
Abstract
Heterogeneous therapeutic responses to leukotriene modifiers (LTMs) are likely due to variation in patient genetics. Although prior candidate gene studies implicated multiple pharmacogenetic loci, to date, no genome-wide association study (GWAS) of LTM response was reported. In this study, DNA and phenotypic information from two placebo-controlled trials (total N=526) of zileuton response were interrogated. Using a gene–environment (G × E) GWAS model, we evaluated 12-week change in forced expiratory volume in 1 second (ΔFEV1) following LTM treatment. The top 50 single-nucleotide polymorphism associations were replicated in an independent zileuton treatment cohort, and two additional cohorts of montelukast response. In a combined analysis (discovery+replication), rs12436663 in MRPP3 achieved genome-wide significance (P=6.28 × 10−08); homozygous rs12436663 carriers showed a significant reduction in mean ΔFEV1 following zileuton treatment. In addition, rs517020 in GLT1D1 was associated with worsening responses to both montelukast and zileuton (combined P=1.25 × 10−07). These findings implicate previously unreported loci in determining therapeutic responsiveness to LTMs.
Similar content being viewed by others
Introduction
The leukotriene pathway is an important therapeutic target for asthma therapy. Leukotriene-modifying agents (LTMs) improve asthma by either selectively preventing leukotriene production from arachidonic acid via 5-lipoxygenase (5-LO) inhibition (e.g., zileuton), or by preventing leukotrienes from binding to the major cysteinyl leukotriene receptor, CysLT1 (e.g., montelukast, pranlukast and zarfirlukast).1, 2, 3 LTMs are effective in improving lung function, asthma symptoms and quality of life in patients with asthma and allergic rhinitis by reducing airway hyper-responsiveness and eosinophilia.4, 5, 6
Despite their general efficacy, patient responses to LTMs are not consistent, with some individuals failing to respond to treatment.7, 8, 9 The presence of significant inter-individual variability in the treatment response to LTMs suggests that variation in patient genetics may underlie this phenomenon.7, 8, 9, 10, 11 Historically, variable response to leukotriene inhibition formed the basis for the first reported asthma pharmacogenetic study.7 In an effort to characterize this pharmacogenetic relationship, researchers initially focused on candidate genes within pharmacological pathways for LTM response, including arachidonate 5-lipoxygenase (ALOX5), the gene encoding 5-LO, in addition to ATP-binding cassette, sub-family C (CFTR/MRP), member 1 (ABCC1),9, 11 cysteinyl leukotriene receptor 1 (CYSLTR1),9, 11, 12 cysteinyl leukotriene receptor 2 (CYSLTR2),9, 11, 12 leukotriene A4 hydrolase (LTA4H),9, 10, 11, 12 leukotriene C4 synthase (LTC4S)9, 10, 11, 12 and others. Studies of ALOX5 have identified multiple single-nucleotide polymorphism (SNP) associations with symptomatic improvement and changes in measures of lung function during LTM therapy.7, 9, 10, 11, 12 Two studies of the LTC4S gene identified promoter and intronic SNPs associated with improved response to LTMs; in addition, regulatory variants in CYSLT2 (but not CYSLT1) that were significantly associated with increased morning peak expiratory flow in patients taking montelukast were identified.9, 10, 11, 12 SNPs associated with LTM plasma levels and differential responsiveness to LTMs have also been reported for solute carrier organic anion transporter family, member 2B1 (SLCO2B1) and ABCC1.9, 10, 13, 14 Importantly, we have previously demonstrated that variants in multiple genes contribute to the response of both leukotriene inhibitors (e.g., zileuton) and leukotriene receptor antagonists (e.g., montelukast).11
Although the initial results of these candidate gene studies are promising, replication of identified associations has been problematic, making it difficult to estimate the degree of contribution of individual genes to LTM response. By necessity, candidate gene studies focus on characterizing the genotype–phenotype relationships within individual genes that are chosen a priori based on biological and clinical information, rather than an agnostic approach that considers data from the entire genome. Recent genome-wide association studies (GWASs) of symptomatic response to asthma medications have identified multiple genes associated with patient responsiveness to inhaled corticosteroids, adding to the growing evidence for a genetic basis for therapeutic efficacy in asthma patients.15, 16, 17 Limited information from candidate gene studies of zileuton and montelukast response indicates that patient genetics has a role in LTM response; however, to date, no genome-wide investigation of LTM response has been reported. For these reasons, we performed a GWAS to identify novel genetic loci associated with leukotriene modifier response in asthmatic patients.
Materials and methods
Overview of study populations
The discovery and replication cohorts evaluated in this study consisted of 526 adult patients with moderate, persistent asthma from two independent, placebo-controlled clinical trials (conducted by Abbott, Chicago, IL, USA)18, 19 evaluating the efficacy of a controlled-release (CR) formulation of zileuton. The primary GWAS cohort (‘Abbott trial 1’) included 160 patients randomized to receive zileuton and 144 patients randomized to receive placebo for 12 weeks (details of the trial are provided in Nelson et al.18). Replication of initial GWAS results was performed in a cohort of 149 patients randomized to receive zileuton and 73 patients receiving placebo for 12 weeks (‘Abbott trial 2’), as adjunctive therapy to usual care (details of the trial are provided in Wenzel et al.19). To evaluate potential overlap between genetic predisposition to zileuton and montelukast responses, additional replications were performed using two clinical trials of montelukast response, which were previously described,20, 21 the American Lung Association Asthma Clinical Research Center (ALA-ACRC) trials: the Leukotriene Modifier Or Corticosteroid or Corticosteroid-Salmeterol trial (LOCCS) and Effectiveness of Low Dose Theophylline as Add On Therapy for the Treatment of Asthma (LODO). All studies were approved by the institutional review boards of their corresponding institutions, and all participants provided informed consent for genetic studies. Demographic characteristics of the cohorts evaluated in this study are shown in Table 1. Each cohort is described in detail below.
Zileuton populations (Abbott)
Full study design details of the two Abbott clinical trials evaluated in this study have been reported elsewhere.18, 19 In both cohorts, ‘moderate asthma’ was defined by the following criteria: (i) forced expiratory volume in 1 s (FEV1) measurement of 40–75% of predicted FEV1 when taken at least 48 h after the last theophylline use, at least 12 h after a long-acting β-agonist and at least 6 h after a short-acting β-agonist, (ii) ⩾15% increase in FEV1 at least 15 min after inhaled albuterol, and (iii) a history of 15% reversibility within the year before study enrollment.
The first Abbott cohort, termed 'Abbott trial 1' in this investigation, was a phase 3, randomized, placebo-controlled, multi-center study evaluating response to treatment with zileuton CR (1200 mg twice daily) vs zileuton immediate release (600 mg four times daily).18 The study population was composed of 786 nonsmoking males and females, aged 12 years and older, who had mild to moderate asthma. Patients who had been hospitalized for asthma within 6 months, or who had taken any investigational drugs, excepting β-agonist inhaler usage, were excluded. The study population included both white (81.4%) and non-white (18.6%) individuals. Following a 2-week-placebo lead-in period, patients with an FEV1 of 40–75% of predicted who used a β-agonist inhaler at least twice per day and showed an improvement of ⩽10% over baseline FEV1 were randomized to zileuton or placebo (1:1 ratio) and evaluated over a 12-week period. In this study, only data from the first 12 weeks following randomization to zileuton or placebo were evaluated. Although the clinical trial evaluated 786 patients, complete genome-wide genotype and phenotype data were only available from 160 patients who received zileuton, and 144 patients who received placebo; these patients represented the discovery GWAS population evaluated in this study.
The second Abbott cohort, termed ‘Abbott trial 2’ in this investigation, was a phase 3, double-blind, randomized, placebo-controlled, multi-center study to evaluate the long-term safety and efficacy of add-on zileuton CR (1200 mg twice daily), or placebo, in addition to usual care (that is, the usual treatment regimens these patients received while they were being treated by their primary care providers). For all patients, this could include treatment regimens including, but not limited to, theophylline, over 6 months.19 The study population included 926 nonsmoking males and females, aged 12 years and older, who had not taken any investigational drugs and who demonstrated an FEV1 of at least 40% of predicted when taken 48 h or more after the last theophylline administration and at least 12 h following long-acting β-agonist use and at least 6 h following short-acting β-agonist use, and demonstrated a 15% or greater increase in FEV1 following inhaled albuterol, or reported a history of 15% reversibility within 1 year before entering into the study.19 Patients were randomized to zileuton CR plus usual care, or placebo plus usual care, in a 2:1 ratio, and were evaluated over 6 months. Both white (85.7%) and non-white (14.3%) subjects were evaluated in this study. Although 926 subjects were included in the clinical population, for the GWAS, complete genome-wide genotype data were available from 149 patients randomized to receive zileuton and 73 patients receiving placebo. In this study, data from the first 12 weeks following randomization to zileuton or placebo were investigated. This population was used to replicate the results from the discovery GWAS.
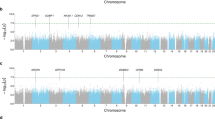
Although both primary and replication cohorts contained comparable numbers of white subjects, and had patients of similar mean age, the discovery cohort included a greater proportion of females (48% for Abbott trial 1 vs 32.4% for Abbott trial 2). In addition, the discovery cohort patients showed a modestly greater mean % change in FEV1 following 12-week zileuton administration (Table 1). Although the cohorts had high percentages of white subjects, the inclusion of non-white patients may have introduced population stratification. However, the genomic inflation factor values for these cohorts suggested that minimal stratification was present, and the quantile-quantile (QQ) plots of both GWAS were well behaved (Figure 1).

Quantile–quantile (Q–Q) plots of Abbott populations. Q–Q plots show results for Abbott trial 1 (discovery) (a) and Abbott trial 2 (replication) (b). Lambda (λ) indicates the genomic inflation factor values.
Montelukast populations (LOCCS and LODO)
The clinical outcomes and results have been reported elsewhere for the LOCCS and LODO trials.20, 21 LOCCS evaluated participants aged 15 years and older with physician-diagnosed asthma, who had been prescribed daily asthma medications for at least 1 year, had an FEV1 of 50% or more of predicted values and had poor asthma control as defined by a score of 1.5 or greater on the Asthma Control Questionnaire.20, 21 One hundred and sixty-six patients taking montelukast (5 or 10 mg daily) were evaluated over 16 weeks, with time to treatment failure as the primary phenotype outcome. Genotype and phenotype data from 210 patients, of which 69 patients were taking montelukast, were available for additional replication of the zileuton GWAS findings.
The LODO trial evaluated patients aged 6 years and older with physician-diagnosed asthma, FEV1 of 60% or greater than predicted, evidence of airway reversibility (defined as 12% or greater β-agonist reversibility using up to four puffs of albuterol within 2 years before enrollment or PC20 FEV1 methacholine (provocative concentration of methacholine producing a 20% fall in FEV1) of 8 mg ml−1 or less within 2 years of enrollment) and poor asthma control as defined by a score of 1.5 or greater on the Asthma Control Questionnaire.20, 21 The data evaluated from LODO included complete genotype and phenotype information from 122 patients, of which 64 patients taking montelukast were available for interrogation as an additional replication population.
This study evaluated data from LOCCS and LODO for the first 12 weeks following randomization to montelukast. Both study populations were used to replicate the results of the zileuton GWAS. The characteristics of LOCCS and LODO were comparable to each other, and to Abbott, although LODO demonstrated a lower % mean change in FEV1 as compared with the other cohorts (Table 1).
Phenotyping
For the zileuton discovery and replication GWAS, we evaluated the difference between the FEV1 during the placebo run-in period at baseline (randomization) and the FEV1 at 12 weeks on zileuton, as the primary outcome phenotype.18, 19 Similarly, for the LOCCS and LODO studies, we also evaluated 12-week ΔFEV1 from baseline (randomization) relative to montelukast administration. For both zileuton cohorts, the mean (±s.d.) FEV1% difference was similar (13.5±25.9 for Abbott trial 1 and 12.5±45.0 for Abbott trial 2) (Table 1).
Genotyping
Genome-wide genotyping of the Abbott patients was conducted using the Illumina HumanHap550 chip (Illumina, San Diego, CA, USA). LOCCS and LODO patients were genotyped using the Infinium HD Human610-Quad BeadChip (Illumina). The software PLINK v.1.07 [ref. 22] was used for quality control (QC) and statistical analysis of genotype data. For QC, SNPs with a study-wise missing data proportion above 0.05 were removed from the analysis. SNPs failing to meet Hardy–Weinberg equilibrium (P<0.0001), in addition to SNPs with >10% missing genotypes were also dropped from the analysis. A total of 542 562 SNPs from the Abbott trials passed QC and were included in the analysis. For LOCCS and LODO, a total of 545 934 SNPs passed QC. To assess population stratification in the primary analysis, the P-values were adjusted for genomic control using PLINK.
Statistical analysis
The zileuton GWAS evaluated 542 562 SNPs in a discovery cohort of 304 patients, of which 160 patients received zileuton and 144 patients received placebo (Abbott trial 1). The association of SNP genotypes with changes in FEV1 related to zileuton or montelukast administration was determined using linear regression (PLINK v.1.0.7). Regression models were adjusted for baseline FEV1, age, race and gender. To enrich for SNP associations related specifically to drug treatment response (i.e., represented true pharmacogenetic (drug x genotype) associations), we performed a primary GWAS that evaluated a gene–environment (G × E) interaction model to jointly evaluate SNP genotype by treatment group (zileuton or placebo) in all individuals in the cohort. Additive, dominant and recessive genetic models were evaluated using PLINK. SNP associations were considered significant if they met criteria for genome-wide significance (P<10−08). In addition, the top 50 SNPs, based on P-values, were carried forward for replication. For replication, SNPs that also met a threshold of P<0.05 in the replication populations, and possessed a shared direction of effect (as indicated by the signs of the β coefficient) in the discovery and replication cohorts, were included in the combined analyses (discovery+replication). Combined P-values were then calculated from the one-sided P-values of the replication populations using R version 2.15 software.23
Results
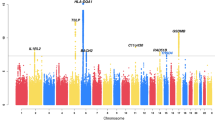
Figure 2 shows the discovery GWAS results. Although none of the SNPs in the discovery cohort achieved genome-wide significance, 10 of the 50 SNPs carried forward for replication approached genome-wide significance and were also at least nominally significant in the replication cohort; the replicated SNPs are shown in Table 2.

Results of the GWAS of zileuton response. Manhattan plots show GWAS results for the discovery cohort (Abbott trial 1) for additive (a) dominant (b) and recessive (c) genetic models. For each plot, the y axis represents –log10(P-values) for each SNP (gray and black dots), grouped by chromosomes 1–22 (x axis). Horizontal lines in the plots indicate the threshold for suggestive genome-wide significance (P=1 × 10−05).
In the combined analysis of the discovery and replication cohorts, one SNP, rs12436663, achieved genome-wide significance and was associated with worsening response to zileuton (that is, significant reduction in mean ΔFEV1) in both the discovery and replication cohorts (combined β=−1.85 L; combined P=6.28x10−08, recessive model; Table 2). Relative to the patients who carried at least one reference allele, patients who were homozygous for rs12436663 demonstrated a mean ΔFEV1 of −0.123 L for the homozygous rs12436663 ‘AA’ genotype vs mean ΔFEV1 of 0.233 L for individuals with either the ‘AG’ or ‘GG’ genotypes (P=0.03) (Figure 3). rs12436663 resides in an intronic region within KIAA0391, also called MRPP3, a gene that encodes a mitochondrial RNase P protein involved in post-translational modification of transfer RNA molecules (Figure 4).

rs12436663 genotype-dependent change in lung function. The boxplots show ΔFEV1 (L) grouped by rs12436663 genotype: variant genotype (‘AA’ (N=11 patients), left), heterozygous genotype (‘AG’ (N=151 patients), center) and reference genotype (‘GG’ (N=362 patients), right).

Regional association (LD) plot for rs12436663. The regional association plot was generated from the GWAS association data (discovery+replication) with Locus Zoom (http://csg.sph.umich.edu/locuszoom/),33 specifying a ±200-kb region from rs12436663 (diamond symbol) for all GWAS SNPs (circle symbols), and using the HapMap Phase II CEU version 18 (no LD) as the reference genome build.
To identify shared SNP associations for treatment responses related to leukotriene inhibition and leukotriene receptor antagonism, the replicated zileuton GWAS results were carried forward for an additional replication in the LOCCS and LODO patients who received montelukast. From this analysis, we identified one SNP, rs517020, that replicated in LOCCS and was associated with reduced responses to both zileuton and montelukast (Table 3). rs517020 is found in an intron of the gene glycosyltransferase 1 domain containing 1 (GLT1D1).
Discussion
This study is the first genome-wide analysis of the clinical response to leukotriene modifiers, and also provides additional evidence of shared pharmacogenetic loci for zileuton and montelukast. We conducted a discovery GWAS evaluating lung function following zileuton treatment, focusing on the interaction of zileuton treatment and genotype (G × E). We performed the G × E GWAS to enrich for associations that represented true pharmacogenetic effects, instead of more general associations with asthma. Although none of the SNPs achieved genome-wide significance in the discovery cohort, when the replicated SNP P-values from discovery and replication analyses were combined, we identified rs12436663, which met genome-wide significance. Furthermore, through replication of the zileuton GWAS SNP associations in the two montelukast treatment arms of the LOCCS and LODO trials, we identified an additional SNP, rs517020, associated with both zileuton treatment response and montelukast treatment response (in LOCCS).
rs12436663 resides in an intronic region of MRPP3, a.k.a. KIAA0391, a gene that has a crucial role in transfer RNA processing and maturation. Complex diseases associated with this gene include myocardial infarction, coronary artery disease, juvenile rheumatoid arthritis and psoriasis, all of which, like asthma, feature dysregulation in multiple immunological genes and pathways as causal mechanisms.24, 25, 26, 27 Multiple SNPs within the same chromosomal 14q13 region occupied by MRPP3 are also associated with cardiovascular disorders, and the region contains microsattelites that are implicated in autoimmune diseases.25 Although there is no direct evidence to suggest that MRPP3 is involved in asthma, variation within the MRPP3-encoding locus may potentially affect leukotriene synthesis and responses in asthma, presumably through altering post-transcriptional modification of transfer RNAs in leukotriene-producing cells. Leukotriene- and histamine-mediated allergic responses are induced through antigen binding to immunoglobulin E receptors on mast cells and basophils. Furthermore, the 14q region contains a cluster of genes (including MRPP3) that encode members of the ubiquitin–proteasome pathway, and markers within these genes are implicated in immunoglobulin E phenotypes and variation within immune response cascades. Consistent with this information, our results suggest that MRPP3 could represent a novel candidate gene for leukotriene modifier responses in asthma.
We also evaluated whether the SNPs that replicated in Abbott also replicated in LOCCS and LODO, that is, represented associations that were shared between the zileuton-treated and montelukast-treated cohorts, and identified a SNP that replicated in Abbott cohorts and LOCCS. This SNP, rs517020, was associated with worsening responses to both medications. rs517020 was present within an intronic region of GLT1D1, whose gene product transfers glycosyl groups such as galactose, N-acetylglucosamine and sialic acid to the HIV-1 protein, gp120.28, 29, 30, 31 Although GLT1D1 has unknown roles in asthma and asthma treatment response, as it was present within a predicted QTL region for allergic/atopic asthma it may have a role in the development or severity of asthma phenotypes, including reversible airflow obstruction.32 In addition, GLT1D1 may also contribute to LTM pharmacological response through its glycosyltransferase activity. For example, a wide range of bioactive lipids in the leukotriene pathway is modulated by glycosylation, and the activity of glycosyltransferases on these lipids could potentially affect multiple diverse functions throughout the body.
Although our study presents novel findings, these results should be interpreted in the context of some important limitations. First, the zileuton replication study population included individuals taking zileuton as ‘add on’ therapy to usual care, whereas the primary zileuton population included individuals on zileuton monotherapy. Although this might bias towards the null, it does not detract from the associations noted. Second, although these associations are generalizable to the overall treatment pathway, it is difficult to discern whether any loci in the zileuton-treated populations that did not replicate in the montelukast-treated populations represent false positives in the initial population, or are actually specific to zileuton response. Achieving replication in the montelukast populations boosts confidence in the generalizability of these findings towards LTM responses. Third, we did not use imputed GWAS data in our initial GWAS, which may have identified additional associations or strengthened existing ones. Fourth, because of the small sample size, replication in a larger cohort would be helpful to validate these findings. Finally, although multiple loci related to LTM response were identified through candidate gene studies (in particular, seven SNPs within ALOX5, ABCC1 and LTC4S11), we were unable to replicate these SNPs in either primary or discovery GWAS cohorts. Five of the seven SNPs were not genotyped in these populations (due to differences in genotyping platforms used), and the remaining two did not achieve nominally significant associations with ΔFEV1 (data not shown). Additional studies are necessary to ascertain the involvement of MLLT3 and GLT1D1 in asthma, and leukotriene modifier responses.
Evidence from an increasing number of genetic association studies implicates genes within leukotriene production and metabolism pathways as involved in clinical response to zileuton. Our results provide insight into both unique and shared clinical effects of LTMs in treatment of asthma, and represent a potential mechanism for the responsiveness to leukotriene antagonists in asthma patients.
References
Salmon JA, Higgs GA . Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull 1987; 43: 285–296.
Hammarström S . Biosynthesis and metabolism of leukotrienes. Monogr Allergy 1983; 18: 265–271.
Hammarström S . Leukotrienes. Annu Rev Biochem 1983; 52: 355–377.
Zileuton for asthma. Med Lett Drugs Ther 1997; 39: 18–19.
Bell RL, Young PR, Albert D, Lanni C, Summers JB, Brooks DW et al. The discovery and development of zileuton: an orally active 5-lipoxygenase inhibitor. Int J Immunopharmacol 1992; 14: 505–510.
DuBuske LM, Grossman J, Dubé LM, Swanson LJ, Lancaster JF . Randomized trial of zileuton in patients with moderate asthma: effect of reduced dosing frequency and amounts on pulmonary function and asthma symptoms. Zileuton Study Group. Am J Manag Care 1997; 3: 633–640.
Drazen JM, Yandava CN, Dubé L, Szczerback N, Hippensteel R, Pillari A et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat Genet 1999; 22: 168–170.
Drazen JM, Silverman EK, Lee TH . Heterogeneity of therapeutic responses in asthma. Br Med Bull 2000; 56: 1054–1070.
Lima JJ . Treatment heterogeneity in asthma: genetics of response to leukotriene modifiers. Mol Diagn Ther 2007; 11: 97–104.
Lima JJ, Zhang S, Grant A, Shao L, Tantisira KG, Allayee H et al. Influence of leukotriene pathway polymorphisms on response to montelukast in asthma. Am J Respir Crit Care Med 2006; 173: 379–385.
Tantisira KG, Lima J, Sylvia J, Klanderman B, Weiss ST . 5-lipoxygenase pharmacogenetics in asthma: overlap with Cys-leukotriene receptor antagonist loci. Pharmacogenet Genomics 2009; 19: 244–247.
Duroudier NP, Tulah AS, Sayers I . Leukotriene pathway genetics and pharmacogenetics in allergy. Allergy 2009; 64: 823–839.
Lima JJ, Blake KV, Tantisira KG, Weiss ST . Pharmacogenetics of asthma. Curr Opin Pulm Med 2009; 15: 57–62.
Mougey EB, Feng H, Castro M, Irvin CG, Lima JJ . Absorption of montelukast is transporter mediated: a common variant of OATP2B1 is associated with reduced plasma concentrations and poor response. Pharmacogenet Genomics 2009; 19: 129–138.
Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med 2011; 365: 1173–1183.
Tantisira KG, Damask A, Szefler SJ, Schuemann B, Markezich A, Su J et al. Genome-wide association identifies the T gene as a novel asthma pharmacogenetic locus. Am J Respir Crit Care Med 2012; 185: 1286–1291.
Tamari M, Tomita K, Hirota T . Genome-wide association studies of asthma. Allergol Int 2011; 60: 247–252.
Nelson H, Kemp J, Berger W, Corren J, Casale T, Dube L et al. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of moderate persistent asthma: a 3-month randomized controlled study. Ann Allergy Asthma Immunol 2007; 99: 178–184.
Wenzel S, Busse W, Calhoun W, Panettieri R, Peters-Golden M, Dube L et al. The safety and efficacy of zileuton controlled-release tablets as adjunctive therapy to usual care in the treatment of moderate persistent asthma: a 6-month randomized controlled study. J Asthma 2007; 44: 305–310.
American Lung Association Asthma Clinical Research Centers. Clinical trial of low-dose theophylline and montelukast in patients with poorly controlled asthma. Am J Respir Crit Care Med 2007; 175: 235–242.
Peters SP, Anthonisen N, Castro M, Holbrook JT, Irvin CG, Smith LJ et al. Randomized comparison of strategies for reducing treatment in mild persistent asthma. N Engl J Med 2007; 356: 2027–2039.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Liptak T . On the combination of independent tests. Magyar Tud Akad Mat Kutato Int Kozl 1958; 3: 171–197.
Sjakste T, Trapina I, Rumba-Rozenfelde I, Lunin R, Sugoka O, Sjakste N . Identification of a novel candidate locus for juvenile idiopathic arthritis at 14q13.2 in the Latvian population by association analysis with microsatellite markers. DNA Cell Biol 2010; 29: 543–551.
Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet 2012; 90: 636–647.
Stuart PE, Nair RP, Ellinghaus E, Ding J, Tejasvi T, Gudjonsson JE et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat Genet 2010; 42: 1000–1004.
Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W . RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008; 135: 462–474.
Kozarsky K, Penman M, Basiripour L, Haseltine W, Sodroski J, Krieger M . Glycosylation and processing of the human immunodeficiency virus type 1 envelope protein. J Acquir Immune Defic Syndr 1989; 2: 163–169.
Mizuochi T, Matthews TJ, Kato M, Hamako J, Titani K, Solomon J et al. Diversity of oligosaccharide structures on the envelope glycoprotein gp 120 of human immunodeficiency virus 1 from the lymphoblastoid cell line H9. Presence of complex-type oligosaccharides with bisecting N-acetylglucosamine residues. J Biol Chem 1990; 265: 8519–8524.
Shimizu H, Tsuchie H, Honma H, Yoshida K, Tsuruoka T, Ushijima H et al. Effect of N-(3-phenyl-2-propenyl)-1-deoxynojirimycin on the lectin binding to HIV-1 glycoproteins. Jpn J Med Sci Biol 1990; 43: 75–87.
Yeh JC, Seals JR, Murphy CI, van Halbeek H, Cummings RD . Site-specific N-glycosylation and oligosaccharide structures of recombinant HIV-1 gp120 derived from a baculovirus expression system. Biochemistry 1993; 32: 11087–11099.
Laulederkind SJ, Hayman GT, Wang SJ, Smith JR, Lowry TF, Nigam R et al. The Rat Genome Database 2013—data, tools and users. Brief Bioinform 2013; 14: 520–526.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–2337.
Acknowledgements
We thank collaborators and research staff at the many study sites, as well as the study participants, for their generous contributions. We also thank Dr Jeffrey Drazen for his significant assistance with this study. This study is supported by the National Institutes of Health - R01 HL092197, U01 HL65899 and R01 NR013391. This study is also supported by the NIH Pharmacogenomics Research Network (PGRN)—RIKEN Center for Genomic Medicine (CGM) Global Alliance and by funding from the BioBank Japan project that was supported by the Ministry of Education, Culture, Sports, Sciences and Technology of the Japanese government. AD is supported by NHLBI K12 HL120004. We acknowledge the American Lung Association (ALA) and the ALA’s Asthma Clinical Research Centers investigators and research teams for the use of LOCCS and LODO data, with additional funding from HL071394 and HL074755 from the NHLBI, and Nemours Children's’ Clinic. GlaxoSmithKline supported the conduct of the LOCCS trial by an unrestricted grant to the ALA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Dahlin, A., Litonjua, A., Irvin, C. et al. Genome-wide association study of leukotriene modifier response in asthma. Pharmacogenomics J 16, 151–157 (2016). https://doi.org/10.1038/tpj.2015.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2015.34
This article is cited by
-
The phosphatidylinositide 3-kinase (PI3K) signaling pathway is a determinant of zileuton response in adults with asthma
The Pharmacogenomics Journal (2018)
