
Abstract
In our previous study, a new compound, octadecanoic acid-3, 4-tetrahydrofuran diester, possessing potent acaricidal activity was obtained from neem oil. This study performed RNA-seq transcriptomics and iTRAQ proteomics to uncover the acaricidal mechanism of the compound against Sarcoptes scabiei var. cuniculi. The results of transcriptomics indicated that after treatment with octadecanoic acid-3, 4-tetrahydrofuran diester, genes related to “Energy metabolism” were significantly up-/down-regulated, including citrate cycle, oxidative phosphorylation pathway and fatty acid metabolism. Proteomics analysis showed accordant changes of proteins related to oxidative phosphorylation pathway. The target proteins of the compound were NADH dehydrogenase, Ubiquinol-cytochrome c reductase, Cytochrome c oxidase, ATP synthase, enolase and superoxide dismutase. In transcriptomics-proteomics correlation analysis, the concordance rate between protein abundances and their corresponding mRNAs was 57%, while others (43%) were discordant changes, suggesting divergent regulating effects of octadecanoic acid-3, 4-tetrahydrofuran diester. These results suggested that the acaricidal mechanism of octadecanoic acid-3, 4-tetrahydrofuran diester attributed to interference with energy metabolism, especially oxidative phosphorylation pathway.
Similar content being viewed by others

Introduction
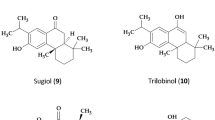
Neem (Azadirachta indica), an Indian medicinal herb, is now widely planted in many Asian and African countries1. Neem oil, derived from the seeds or fruits of the neem tree through pressing or extraction, exhibits broad spectrum of pharmacological properties, including antiparasitic, antifungal, antibacterial, antimalarial, and anti-inflammatory activities2,3,4,5,6,7. Our previous studies obtained a new compound from neem oil, which was identified as octadecanoic acid-3, 4-tetrahydrofuran diester8,9 (Fig. 1). The acaricidal activity of the compound against Sarcoptes scabiei var. cuniculi was significantly higher than that of natural pyrethrins and abamectin, the median lethal concentration (0.082 mg/mL) is only approximately 1/33 of that of neem oil10,11. In our previous work, the activity of superoxide dismutase, peroxidase, Ca2+-ATPase, glutathione-s-transferases, and peroxidase of mites were significantly changed after compound treatment, prompting the hypothesis that octadecanoic acid-3, 4-tetrahydrofuran diester could regulate energy metabolism of mites12. However, which proteins and pathways in energy metabolism were the targets of the compound and whether the related gene expressions were regulated by the compound are still unknown.

The structure of octadecanoic acid-3, 4 - tetrahydrofuran diester.
Transcriptional profiling based on total RNA sequencing (RNA-Seq) is a powerful tool for analyzing changes of gene expression in respond to various environmental stresses13. Isobaric tags for relative and absolute quantification (iTRAQ) is a new protein quantification technology based on isotope labeling combined with multidimensional liquid chromatography and tandem mass spectrometry (LC-MS/MS)14. In this study, parallel analysis of iTRAQ-LC-MS/MS proteomics and RNA-seq transcriptomics of S. scabiei treated with or without octadecanoic acid-3, 4-tetrahydrofuran diester were performed for identifying changes of proteins and transcript levels for genes and revealing the acaricidal mechanism of octadecanoic acid-3, 4-tetrahydrofuran diester.
Results
RNA-seq transcriptomic
Illumina sequencing generated 22,473,816 clean reads. The value of Q20, a standard parameter used to assess the sequencing quality, was above 95.0% in this study, indicating the high reliability of the sequencing data. Due to the absence of reference genomic sequences, a de novo RNA-seq assembly was performed using Trinity15 which produced 95,306 contigs with lengths >200 bp. The transcriptome annotation showed that the S. scabiei unigenes did not have high similarity in the NR database and the main species distribution was in Stegodyphus mimosarum (22.10%). Functional characterization of the S. scabiei contigs was performed by assigning EggNOG annotation with BLAST+. A total of 14,123 contigs could be assigned to three functional categories: cellular processes and signaling (49.67%), information storage and processing (25.61%), metabolism (24.72%). Gene ontology (GO) was also employed to annotate the S. scabiei contigs. In total, 20,166 were retrieved, including biological process (37.59%), molecular function (52.36%) and cellular component (10.05%).
The differentially expressed genes were identified using an R package with edgeR16 (q-value < 0.05). After the compound treatment, we found that 35,792 genes were significantly changed, including 10,541 down-regulated genes and 20,751 up-regulated genes. The GO annotation with BLAST2GO obtained 11,097 GO terms consisted of 40.00% “Biological process”, 40.47% “Molecular function” and 19.53% “Cellular component” (Fig. 2). The network topology of GO annotation indicated that the function of differentially expressed genes mainly distributed in several categories, such as “Glucosamine metabolism” and “Carbohydrate metabolism”. The results of KEGG pathway annotation showed that 6,366 differentially expressed genes can be enriched in 259 pathways, and 28 pathways (9 pathways in “Metabolism”) were significantly enriched (P < 0.05), including “Citrate cycle”, “Propanoate metabolism”, “biosynthesis of amino acids” and “fatty acid metabolism” (Table 1). 62 differentially expressed genes were enriched in “Oxidative phosphorylation” pathway (P > 0.05). These results suggested that octadecanoic acid-3, 4-tetrahydrofuran diester could regulate the gene expressions related to metabolism.

The GO annotation of differentially expressed genes.
Confirmation of differentially expressed genes by quantitative real-time PCR
We used quantitative real-time PCR to validate the transcriptional pattern of randomly selected eight genes related to oxidative phosphorylation pathway in S. scabiei. The c73281, c18049, c54526, c21888, c3910, c21419, c21529 and c4288 unigenes were mostly involved in energy metabolism. All the genes displayed a down-regulated pattern that was in agreement with the RNA-seq analysis (Fig. 3).

Relative expression levels of selected eight genes related to oxidative phosphorylation pathway by quantitative real-time PCR.
Quantitative proteomics
In order to compare the alternations of gene expression at protein level, iTRAQ proteomic analysis was performed. A total of 3,406 proteins were identified. A p-value of less than 0.05 and a 1.2-fold change of abundance were used to identify proteins that were differentially expressed between the treated group and control group. 184 differentially expressed proteins (102 up-regulated and 82 down-regulated) were detected. GO annotation obtained 149 differentially expressed proteins in biological process, 70 in cellular component and 128 in molecular function (Fig. 4). In biological process, 51 differentially expressed proteins belong to metabolic process.

The GO annotation of differentially expressed proteins.
KEGG pathway enrichment analysis was also performed to investigate the main effected pathways by the compound treatment. 150 differentially expressed proteins were enriched in 88 pathways (Table 2). Based on the transcriptomic data, 4 KEGG pathways were significantly regulated after treatment for 8 h, including ‘Basal transcription factors” (ko03022), “Collecting duct acid secretion” (ko04966), “Oxidative phosphorylation” (ko00190) and “Lysosome” (ko04142). In general, proteomic results demonstrated that the main acaricidal mechanism of octadecanoic acid-3, 4-tetrahydrofuran diester was to affect metabolism, especially energy metabolism.
Correlation analysis between proteome and transcriptome
After compound treatment, 100 differentially expressed genes and corresponding changes of protein abundance were obtained (Fig. 5). The concordance rate between protein abundances and their corresponding mRNAs was 57% (6 up-regulation, 51 down-regulation), while others (43%) were discordant changes. In energy metabolism, NADPH ubiquinone oxidoreductase (F subunit) and its mRNA were down-regulated; ATPase (E subunit) and F1-ATPase (α/β subunit) were up-regulated both in the protein and mRNA level.

Up and Down represent that the differentially expressed genes and corresponding changes of protein abundances were up- and down- regulated, respectively. Protein-up represents that the protein abundances were up-regulated, but their corresponding mRNAs was down-regulated. Protein-down represents that the protein abundances were down-regulated, but their corresponding mRNAs was up-regulated.
Discussion
It had been shown that there were several insecticidal ways of pesticides, mainly through interfering with insect metabolism. First, pesticides can alter the membrane structure and function causing damage of metabolic transit, such as ivermectin17,18. Second, pesticides can affect energy metabolism, such as α-Viniferin suppressing the malate dehydrogenase activity19. Third, pesticides can affect protein metabolism, such as chloroquinum inhibiting protein synthesis20. Fourth, pesticides can affect enzyme activity related to nucleotide synthesis, such as pyrazofurin inhibiting activities of lactate dehydrogenase and orotidylic acid pyrophosphorylase21. It has been over eight years since the new acaricidal compound, octadecanoic acid-3, 4-tetrahydrofuran diester, had been obtained from neem oil, however, we still know little about its insecticidal mechanism. Therefore, in the present study, we used RNA-seq transcriptomics and iTRAQ proteomics to identify the target of the compound.
Mitochondria, known as “the power center of the cell”, play a crucial role in cell survival, because they were major suppliers of adenosine triphosphate (ATP) which was used as a source of chemical energy. Mitochondria are the place where protein, fat and carbohydrate are oxidized mainly through tricarboxylic acid cycle and oxidative phosphorylation. Mitochondrial dysfunction could induce many diseases22. Therefore, mitochondrial could be the target of acaricidal drug which could produce adverse effects on mitochondrial function, finally leading to mite death. In this study, the results of RNA-seq transcriptomics showed that there were 45 differentially expressed down-regulated genes in the tricarboxylic acid cycle paths and 62 differentially expressed genes (60 down-regulated and 2 up-regulated) in oxidative phosphorylation pathway after treatment. These results suggested that octadecanoic acid-3, 4-tetrahydrofuran diester may affect the activity of related enzymes in the process of glycolysis, leading to the energy metabolic block. Besides, the differentially down-regulated genes linked to the oxidative phosphorylation included cytochrome C oxidase cbb3 subunit, cytochrome C reductase b/c1 subunit, ATP synthase and NADH dehydrogenase.
NADH-dehydrogenase, also called “Complex I”, was the first protein known as electron carriers in the transport chain, which can be oxidized by Coenzyme Q1023. Some pesticides, such as rotenone, could inhibit the activity of NADH-dehydrogenase24. Succinate dehydrogenase, also called “Complex II”, is the second protein known as electron carriers in the transport chain, which also belongs to the tricarboxylic acid cycle25. Our study showed that the genes of NADH-dehydrogenase and succinate dehydrogenase were down-regulated after treatment, suggesting that Octadecanoic acid-3, 4-tetrahydrofuran diester could inhibit the NADH-dehydrogenase activity and production of fumk5arate, resulting in electron transport inhibition.
Ubiquinol-cytochrome C reductase, also called “Complex III”, can catalyze coenzyme Q oxidation and reduction process of cytochrome c. Besides, the enzyme plays an irreplaceable role in donating electron from QH2 to cytochrome c receptor26. Cytochrome c oxidase, also called “Complex IV”, is the last protein known as electron carriers in the transport chain26. After treatment, the down-regulated gene expressions of Cytochrome C reductase subunit b/c1and oxidase cbb3 subunit may reduce the activity of Cytochrome C reductase and oxidase and disturb proton transport, indirectly inducing abnormal ATP synthesis.
ATP synthase, also called “Complex V”, is the terminase of oxidative phosphorylation pathway. It drives ADP and phosphate (Pi) to produce ATP by using the energy stored in the transmembrane proton gradient27. It is reported that after mutation of F ATP synthase subunit α, F ATP synthase could only passively transfer H+ and was unable to coupling with ATP, leading to losing the ability to synthesize ATP28. After treatment, the gene expression of F ATP synthase was up-regulated, indicating that F ATP synthase may be involved in detoxification of xenobiotics or repairing mitochondrial damage.
In accordance with the results of RNA-seq transcriptomics, iTRAQ proteomics study also showed that octadecanoic acid-3, 4-tetrahydrofuran diester could interfere with mite metabolism, especially energy metabolism. The changes of protein abundances of NADH ubiquinone oxidoreductase subunit F and ATPase linked to energy metabolism were the same as their corresponding mRNAs. Besides, enolase, one of the important glycolytic enzymes for energy metabolism, could lower host immunity for helping parasites escaping immune surveillance29. After treatment, the protein abundance of enolase was reduced. These results accumulated evidences for our finding that the enzymes belonging to energy metabolism were the main target of octadecanoic acid-3, 4-tetrahydrofuran diester.
In addition to these enzymes of energy metabolism, this study also found that other proteins of mites could be the target of octadecanoic acid-3, 4-tetrahydrofuran diester. Superoxide dismutase (SOD), an important protective enzyme, can eradicate the oxygen free radicals for preventing radical-related diseases30. In accordance with our previous studies12, we found that the protein abundance of SOD was down-regulated after treatment, suggesting that octadecanoic acid-3, 4-tetrahydrofuran diester had an inhibitory effect on the enzymatic activity of SOD, reducing the free radical-scavenging activity, which could eventually lead to mite death. Cathepsin, a kind of proteinase found in all animals, can degrade host proteins as its nutrients31. We showed that the compound could reduce the protein abundance of cathepsin, suggesting that cathepsin could be the new target of acaricidal drugs. Vitellogenin, a precursor of phospholipoprotein, is the key factor in vitellogenesis in insects32. It was reported that abamectin had an adverse effect on the growth and development of haemaphysalis longicornis and decreased ovary weight33. This study showed that the expression of vitellogenin was inhibited after treatment, suggesting that the compound could inhibit the development of mite ovary.
The lysosomes commonly act as waste bags to digest unwanted macromolecules in the cytoplasm, both from outside the cell and obsolete components inside the cell 34. Phagosomes in the fusion with lysosomes form phagolysosomes, which not only promote killing and degradation of microorganisms and apoptotic bodies, but also enable them to conduct autophagy to clear damaged structures34,35. In this study, transcriptomics and proteomic studies found that part of differentially expressed genes/proteins were enriched in “Phagosome” and “lysosome” pathways. The occurrence of this phenomenon may attribute to treatment with octadecanoic acid-3, 4-tetrahydrofuran diester, which could cause damages to mites leading to production of impaired macromolecules, such as proteins and organelles, finally facilitates the functions of “Phagosome” and “lysosome” pathways.
In conclusion, the acaricidal mechanism of octadecanoic acid-3, 4-tetrahydrofuran diester mainly attributed to interference with energy metabolism, especially oxidative phosphorylation pathway (Summarized in Fig. 6). The target proteins were NADH dehydrogenase, Ubiquinol-cytochrome c reductase, Cytochrome c oxidase, ATP synthase, enolase and superoxide dismutase.

The subunit of each protein in green box represents down-regulation and in red box represents up-regulation.
Methods
Treatment
S. scabiei larvae were isolated from infested rabbits according to the methods previously described12. Briefly, the octadecanoic acid-3, 4-tetrahydrofuran diester was diluted with liquid paraffin to 10 mg/mL. Then, 10 μL of solution was added into Petri dishes (8.5 mm in diameter). The mites were then placed into the solution with a needle (20 mites for each drop of the compound8). The liquid paraffin was served as control. All plates were incubated at 25 °C and 75% relative humidity. After treatment for 8 h, the mites were washed thrice with phosphate buffered saline (pH 7.4) and cryopreserved with liquid nitrogen for isolation of RNA and protein.
RNA extraction, Illumina sequencing and data analysis
Total RNA was extracted from the mites using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The concentration and quality of RNA were determined using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). The construction of cDNA library and sequencing were performed as previous z6IIFFE6ed using the oligo (dT) magnetic beads. The mRNA was interrupted to short fragments after fragmentation buffer was added, followed by cDNA synthesis. The double-stranded cDNA was purified using the QiaQuick PCR Purification Kit (Qiagen, Germany) and subjected to library preparation for sequencing analysis via Illumina HiSeqTM 2000.
The transcriptomic data were analyzed using a previously described protocol with modification24. Cleaned reads of each sample were mapped to the sequenced genome of S. scabiei using Bowtie36. Reads aligned using Bowtie were assembled into transcripts using Cufflinks, and then merged with Cuffmerge. Differential expression profiles were determined using Cuffdiff (Version: 2.1.1) with default parameters. Hierarchical clustering analysis was performed using Cluster 3.037. The gene expression level is calculated by using RPKM method (Reads Per kb per Million reads)38.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was employed to verify the results of RNA-Seq. Total RNA was extracted using the RNAprep pure Tissue Kit (DP431, Tiangen, China) and immediately subjected to reverse transcription using the PrimeScript™ RT reagent Kit with gDNA Eraser (RR047, TaKaRa, China). The specific primers (Table 3) for randomly selected eight genes related to oxidative phosphorylation pathway were designed using the Primer software (Version 5.0). qRT-PCR was performed at 95 °C for 30 s followed by 39 cycling of 95 °C for 5 s, 60 °C for 10 s and 72 °C for 15 s by using the iQ SYBR Green Supermix (Bio-rad, CA) on a CFX Connect™ System (Bio-rad, CA). GAPDA was used as a reference gene. The relative expression levels of target genes were analyzed using the 2−ΔΔCT method39.
Protein extraction and iTRAQ labeling
The mites for iTRAQ analysis were homogenized in a lysis buffer (C006225, Sangon Biotech Co., Ltd). The homogenates were centrifuged at 15,000 g for 10 min. The supernatant was purified by using 2D clean-up kit (Bio-Rad, CA). Protein concentrations and quality were determined using a Protein Assay Kit (Bio-Rad, CA).
iTRAQ analysis was carried out at Honor Tech Co. Ltd (Beijing, China) as previous reports40. Brief, after adjusting the pH to 8.5 with 1 M ammonium bicarbonate, total protein from each sample was reduced for 1 h at 56 °C by adding DTT to 10 mM and alkylated with 55 mM iodoacetamide for 45 min at room temperature in dark place. The protein was digested by adding trypsin (1:20, w/w). The digestion was incubated at 37 °C for overnight. A total of four samples were then labeled using iTRAQ Reagent-8plex Multiplex Kit according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA,USA). Two test samples were labeled with iTRAQ tags 113 and 115, two control samples labeled with tags 117 and 119.
The labeled samples were mixed and lyophilized. They were then resuspended in 4 mL buffer containing 25% v/v acetonitrile and 25 mM NaH2PO4 (pH 2.7), and followed by fractionating using a Ultremex SCX column (4.6 × 250 mm) by HPLC system (Shimadzu LC-20AB). A total of twenty fractions obtained were desalted using a Strata X C18 column (Phenomenex, USA) and then vacuum-dried.
Liquid chromatography-mass spectrometry
Each of the dried fractions was dissolved with buffer containing 5% v/v acetonitrile and 0.1% Formic acid and centrifuged at 20,000 g for 10 min. The peptides were separated by a 2 cm C18 trap column (inner diameter 200 μm) on an nano HPLC (Shimadzu LC-20AD). The eluted peptides were analyzed using nanoelectrospray ionization, followed by tandem mass spectrometry (MS/MS) in an Q-Exactive (Thermo Fisher Scientific, San Jose, USA) coupled with an online HPLC system. Intact peptides were detected in the Orbitrap with a resolution of 70,000. Peptides were selected for MS/MS using higher energy collision dissociation (HCD) operating mode with a normalized collision energy setting of 27%. A data-dependent procedure was applied for the three most abundant precursor ions above a threshold ion count of 20,000 in the MS survey scan.
Proteomic data analysis
The MS spectra were analyzed by a thorough search using Mascot software (version 2.3.02, Matrix Science Inc, Boston, MA) against S. scabiei database. To reduce false positive results, all data were reported based on a 95% confidence and false discovery rate (FDR) less than 1%. For quantitative analysis, a protein must have at minimum one unique peptide matches with iTRAQ ratios. A 1.2-fold cutoff value was used to identify up-regulated and down-regulated proteins with a p-value of less than 0.05.
Functional annotation
Interproscan 4.5 was used to identify conserved domains from peptide translations and database for protein sequences matching to MS peptides41. Gene ontology (GO) annotations for identified proteins based on BLAST results were performed using Blast2GO (http://www.blast2go.de/). Sequences were assigned to KEGG orthologous groups by comparison with the KEGG protein database42,43,44. Custom perl scripts were used to bin orthologous groups into broad categories and KEGG modules.
Additional Information
How to cite this article: Song, X. et al. Transcriptomics and proteomic studies reveal acaricidal mechanism of octadecanoic acid-3, 4 - tetrahydrofuran diester against Sarcoptes scabiei var. cuniculi. Sci. Rep. 7, 45479; doi: 10.1038/srep45479 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kumar, V. S. & Navaratnam, V. Neem (Azadirachta indica): prehistory to contemporary medicinal uses to humankind. Asian. Pac. J. Trop. Biomed. 7, 505–514 (2013).
Remedio, R. N., Nunes, P. H., Anholeto, L. A., Oliveira, P. R. & Camargo-Mathias, M. I. Morphological effects of neem (Azadirachta indica A. Juss) seed oil with known azadirachtin concentrations on the oocytes of semi-engorged Rhipicephalus sanguineus ticks (Acari: Ixodidae). Parasitol. Res. 2, 431–444 (2015).
Kelsey, D. J., Nieto-Delgado, C., Cannon, F. S. & Brennan, R. A. Organic neem compounds inhibit soft-rot fungal growth and improve the strength of anthracite bricks bound with collagen and lignin for use in iron foundry cupolas. J. Appl. Microbiol. 1, 65–75 (2015).
Del Serrone, P., Failla, S. & Nicoletti, M. Natural control of bacteria affecting meat quality by a neem (Azadirachta indica A. Juss) cake extract. Nat. Prod. Res. 10, 985–987 (2015).
Umar, M. I. et al. Multi-constituent synergism is responsible for anti-inflammatory effect of Azadirachta indica leaf extract. Pharm. Biol. 11, 1411–1422 (2014).
Tapanelli, S. et al. Transmission blocking effects of neem (Azadirachta indica) seed kernel limonoids on Plasmodium berghei early sporogonic development. Fitoterapia. 114, 122–126 (2016).
Patel, S. M. et al. Potential of neem (Azadirachta indica L.) for prevention and treatment of oncologic diseases. Semin. Cancer. Biol. 40–41, 100–115 (2016).
Du, Y. H. et al. Acaricidal activity of extracts of neem (Azadirachta indica) oil against the larvae of the rabbit mite Sarcoptes scabiei var. cuniculi in vitro . Vet. Parasitol. 157, 144–148 (2008).
Du, Y. et al. Acaricidal activity of neem oil against Sarcoptes scabiei var. cuniculi larvae in vitro . Vet. Sci. China. 37, 1086–1089 (2007).
Du, Y. H. et al. Acaricidal activity of four fractions and octadecanoic acid-tetrahydrofuran-3, 4-diyl ester isolated from chloroform extracts of neem (Azadirachta indica) oil against Sarcoptes scabiei var. cuniculi larvae in vitro . Vet. Parasitol. 163, 175–178 (2009).
Xu, J. et al. The preparation of neem oil microemulsion (Azadirachta indica) and the comparison of acaricidal time between neem oil microemulsion and other formulations in vitro . Vet. Parasitol. 169, 399–403 (2010).
Chen, Z. et al. Studies on the acaricidal mechanism of the active components from neem (Azadirachta indica) oil against Sarcoptes scabiei var. cuniculi. Vet. Parasitol. 204, 323–329 (2014).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Zhu, G. et al. Quantitative iTRAQ LC-MS/MS Proteomics Reveals Transcription Factor Crosstalk and Regulatory Networks in Hypopharyngeal Squamous Cell Carcinoma. J. Cancer. 5, 525–536 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 26, 136–138 (2010).
Horn, D. & Duraisingh, M. T. Antiparasitic chemotherapy: from genomes to mechanisms. Annu Rev. Pharmacol. Toxicol. 54, 71–94 (2014).
Dent, J. A., Smith, M. M., Vassilatis, D. K. & Avery, L. The genetics of ivermectin resistance in Caenorhabditis elegans. PNAS. 97, 2674–2679 (2000).
Roy, B. & Giri, B. R. α-Viniferin and resveratrol induced alteration in the activities of some energy metabolism related enzymes in the cestode parasite Raillietina echinobothrida. Acta. Trop. 154, 102–106 (2016).
Baragaña, B. et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 522, 315–20 (2015).
Li, Y. 2011. The Research and Latest Progress of the Mechanisms of Antihelmintics. J. Liaoning. Medical. University. 32, 563–567 (2011).
Brand, M. D. & Nicholls, D. G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2, 297–312 (2011).
Fromm, S., Senkler, J., Eubel, H., Peterhänsel, C. & Braun, H. P. Life without complex I: proteome analyses of an Arabidopsis mutant lacking the mitochondrial NADH dehydrogenase complex. J. Exp. Bot. 67, 3079–3093 (2016).
Sun, Q. et al. Transcriptome analysis of stem development in the tumourous stem mustard Brassica juncea var. tumida Tsen et Lee by RNA sequencing. BMC. Plant. Biol. 12, 53 (2012).
Williamson, S. R. et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 1, 80–94 (2015).
Esser, L. et al. Hydrogen Bonding to the Substrate Is Not Required for Rieske Iron-Sulfur Protein Docking to the Quinol Oxidation Site of Complex III. J. Biol. Chem. 291, 25019–25031 (2016).
Stock, D., Leslie, A. G. & Walker, J. E. Molecular architecture of the rotary motor in ATP synthase. Science. 286, 1700–1705 (1999).
Fillingame, R. H. Molecular mechanics of ATP synthesis by F1F0-type H+-transporting ATP synthases. The bacteria. 12, 345–391 (1990).
Avilán, L. et al. Enolase: a key player in the metabolism and a probable virulence factor of trypanosomatid parasites—perspectives for its use as a therapeutic target. Enzyme. Res. 2011, 932549 (2011).
Zhao, L., Xu, X., Xu, Z., Liu, Y. & Sun, S. Diapause Induction, Color Change, and Cold Tolerance Physiology of the Diapausing Larvae of the Chouioia cunea (Hymenoptera: Eulophidae). J. Insect. Sci. 14, 294 (2014).
Maldonado-Aguayo, W., Chávez-Mardones, J., Gonçalves, A. T. & Gallardo-Escárate, C. Cathepsin Gene Family Reveals Transcriptome Patterns Related to the Infective Stages of the Salmon Louse Caligus rogercresseyi. PLoS One. 10, e0123954 (2015).
Tufail, M. & Takeda, M. Molecular characteristics of insect vitellogenins. J. Insect Physiol. 54, 1447–1458 (2008).
Doan, H. T. T. et al. The efficacy of avermectins (ivermectin, doramectin and abamectin) as treatments for infestation with the tick Haemaphysalis longicornis on rabbits in Korea. Vet. Parasitol. 198, 406–409 (2013).
Appelqvist, H., Wäster, P., Kågedal, K. & Öllinger, K. The lysosome: from waste bag to potential therapeutic target. J. Mol. Cell. Biol. 4, 214–226 (2013).
Kissing, S. et al. Vacuolar ATPase in phagosome-lysosome fusion. J. Biol. Chem. 22, 14166–14180 (2015).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome. Biol. 10, R25 (2009).
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. PNAS. 95, 14863–14868 (1998).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 5, 621–628 (2008).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25, 402–408 (2001).
Yang, L. T. et al. iTRAQ protein profile analysis of Citrus sinensis roots in response to long-term boron-deficiency. J. Proteomics. 93, 179–206 (2013).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847–848 (2001).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic. Acids. Res. 28, 27–30 (2000).
Kanehisa, Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic. Acids. Res. 45, D353–D361 (2017).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic. Acids. Res. 44, D457–D462 (2016).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 31372477), the International Cooperation Projects of Sichuan Province (2014HH0058), and the Agricultural Technology Research and Development Project of Chengdu (2015-NY02-00266-NC).
Author information
Authors and Affiliations
Contributions
X.S., Z.C. and Y.Z. conceived the experiments and wrote the main manuscript. X.S., M.C. and R.J. conducted the experiments. Z.Y. supervised the experimental works. X.L., L.L., L.Y., C.H., G.Y. and Y.Z. analyzed the results. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Song, X., Chen, Z., Jia, R. et al. Transcriptomics and proteomic studies reveal acaricidal mechanism of octadecanoic acid-3, 4 - tetrahydrofuran diester against Sarcoptes scabiei var. cuniculi. Sci Rep 7, 45479 (2017). https://doi.org/10.1038/srep45479
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45479
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
