and 2, respectively. Using the vdW-DF2 energies, we found that at T = 60 K, the atop and hollow sites are almost equally occupied, with probabilities of 52% and 48%, respectively. At T = 90 K the occupation probabilities are 46% and 54%, respectively. This shows that the hollow site has a substantial probability to be occupied in this temperature range, despite its higher energies. Even if our calculations are off by a factor of two due to density functional inaccuracies (e.g., if the difference in energy was 8 meV), the hollow site would still have significant occupation. In contrast, the atop to hollow site energy difference of 36 meV predicted from LDA would result in 98% atop site occupation at 90 K. The same procedure gives similar results for the other systems investigated in this study. This analysis indicates that hollow sites should also be observed with a certain probability, although atop sites have lower energy based on different functional calculations. Thus, the energy differences computed from vdW-DF cannot explain the experimental observation of only atop site adsorption occupation for many different systems. To understand the real reason for the experimentally observed adsorption site preference we must consider the two-dimensional potential energy surface, which will be discussed below.
° structure was reported to be stable [58, 59, 7]. We assumed a degeneracy for atop and hollow sites of 1 and 2, respectively. Using the vdW-DF2 energies, we found that at T = 60 K, the atop and hollow sites are almost equally occupied, with probabilities of 52% and 48%, respectively. At T = 90 K the occupation probabilities are 46% and 54%, respectively. This shows that the hollow site has a substantial probability to be occupied in this temperature range, despite its higher energies. Even if our calculations are off by a factor of two due to density functional inaccuracies (e.g., if the difference in energy was 8 meV), the hollow site would still have significant occupation. In contrast, the atop to hollow site energy difference of 36 meV predicted from LDA would result in 98% atop site occupation at 90 K. The same procedure gives similar results for the other systems investigated in this study. This analysis indicates that hollow sites should also be observed with a certain probability, although atop sites have lower energy based on different functional calculations. Thus, the energy differences computed from vdW-DF cannot explain the experimental observation of only atop site adsorption occupation for many different systems. To understand the real reason for the experimentally observed adsorption site preference we must consider the two-dimensional potential energy surface, which will be discussed below.
It is interesting to explore why there is such a large difference in the site preference energies computed from the LDA, PBE, and vdW-DF methods. To this end, we have compared the differences between the atop and fcc energies for Xe/Pt(111) computed from the exchange and correlation functionals for LDA, PBE, and vdW-DF2. We found that the differences between the correlation energies for atop and fcc sites are very small (much less then 1 meV) at all distances examined, and for all three of the functionals. There is also very little difference in the exchange energies for distances greater than about 4.0 Å. However, the LDA exchange repulsion is weaker for the atop site compared with the hollow site by about 60 meV at the LDA equilibrium distance of 2.9 Å. This agrees with the previous observations of Da Silva et al [17] who used this fact to ascribe the atop adsorption site preference to weaker Pauli repulsion at the atop site. In contrast, exchange energies computed from the vdW-DF2 functional near the equilibrium distance of 3.7 Å are almost identical for atop and fcc sites (difference < 0.2 meV ). Decomposition of the interaction energy indicates that the slight lower energy at atop site is due to a delicate balance between the kinetic energy and electrostatic energy. Specifically, Xe adsorbed at the atop site of Pt(111) has a lower kinetic energy than when adsorbed on the fcc site by about 50 meV, while the electrostatic energy at the atop site is higher by about 43 meV [19]. The difference of these two quantities determines the site preference.
3.3. Perpendicular vibrational energy
Vibrational energies are sensitive probes of the curvature of the potential energy surface near the minimum. We have used the following physically motivated function [16, 64] to model the gas–metal surface interactions:
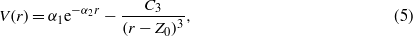
where V(r) is the potential energy between Xe and the metal surface at a distance r from the surface. The parameters α1,α2,C3, and Z0 are determined by fitting to the DFT results. For LDA and PBE calculations, we limited the above fit to energies around the equilibrium distance because the adsorption potential in this case decays exponentially fast and the long-range-limit of equation (5) will not adequately describe the calculated data. The vibrational energy is calculated from

where Evib is the vibrational energy, h is the Planck constant, Mgas is the mass of the gas atom, and ke is the force constant that is the second derivative of the potential at the minimum. We also used a Morse potential to fit the potential energy surfaces around the minimum and obtained similar frequencies to those obtained using equation (5).
The perpendicular vibrational energies are presented in table 1. For example, the LDA vibrational energies for Xe/Pd(111) are 6.5 meV at the atop site, which is similar to the value of 8.2 meV from Da Silva et al [16]. To the best of our knowledge, experimental vibrational energy data are only available for Ar, Kr, and Xe on Pt(111), as well as Xe monolayer adsorption on Cu(111). Bruch et al [4] reported a value of 3.5 meV for the perpendicular vibrational energy of Xe/Pt(111), and Hall et al [66] gave a value of 3.70 meV for Xe/Pt(111). The vibrational energies computed from vdW-DF2 for Xe/Pt(111) is 3.6 meV at atop site, in excellent agreement with reported experimental values. In addition, the calculated vibrational energies are 4.1 and 5.4 meV for atop site of Kr/Pt(111) and Ar/Pt(111), respectively, in good agreement with experimental data [66] of 3.90 and 4.85 meV. The DFT-D2 data based vibrational energies are also calculated. These are in worse agreement with experiment than the vdW-DF results. For example, the vibrational energy computed from DFT-D2 is 8.2 meV for Ar/Pt(111), while the experimental value is 4.9 meV. In general, the vibrational energies computed from the PBE functional are the smallest, LDA or DFT-D2 values are typically the largest, and vdW-DF1 and vdW-DF2 data lie in between the PBE and LDA values. This trend correlates well with the curvature of the potential energy curves from different functionals. The vibrational energies computed from vdW-DF1 are smaller than those from vdW-DF2 by about 0.5–0.8 meV, which is consistent with the fact that the vdW-DF2 potential energy surface has a higher curvature near the minimum (see figure 2). For Xe/Cu(111), the perpendicular vibrational energy of 2.89 meV is close to experimental value of 2.6 meV [68]. The good agreement between the vdW-DF2 vibrational energies and available experimental data provides an independent assessment of the accuracy of vdW-DF2 potential energy curve, especially around the minimum.
3.4. Two-dimensional potential energy surfaces
As discussed above, the vdW-DF adsorption energies have very small energy differences of only a few meV between low- and high-coordination sites for all of the systems investigated. This small energy difference does not appear to be in accord with the experimental observations that only low-coordination sites are occupied in the temperature range of 60–90 K. The two-dimensional potential energy surfaces (PES) provide an explanation for the experimentally observed adsorption site preference. Figure 5 shows the LDA, PBE, and vdW-DF2 PES for Xe/Pt(111), and the conclusions are applicable to other (111) surfaces. We see that the atop site is a true minimum on the PES computed from all three functionals. Moreover, the bridge site between the hcp and fcc sites is a transition state, while both hollow sites (hcp and fcc) are local maxima. Hence, both hcp and fcc sites are saddle points of index 2, i.e., they have two eigenvectors with imaginary frequencies parallel to surface plane. This indicates that the hollow sites are ephemeral transient states rather than true stable states and thus are not easily observed experimentally. The bridge site is a transition state (saddle point of index 1), connecting two neighboring minima (atop sites). Therefore, the minimum energy pathway for the motion of the Xe monolayer is atop–bridge–atop. Although Cu(110) surface is topologically different from the (111) surface, the four-fold hollow site (in the trough) is also an index 2 saddle point and the low-coordination atop site (on the row tops) is a global minimum, according to our calculations. The Xe/Cu(110) model used in this study is different from the experimentally observed Cu(110)-(2 × 2)-14 Xe [10]. We used a smaller supercell with slightly lower Xe coverage for the sake of computational efficiency. The experiments found that Xe atoms adsorb on row tops rather than troughs, but because of the higher loading the Xe atoms were not all adsorbed on atop sites. However, the low-coordination site preference from our calculations agrees qualitatively with experiments. Overall, the experimental observation of atop adsorption site preference is due to the presence of high-index saddle points rather than large differences in the adsorption energies between competing adsorption sites.
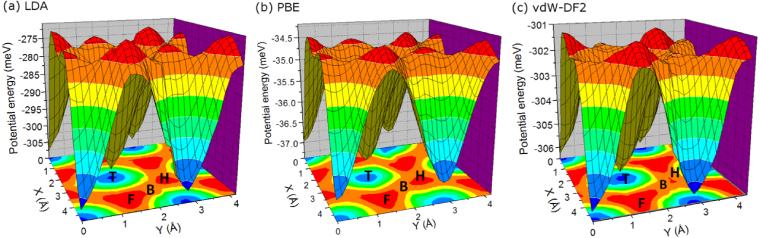
Figure 5. Two-dimensional potential energy surfaces of Xe/Pt(111), calculated from (a) LDA, (b) PBE, and (c) vdW-DF2. The orientation of the projected XY plane is the same as the surface model in figure 1. The atop, hcp, and fcc sites are labeled as T, H, and F, respectively, while B represents the bridge site.
Download figure:
Standard imageThe shapes of PESs based on different functionals are very similar to each other, although the magnitude of the potential energies are very different. The energy barriers to diffusion, i.e., energy differences between atop and bridge sites, are 30.7, 4.8, and 2.3 meV, as computed from LDA, PBE, and vdW-DF2, respectively. The features of the PES computed from LDA, PBE, and vdW-DF for the other rare gases on the metal surfaces are similar to those of the Xe/Pt(111) system. We have verified this by computing the vibrational frequencies at the high symmetry points on the PES. However, the PES computed from DFT-D2 is qualitatively different for several of the systems. As we discussed above, fcc sites are energetically favored for four of nine systems based on DFT-D2 computations. Vibrational frequency calculations confirm that the fcc sites have all real frequencies, while atop sites have two imaginary frequencies for these four systems.
Recently, Zhang et al [20] investigated Ne, Kr, and Xe adsorption on the main group Pb(111) metal surface using LDA and vdW-DF. Surprisingly they found that the hollow sites are more favorable than atop site, which is opposite to the results from our studies of transition metal surfaces. Note that the hybridization between the p electrons of noble gases and d electrons of transition metals plays a key role in determining the lower energy of atop site adsorption [16, 19]. In contrast, Pb has s and p electrons involved in hybridization with electrons from noble gases, but no d electrons. The differences in orbital interactions likely underlie the difference of adsorption site preference between transition metal surfaces in our study and main group metal Pb(111) studied by Zhang et al [20]. However, an in-depth investigation of this phenomenon is beyond the scope of this work.
4. Conclusions
We have studied the adsorption of Ar, Kr, and Xe on a series of transition metal surfaces using several different DFT functionals, namely, LDA, PBE, DFT-D2, and the recently developed vdW-DF. The vdW-DF2 functional was found to give the best agreement with experimental data for adsorption energies, equilibrium distances, and vibrational energies. Overall, the vdW-DF2 predicted equilibrium distances are slightly larger than experimental data; the agreement could be improved by using a different exchange functional [52, 57]. The vdW-DF2 approach offers significant improvement over the LDA functional for these systems. Generally, the equilibrium distances predicted from LDA are too small compared with experiments. The DFT-D2 results, with the recommended parameterization, generally overpredict the adsorption energy and underestimate the equilibrium distance, due to the overestimated dispersion contribution.
Adsorption energy differences between low- and high-coordination sites for Ar, Kr, and Xe on different metal surfaces were computed using different functionals. The low-coordination site was found to be energetically favorable for all of the systems studied with all functionals except DFT-D2. Calculations using DFT-D2 give the opposite adsorption site preference for four of nine systems we investigated, indicating that the standard parameterization of DFT-D2 may be inaccurate for metallic systems. In all cases the vdW-DF functional predicts that the energy differences between low- and high-coordination sites is only a few meV. In contrast, the LDA calculations give much larger differences (as much as an order of magnitude) with the atop site being favored. Analysis of the LDA calculations indicates that the reason for this preference is primarily due to weaker exchange repulsion at atop sites compared to fcc sites. In contrast, differences in exchange repulsion do not play a determining role in the site preference as computed from the vdW-DF approach. Different density functionals (LDA, PBE and vdW-DF) give similar shapes of two-dimensional potential energy surfaces, although the magnitude of potential energies are very different. The hollow sites of the metal surfaces we examined are found to be local maxima on the PES and therefore kinetically unstable. This is a general phenomenon for various transition metal surfaces, and explains the experimental observation for atop adsorption site preference.
Acknowledgments
We gratefully acknowledge funding from the US Department of Energy under grant number DE-FG02-10ER16165. We thank L W Bruch and R D Diehl for helpful discussions. Calculations were performed at the University of Pittsburgh Center for Simulation and Modeling.