
Abstract
Variation in obsessive–compulsive symptoms (OCS) has a heritable basis, with genetic association studies starting to yield the first suggestive findings. We contribute to insights into the genetic basis of OCS by performing an extensive series of genetic analyses in a homogeneous, population-based sample from the Netherlands. First, phenotypic and genetic longitudinal correlations over a 6-year period were estimated by modeling OCS data from twins and siblings. Second, polygenic risk scores (PRS) for 6931 subjects with genotype and OCS data were calculated based on meta-analysis results from IOCDF-GC, to investigate their predictive value. Third, the contribution of measured single nucleotide polymorphisms (SNPs) to the heritability was estimated using random-effects modeling. Last, we performed an exploratory genome-wide association study (GWAS) of OCS, testing for SNP- and for gene-based associations. Stability in OCS (test–retest correlation 0.63) was mainly explained by genetic stability. The PRS based on clinical samples predicted OCS in our population-based twin-family sample. SNP-based heritability was estimated at 14%. GWAS revealed one SNP (rs8100480), located within the MEF2BNB gene, associated with OCS (P=2.56 × 10−8). Additional gene-based testing resulted in four significantly associated genes, which are located in the same chromosomal region on chromosome 19p13.11: MEF2BNB, RFXANK, MEF2BNB-MEF2B and MEF2B. Thus, common genetic variants explained a significant proportion of OCS trait variation. Genes significantly associated with OCS are expressed in the brain and involved in development and control of immune system functions (RFXANK) and regulation of gene expression of muscle-specific genes (MEF2BNB). MEF2BNB also showed a suggestive association with OCD in an independent case–control study, suggesting a role for this gene in the development of OCS.
Similar content being viewed by others
Introduction
Obsessive–compulsive disorder (OCD) is a neuropsychiatric disorder characterized by recurrent, persistent and intrusive anxiety-provoking thoughts or images (obsessions) and subsequent repetitive behaviors (compulsions).1 The lifetime prevalence of OCD has been estimated between 0.5 and 2.0%,1 and among all anxiety disorders, it is known as a major cause of social impairment and a leading cause of non-fatal disease burden worldwide.2
It is clear that genetic factors are important in the etiology of obsessive–compulsive symptoms (OCS), with heritability estimated ~40%.3, 4 These results did not vary with sex or symptom severity.3 Consistent with what is expected for individual single nucleotide polymorphism (SNP) effect sizes in highly polygenic traits, the first molecular association studies for OCD have not identified large effect variants.5, 6, 7 Taylor et al.7 performed a comprehensive meta-analysis of genetic association studies in OCD, including 113 relevant studies. Their main meta-analysis showed that OCD was associated with serotonin-related polymorphisms (5-HTTLPR and HTR2A) and, in males only, with polymorphisms involved in catecholamine modulation (COMT and MAOA). A secondary meta-analysis by the same group, which targeted polymorphisms that were investigated in fewer than 5 datasets, identified another 18 polymorphisms with significant odds ratios. These polymorphisms might be useful candidates for further investigation, although most results were based on candidate gene studies and must be treated with care due to the possible confounding effects of population stratification.
The international OCD foundation Genetics Collaborative (IOCDF-GC; Stewart et al.),5, 7 conducted an ancestry-stratified case–control genome-wide association (GWA) analysis, in 1465 cases, 5557 ancestry-matched controls and 400 trios, consisting of 1 affected offspring with 2 parents. The trio analysis revealed a significant SNP near the BTBD3 gene (rs6131295). However, this variant lost genome-wide significance when meta-analyzed with the case–control data. The meta-analysis showed a significant enrichment of methylation quantitative trait loci (P<0.001) and frontal lobe expression for the top-ranked SNPs (P<0.01).5 Recently, a second multinational collaboration (OCD Collaborative Genetics Association Study (OCGAS); Mattheisen et al.).8 performed a GWA study (GWAS) in 1598 patients and 3473 controls. The smallest P-value was observed for a marker near the PTPRD gene on chromosome 9 with P=4.13 × 10−7.8 Gene-based testing for associations at the gene instead of SNP level, revealed a significant association of IQCK, C16orf88 and OFC11.8 These findings await replication.
With this study, we aim to gain further insights into the genetic basis of OCS by performing a series of analyses in a homogeneous, population-based sample of twin families, registered with the Netherlands Twin Registry (NTR). Well-phenotyped population cohorts may contribute to the understanding of the architecture of common complex disorders. We first performed genetic structural equation modeling (SEM)9 to estimate twin–twin and twin–sibling correlations and heritability for OCS in adults as measured with the Padua Inventory Revised Abbreviated (PI-R-ABBR).10 Information on the PI-R-ABBR was available for two time points (2002 and 2008), which allowed calculation of the stability of OCS across a 6-year period. The long-term stability of the phenotype puts an upper limit to heritability—that is, reveals the proportion of total variation across time that is due to differences among individuals, and puts genetic association studies into perspective. Next, we investigated whether polygenic risk scores (PRS) based on a GWA analysis of clinical OCD cases5 significantly predict OCS in our population-based sample. If so, this indicates genetic overlap between the two types of samples, and suggests that future GWA studies can benefit from combining both population-based and case–control samples. We estimated the proportion of phenotypic variance explained by all autosomal SNPs using the Genome-wide Complex Trait Analysis (GCTA) software (Brisbane, QLD, Australia) in a sample of related and unrelated individuals. Further, we performed an explorative GWAS on OC symptom scores from 6931 subjects and entered the GWAS output into a gene-based analysis, to test for associations at the gene rather than the single SNP level.11
Materials and methods
Participants and measures
Study data were collected in participants of the NTR.12, 13 We analyzed data collected in 2002 and 2008 using the PI-R-ABBR.14 The total sample with phenotype data from either the 2002 and/or 2008 data collection contained 20 376 individuals from 7812 families. All individuals with information from at least one survey were included in the analysis. A total of 10 134 individuals had sum scores available for the PI-R-ABBR collected in 2002 and 15 720 individuals had sum scores for the PI-R-ABBR collected in 2008. Longitudinal data were available for 5478 individuals. The distribution of the OC symptom scores, from the PI-R-ABBR collected in 2002 and 2008, is provided in Supplementary Figure 1.
For more information on the PI-R-ABBR OCS measures, please see Supplementary Methods ‘participants and measures’.
On 6931 subjects (twins, their siblings, parents and spouses), phenotypic and genetic data were available. These subjects were entered into the genetic analyses (PRS, GWAS). For GCTA analysis, from these 6931, 6881 subjects were included in the Genetic Relationship Matrix. If a subject filled out both surveys we choose to enter the score of the last survey into GWAS and GCTA. Table 1 gives an overview of subjects per analysis and their demographics (see also Supplementary Methods ‘Subjects entered into different genetic analyses’ for more details and the study by Zilhão et al.).15
This study has been approved by the Central Ethics Committee on Research involving human subjects of the VU University Amsterdam. Informed Consent was obtained from all subjects.
Genotyping and imputation
DNA was extracted from either blood or buccal cell samples that were collected for various projects done by the NTR.12, 13 A total of 31,265,038 SNPs were included in the analysis using the 1000 Genome phase 1 version 3 as a reference panel. For further details on genotyping, quality control and imputation methods see Supplementary Methods ‘genotyping and imputation’.
Heritability estimates from SEM
To estimate the total contribution of genetic factors to trait variance and to the longitudinal covariance, the resemblance among twins and siblings was compared. Monozygotic (MZ) twins share (nearly) all their genes, whereas dizygotic (DZ) twins, just like non-twin siblings, share on average half of their genetic variation. In quantitative genetics, this information is used to decompose the total variance of a trait into additive genetic (A), non-additive genetic or dominance (D), and unshared environmental variance (E). The greater the phenotypic similarity between MZ twins, when compared with DZ twins and non-twin siblings, the more of the variance of the trait is attributed to genetic factors. Genetic analyses were carried out by SEM as implemented in OpenMx.9 Further details on SEM analysis may be found in Supplementary Methods ‘SEM methods’.
Polygenic risk scores
To examine shared polygenic risk at an aggregate level between two independent GWAS samples, we used genetic risk score profiling as described by Purcell et al.16 GWAS results (SNP, effect allele, effect size as represented by a Beta-value and P-value) obtained from the analysis in the European case–control sample by Stewart et al.5 were used as a discovery dataset for calculating PRS within our NTR target sample in Plink. PRS were then regressed against the OCS scores from the NTR dataset (n=6931) to calculate the proportion of variance in this target set explained by PRS obtained from the discovery set, with 15 statistical cutoffs for SNP inclusion in the score (cutoffs: P<0.00001, P<0.0001, P<0.001, P<0.01, P<0.05, P<0.1, P<0.2, P<0.3, P<0.4, P<0.5, P<0.6, P<0.7, P<0.8, P<0.9, P⩽1). To correct for family relatedness in the NTR sample, generalized estimating equations in SPSS with the exchangeable and robust function were used.17 Sex, age, principal components to correct for the population substructure18 and genotyping platform were included as covariates. As an additional test, we regressed the PRS on a non-psychiatric, and OCS-uncorrelated trait (height), from the NTR (n=6715); we sought to rule out a possible spurious association consequent of incomplete correction for population stratification and/or cryptic relatedness between the discovery and target datasets. Finally, to test for concordance of effect directions across both datasets, we performed Fisher’s exact statistical test using the online-based application SECA (SNP effect concordance analysis)—http://neurogenetics.qimrberghofer.edu.au/SECA/.19 For 12 subsets of SNPs with P<(0.01, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.8, 0.9, 1.0) in both datasets, Fisher’s exact tests were performed to evaluate if there is an excess of SNPs in the first dataset (European case–control study) with same direction of effect in the NTR dataset across the total 144 SNP subsets. An empirical P-value was generated by permutations (1000) for observing the number of SNP subsets with nominally significant concordance.
Estimations of variance explained by common SNPs (GCTA)
The variance of OCS explained by measured and imputed SNPs was estimated with GCTA using the Restricted Maximum Likelihood procedure.20, 21 This method gives insight into the contribution to the additive genetic variance of all genotyped and imputed SNPs. This provides an upper bound on the variance that can be explained by the set of SNPs. To analyze all available data we followed the method proposed by Zaitlen et al,22 in which the SNP-based and kinship-based heritability can be estimated simultaneously. For further details on the method followed and quality control, refer to Supplementary Methods ‘estimations of variance explained by common SNPs (GCTA)’.
GWAS
GWA analysis was conducted with linear regression under an additive model with adjustment for age, age-squared, principal components of genetic ancestry, genotyping platform and sex. SNPs with values of P<5.00E−08 were declared genome-wide significant. For further details on this analysis, refer to Supplementary Methods ‘GWAS’.
Gene-based analysis
GATES, as implemented in the open-source tool Knowledge-Based Mining System for Genome-Wide Genetic Studies (KGG, version 3.0, Hong Kong, China), was used to perform a gene-based genome-wide analysis.11 GATES employs an extension of the Simes procedure23 to assess the significance of a statistical association at the gene level, by combining the individual genotype–phenotype association tests applied at each single marker. In short, it sums all the individual SNP P-values, available from GWAS summary data, within a gene to output a gene-based P-value. The effective number of independent P-values is given by appropriately controlling for the Linkage Disequilibrium structure on the specified SNPs. SNPs were allocated to genes including gene boundaries of ±5 kb from the 5′and 3′ untranslated region. To correct for multiple testing false discovery rate was set at Q<0.05.
Results
Demographics
For subjects who completed the PI-R-ABBR in 2002 and in 2008 the average age was 33.0 (s.d.=11.5), and 34.7 (s.d.=14.6). Retest stability for OCS scores over a time span of ~6 years was 0.63. The effect of age on OCS scores was a drop of 0.03 per year for both PI-R-ABBR collected in 2002 and 2008.
Genetic modeling (SEM)
MZ, DZ/sibling, test–retest and cross-correlations are summarized in Table 2. Twin correlations for MZ males and MZ females were equal, as were twin/sibling correlations for DZ males, DZ females, DZ opposite-sex twins and siblings. This indicates that there is no evidence for qualitative sex differences in the heritability of OCS and that to a large extent the same genes influence these symptoms in males and females. Twin correlations were more than twice as large in MZ as compared with DZ/sib pairs, indicating that phenotypic similarity is predominantly accounted for by genetic effects rather than shared environment. The same pattern was observed for cross-twin–cross-time correlations, indicating that the observed stability is also mainly caused by genetic factors. SEM showed a significant heritability (P<1.0 × 10−10) for OCS at both time points of 0.42 (95% confidence interval (CI) PI-R-ABBR 2002=0.371–0.467 for, and 95% CI PI-R-ABBR 2008 =0.379–0.456). The estimation of the bivariate (broad sense) heritability found that 56% (95% CI=0.497–0.619) of the stability of OC symptom was due to genetic factors (both additive and dominant components), and the longitudinal additive genetic correlation, that is, the degree of overlap between additive genetic influences at both time points was 0.58.
PRS regressed on PI-R-ABBR sum scores
The proportions of variance of OCS in the NTR sample explained by PRS obtained from the discovery set (European case control sample Stewart et al.),5 using the 15 statistical cutoffs for SNP inclusion in the score, are summarized in Figure 1. Results show that when including more SNPs in the analyses an increasing amount of variance in OCS is explained, reaching a plateau of 0.2% explained variance (P<0.001) at PRS12 (includes all SNPs from Stewart et al.5 with a P<0.7 (N=4,288,152)). Supplementary Figure 2 provides the results obtained when using height as the outcome phenotype for the target set for the same PRS. For the same 15 statistical cutoffs, no significant result was observed (P<0.01), that is, the PRS did not explain any variance in height, hence excluding the confounding effect due to cryptic relatedness across sample sets and possible residual genetic stratification effects present in both populations. Finally, Supplementary Figure 3 presents the results in a heat map plot from analysis of concordance using SECA. The permuted P-value for the number of SNP subsets nominally significant was P=0.002 thus indicating significant concordance of genetic risk across the datasets.

Proportion of variance in OC symptom scores, as measured in the NTR sample, explained by polygenic scores (PRS) obtained from European case–control sample by Stewart et al.5, with a range of 15 statistical cutoffs for SNP inclusion in the score (PRS1; P<0.00001, PRS2; P<0.0001, PRS3; P<0.001, PRS4; P<0.01, PRS5; P<0.05, PRS6; P<0.1, PRS7; P<0.2, PRS8; P<0.3, PRS9; P<0.4, PRS10; P<0.5, PRS11; P<0.6, PRS12; P<0.7, PRS13; P<0.8, PRS14; P<0.9, PRS15; P⩽1). *P<0.05; **P<0.01. NTR, Netherlands Twin Registry; OC, obsessive–compulsive.
SNP-based heritability
GCTA results showed a significant SNP-based heritability estimate of 0.14 (s.e.=0.05, P=0.003), and a total narrow-sense heritability of 0.34 (s.e.=0.02, P<0.001). Thus 14% of the total phenotypic variance in OCS can be accounted for by the SNPs in the genotyping platform, and the total set of SNPs included in an additive genetic model account for 34% of the total heritability.
GWAs analysis
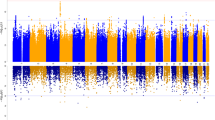
Top associated variants in GWAS analysis (top 20 SNPs) are summarized in Table 3 (a more comprehensive overview of these results is present in Supplementary Table 1). The Manhattan plot, showing the −log(P) plotted against genomic location, and QQ plot of observed versus expected −log(P) statistics for the OC symptom GWAS, are illustrated in Figure 2 and Supplementary Figure 4, respectively. Of the top associated variants, one SNP, rs8100480 (19299079, 19p13.11 (hg19), P=2.56 × 10−8), exceeded the threshold for genome-wide significance and showed a positive association with OCS. This SNP is located within the MEF2BNB gene. A more detailed look into this region is provided by the regional association plot in Supplementary Figure 5.

Manhattan plots of all genotyped single nucleotide polymorphisms (SNPs). Red and blue lines indicate significance thresholds of 5.00E−08 and 1.00E−05, respectively.
Further, we searched in our results whether they replicate top SNPs reported by Stewart et al.5 and Mattheisen et al.8 Supplementary Table 2 summarizes results for the strongest associated GWAS variants from Stewart et al.5 From their table of 43 SNPs (with P<1.0 × 10−5), 16 were found to be independent.8 None of these SNPs were significantly replicated in our sample when correcting for multiple comparison (0.05/16=0.003). However, a trend (P=0.0049) for replication was found for rs4868342, located on chromosome 5. This SNP is located within the HMP19 gene.
Supplementary Table 3 summarizes results for the strongest associated GWAS variants from Mattheisen et al.8 None of the 32 suggestive associations (with P<1.0 × 10−4) were replicated in our sample after correcting for multiple comparison (0.05/32=0.001). Neither were the significant results obtained in our study replicated in the Stewart sample (data not shown).
Gene-based analysis
A total of 2,644,694 SNPs were mapped to 22 759 genes. The QQ plots with the observed versus expected –log(P) of the association tests are presented in Supplementary Figure 6. Table 4 depicts all the genes with a significant association. Although these are all nominal significant, the Benjamin–Hochberg procedure was set to control for the q<0.05.23 After correction, four genes remained significant, the regulatory factor X-associated ankyrin-containing protein (RFXANK), the myocyte enhancer factor 2B (MEF2B), the MEF2B neighbor (MEF2BNB) and the MEF2BNB-MEF2B read through (MEF2BNB-MEF2B). All these genes are located in the same chromosomal region (19p13.11), and share the SNP with the lowest P-value annotated to the gene (rs8100480, P=2.56E−08). The Manhattan plot for the gene association P-values is present in Supplementary Figure 7.
Discussion
This study aimed at getting a better insight into the genetic basis of OCS in a population-based sample.
First, in line with previous studies, we estimated the heritability for OCS at 0.42.4 Stability of OCS over a 6-year time period was 0.63, and cross-twin–cross-time correlations were found to be twice as high in MZ compared with DZ/sib pairs, indicating that the observed stability is mainly caused by genetic factors. Bivariate analyses showed a longitudinal genetic correlation of 0.58. Second, polygenic scores based on a GWA analysis of clinical OCD cases significantly predicted OCS in the independent population-based sample. Analysis of concordance of results indicate that the genetic risk between the two datasets are concordant. These results indicate a genetic overlap between OCD assessed as a categorical disorder and OCS assessed as a continuous trait. Furthermore, the fact that the proportion of variance explained increases as more SNPs are included in the analysis indicates that many small effect variants contribute to the trait. Therefore, future GWA studies can benefit from the inclusion of both population-based and case–control studies, and by analyzing OCD as a quantitative rather than a categorical trait.
Third, we estimated the SNP-based heritability. We were able to find significant explained variance at 0.34. More importantly we were able to partition the heritability into two components, revealing a SNP heritability of 0.14 and the heritability not accounted for by SNPs at ~0.20. These results corroborate well with our findings from our twin model (broad heritability at 0.42). In a previous study by Davis et al.24 using a case–control design, the heritability of OCD was estimated at 0.37. Although our results do not completely agree, different study-designs can render somewhat different estimates, as was shown by Golan et al.25
Fourth, the GWAS performed on the continuous OC symptom scores resulted in one significant SNP (rs8100480), which is located in the MEF2BNB-MEF2B gene. In addition, we sought to calculate gene-based P-values, to determine whether there are genes with associated SNPs, which can collectively achieve statistical significance. Implementing a gene-based analysis as a follow-up complementary analysis is of additional value over traditional GWAS results. Gene-based analysis takes the number of SNPs per gene and gene size into account, considering genes as functional units and informing on the underlying genetic architecture of the phenotype. Furthermore, since for most psychiatric disorders we do not expect large effect causal variants, replication analysis from underpowered GWAS would not reflect these findings. Gene-based tests, by providing an aggregated analysis, may successfully capture and replicate those aggregate effects. We found significant associations for four genes. RFXANK encodes a protein that belongs to the MHC class II molecules, which has an important role in the development and control of the immune system. Bare lymphocyte syndrome 2, an immunodeficiency disorder, has been linked with mutations in this gene. Although RFXANK has not been identified in OCD previously, several lines of research have indicated a role of immune system alterations and of immune system genes, including TNFα, and SLC1A1, in combination with cerebral immunopathological reactions to among others group A ß-hemolytic streptococcus infections26 and Bornea virus infections27, 28 in the onset (and expression) of OCD. This has led to the concept of ‘PANDAS’ and ‘non-PANDAS’ OCD.29, 30, 31 However, very little research has been performed on direct gene–gene interactions/pathways or on gene–environment interactions to better understand immunopathological pathways related to OCS. MEF2B is a protein-coding gene belonging to the DNA-binding protein family MADS/MEF2, that regulates gene expression, specifically in the smooth muscle tissue. Both MEF2BNB and MEF2BNB-MEF2B are closely related to MEF2B, and have mostly regulatory functions. Additional support for a relation between MEF2BNB and OCD comes from a gene-based analysis in a recently published GWAS of OCD8 where the gene was ranked 21st of the 21 567 genes tested (gene-based P=8.09E−04, not significant after correction for multiple testing). All four genes are overlapping, and span a region of 56302 bp located in the 19p13.11 cytogenetic band, on the short arm of chromosome 19. Perhaps the best interpretation for these results, therefore, is in the implication of this genomic location (19p13.11) as a susceptibility locus for OCS. Several SNP–trait associations have been linked to this locus. For example, rs1064395 (NCAN) has been reported as a susceptibility factor for bipolar disorder in a GWAS. The NCAN gene is located just 10095 bp at 5′ from RFXANK, and is one of the few genetic variants that has been genome-wide replicated as a risk factor in both bipolar disorder and schizophrenia.32, 33 In addition, a recent study focusing on the cortical thickness and folding in schizophrenia patients found evidence for association of the NCAN genetic variant in the occipital and prefrontal cortex.34 The SNP rs874628 (MPV17L2 gene), located in this locus, was implicated in multiple sclerosis, an inflammatory disease with disruptions in the nervous system.35
Overall, our results, combined with previous genetic studies in OCD, suggest a possible role for the 19p13.11 region (MEF2BNB gene) in OCS. It might be of interest for future genetic studies to investigate this area in association with OCD into depth. Further, our data shows that well-phenotyped population cohorts could contribute to the understanding of the underlying architecture of common complex disorders such as OCS, and that these partly overlap with results from case–control studies. Therefore, future studies could benefit from combining case–control and population-based samples.
References
American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorder: DSM-IV, 4th edn, American Psychiatric Association: Washington, DC, 1994.
Ayuso Mateos JL . Global Burden of Obsessive-compulsive Disorder in the Year 2000. World Health Organization World Health Statistics: Geneva, Switzerland, 2006.
Taylor S . Etiology of obsessions and compulsions: a meta-analysis and narrative review of twin studies. Clin Psychol Rev 2011; 31: 1361–1372.
Mathews CA, Delucchi K, Cath DC, Willemsen G, Boomsma DI . Partitioning the etiology of hoarding and obsessive-compulsive symptoms. Psychol Med 2014; 44: 2867–2876.
Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry 2013; 18: 788–798.
Pauls DL . The genetics of obsessive-compulsive disorder: a review. Dialogues Clin Neurosci 2010; 12: 149–163.
Taylor S . Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol Psychiatry 2013; 18: 799–805.
Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry 2014; 20: 337–344.
Boker S, Neale M, Maes H, Wilde M, Spiegel M, Brick T et al. OpenMx: an open source extended structural equation modeling framework. Psychometrika 2011; 76: 306–317.
den Braber A, van 't Ent D, Boomsma D, Cath D, Veltman D, de Geus E . Brain activation during cognitive planning in twins discordant or concordant for obsessive-compulsive symptoms. Brain 2010; 133: 3123–3140.
Li MX, Gui HS, Kwan JS, Sham PC . GATES: a rapid and powerful gene-based association test using extended Simes procedure. Am J Hum Genet 2011; 88: 283–293.
Boomsma DI, de Geus EJ, Vink JM, Stubbe JH, Distel MA, Hottenga JJ et al. Netherlands Twin Register: from twins to twin families. Twin Res Hum Genet 2006; 9: 849–857.
Willemsen G, Vink JM, Abdellaoui A, den BA, van Beek JH, Draisma HH et al. The Adult Netherlands Twin Register: twenty-five years of survey and biological data collection. Twin Res Hum Genet 2013; 16: 271–281.
Cath DC, van Grootheest DS, Willemsen G, van Oppen P, Boomsma DI . Environmental factors in obsessive-compulsive behavior: evidence from discordant and concordant monozygotic twins. Behav Genet 2008; 38: 108–120.
Zilhao NR, Smit DJ, den BA, Dolan CV, Willemsen G, Boomsma DI et al. Genetic and environmental contributions to stability in adult obsessive compulsive behavior. Twin Res Hum Genet 2015; 18: 52–60.
Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Minica CC, Dolan CV, Kampert MM, Boomsma DI, Vink JM . Sandwich corrected standard errors in family-based genome-wide association studies. Eur J Hum Genet 2014; 23: 388–394.
Abdellaoui A, Hottenga JJ, de KP, Nivard MG, Xiao X, Scheet P et al. Population structure, migration, and diversifying selection in the Netherlands. Eur J Hum Genet 2013; 21: 1277–1285.
Nyholt DR . SECA: SNP effect concordance analysis using genome-wide association summary results. Bioinformatics 2014; 30: 2086–2088.
Yang J, Lee SH, Goddard ME, Visscher PM . Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations. Methods Mol Biol 2013; 1019: 215–236.
Yang J, Lee SH, Goddard ME, Visscher PM . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S et al. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 2013; 9: e1003520.
Benjamini Y, Hochberg Y . Multiple hypotheses testing with weights. Scand J Stat 1997; 24: 407–418.
Davis LK, Yu D, Keenan CL, Gamazon ER, Konkashbaev AI, Derks EM et al. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet 2013; 9: e1003864.
Golan D, Lander ES, Rosset S . Measuring missing heritability: inferring the contribution of common variants. Proc Natl Acad Sci USA 2014; 111: E5272–E5281.
Swedo SE, Leonard HL, Garvey M, Mittleman B, Allen AJ, Perlmutter S et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry 1998; 155: 264–271.
Marazziti D, Presta S, Pfanner C, Gemignani A, Rossi A, Sbrana S et al. Immunological alterations in adult obsessive-compulsive disorder. Biol Psychiatry 1999; 46: 810–814.
Marazziti D, Mungai F, Masala I, Baroni S, Vivarelli L, Ambrogi F et al. Normalisation of immune cell imbalance after pharmacological treatments of patients suffering from obsessive-compulsive disorder. J Psychopharmacol 2009; 23: 567–573.
Da Rocha FF, Correa H, Teixeira AL . Obsessive-compulsive disorder and immunology: a review. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32: 1139–1146.
Cappi C, Muniz RK, Sampaio AS, Cordeiro Q, Brentani H, Palacios SA et al. Association study between functional polymorphisms in the TNF-alpha gene and obsessive-compulsive disorder. Arq Neuropsiquiatr 2012; 70: 87–90.
Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D . The glutamate-based genetic immune hypothesis in obsessive-compulsive disorder. An integrative approach from genes to symptoms. Neuroscience 2010; 165: 408–417.
Cichon S, Muhleisen TW, Degenhardt FA, Mattheisen M, Miro X, Strohmaier J et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet 2011; 88: 372–381.
Muhleisen TW, Mattheisen M, Strohmaier J, Degenhardt F, Priebe L, Schultz CC et al. Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr Res 2012; 138: 69–73.
Schultz CC, Muhleisen TW, Nenadic I, Koch K, Wagner G, Schachtzabel C et al. Common variation in NCAN, a risk factor for bipolar disorder and schizophrenia, influences local cortical folding in schizophrenia. Psychol Med 2014; 44: 811–820.
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219.
Acknowledgements
We are grateful to the twins and siblings for their participation. This project has been financed by FP7-People-2012-ITN, project: TS-Eurotrain, grant number 316978; BBR Foundation (NARSAD) 21668; ZonMW (Addiction) 31160008 and European Research Council (ERC-230374). Part of the genotyping was funded by the Genetic Association Information Network (GAIN) of the Foundation for the National Institutes of Health, Rutgers University Cell and DNA Repository (NIMH U24 MH068457-06), the Avera Institute, Sioux Falls, South Dakota (USA) and the National Institutes of Health (NIH R01 HD042157-01A1, MH081802, Grand Opportunity grants 1RC2 MH089951 and 1RC2 MH089995). We acknowledge the International OCD Foundation Genetics Collaborative (IOCDF-GC) for making available the meta-analysis results from Stewart et al.8 for the computation of polygenic scores. Dr den Braber has received financial support from the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement nr 115372, resources of which are composed of financial contribution from the European Union's Seventh Framework Program (FP7/2007-2013) and EFPIA companies’ in kind contribution.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
den Braber, A., Zilhão, N., Fedko, I. et al. Obsessive–compulsive symptoms in a large population-based twin-family sample are predicted by clinically based polygenic scores and by genome-wide SNPs. Transl Psychiatry 6, e731 (2016). https://doi.org/10.1038/tp.2015.223
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.223
This article is cited by
-
A dimensional perspective on the genetics of obsessive-compulsive disorder
Translational Psychiatry (2021)
-
CAPS1 is involved in hippocampal synaptic plasticity and hippocampus-associated learning
Scientific Reports (2021)
-
Polygenic risk for obsessive-compulsive disorder (OCD) predicts brain response during working memory task in OCD, unaffected relatives, and healthy controls
Scientific Reports (2021)
-
Genome-wide association study of pediatric obsessive-compulsive traits: shared genetic risk between traits and disorder
Translational Psychiatry (2021)
-
Genome-wide association study identifies novel risk variants from RPS6KA1, CADPS, VARS, and DHX58 for fasting plasma glucose in Arab population
Scientific Reports (2020)
