
Abstract
Sinorhizobium fredii HH103 is a rhizobial soybean symbiont that exhibits an extremely broad host-range. Flavonoids exuded by legume roots induce the expression of rhizobial symbiotic genes and activate the bacterial protein NodD, which binds to regulatory DNA sequences called nod boxes (NB). NB drive the expression of genes involved in the production of molecular signals (Nod factors) as well as the transcription of ttsI, whose encoded product binds to tts boxes (TB), inducing the secretion of proteins (effectors) through the type 3 secretion system (T3SS). In this work, a S. fredii HH103 global gene expression analysis in the presence of the flavonoid genistein was carried out, revealing a complex regulatory network. Three groups of genes differentially expressed were identified: i) genes controlled by NB, ii) genes regulated by TB and iii) genes not preceded by a NB or a TB. Interestingly, we have found differentially expressed genes not previously studied in rhizobia, being some of them not related to Nod factors or the T3SS. Future characterization of these putative symbiotic-related genes could shed light on the understanding of the complex molecular dialogue established between rhizobia and legumes.
Similar content being viewed by others
Introduction
The symbiotic relationship between legumes and nitrogen-fixing rhizobia involves reciprocal communication by means of chemical signals produced by the plant and the bacterium. Plant root secreted flavonoids are phenolic compounds that act as inducers of the bacterial nod genes, which encode enzymes for the production of specific lipochitooligosaccharidic signal molecules or Nod factors (NFs) that, in turn, are responsible for root infection and induction of nodule development. Within this new organ, differentiated bacteria reduce atmospheric nitrogen to ammonia, which is assimilated by the host plant in exchange of a carbon source and an appropriate environment that promotes bacterial growth1,2.
The regulatory protein NodD is constitutively expressed and codes for a LysR-type transcriptional activator that, in the presence of specific flavonoids, recognizes and binds to nod boxes (NB), promoter sequences located upstream of nodulation genes, triggering their transcription3,4,5. Interestingly, many other symbiotic-related traits such as polysaccharide production, phytohormone synthesis, motility, quorum sensing and the activation of the type 3 secretion system (T3SS) are regulated, depending on the rhizobial strain, by specific inducer flavonoids and NodD1,6,7,8,9,10,11. However, to our knowledge, only a few genome-wide transcriptomic analyses of the effect of flavonoids on rhizobial gene expression have been performed so far. In Bradyrhizobium japonicum USDA110 about 100 genes were induced with genistein, including all nod box-associated genes, type 3 secreted effectors, the flagellar cluster and several genes likely involved in transport processes12. Recently Huyghe et al.13 reported that many Sinorhizobium fredii NGR234 genes responded to the presence of daidzein: a total of 754 genes with a cutoff [fold-change value] ≥ 2. Although nod box- and T3SS-associated genes were those showing the highest levels of induction, these results are in agreement with those reported for B. japonicum, that indicate that flavonoids have a much broader function than the mere induction of the nod genes.
Sinorhizobium fredii nodulates more than a hundred genera of legumes, including plants forming determinate and indeterminate nodules such as Glycine max and Glycyrrhiza uralensis, respectively14,15,16. There is wide genomic information of the three most studied S. fredii strains: NGR234, USDA257 and HH10317,18,19,20. The HH103 genome is composed of 7 different replicons which harbour 6960 coding sequences (CDSs)20: the chromosome (4,305,723-bp, 4014 CDSs) and 6 plasmids: pSfHH103e/plasmid e (2,096,125-bp, 1991 CDSs), pSfHH103d/plasmid d/symbiotic plasmid (pSym, 588,797-bp, 667 CDSs), pSfHH103c/plasmid c (144,082-bp, 169 CDSs), pSfHH103b/plasmid b (61,880-bp, 62 CDSs), pSfHH103a1/plasmid a1 (24,036-bp, 19 CDSs) and pSfHH103a2/plasmid a2 (25,081-bp, 38 CDSs). HH103 harbours two copies of nodD, nodD1 and nodD2, although only the former has been related to nod gene expression10. HH103 also possesses a copy of the ttsI gene, which codes for the T3SS transcriptional regulator TtsI6.
In 1999, Perret et al.21 defined 19 functional NB in the symbiotic plasmid of NGR234. Sequencing of the HH103 genome revealed that 4 of these NB (NB4, NB6, NB7 and NB11) are not present in this strain20. In addition, there are differences between NGR234 and HH103 in the genes driven by other 4 NB. Furthermore, 18 potential T3SS-promoter sequences or tts boxes (TB) were identified in the genome of HH10320. Many of the genes located downstream of all these regulatory sequences have not been studied in HH103 and some of them neither in other rhizobia.
In this work, RNA-seq and qRT-PCR (qPCR) studies were addressed to analyse the S. fredii HH103 global gene expression in the presence and absence of genistein, an effective nod gene inducer in this strain10. Three groups of genes differentially expressed upon treatment with genistein were identified: i) genes controlled by NB, ii) genes regulated by TB and iii) genes not preceded by a NB or a TB. Consequently, further studies were performed with mutants affected in the regulatory genes nodD1 and ttsI. Thus, an expression map of the S. fredii HH103 whole genome in the presence of genistein is provided, with special emphasis on the role of NodD1 and TtsI in the regulation of genes whose expression was affected by the presence of this flavonoid.
Results and Discussion
Identification of the Sinorhizobium fredii HH103 genes differentially expressed upon induction with genistein.
In order to identify genes induced with genistein, RNA-seq libraries were generated from HH103 cultures grown in the presence and absence of this flavonoid. To determine the role of NodD1 and TtsI in the regulation of the expression of the HH103 genes affected by genistein, HH103 nodD1 and ttsI mutant derivatives were also included in these studies. Two biological independent replicates were obtained and analyzed for each condition. Thus, 12 RNA-seq libraries were generated and sequenced, obtaining between 39 and 49 million reads in each condition, which indicates that similar amounts of data were generated in all cases. The general features of each run are shown in Supplementary Data 1. In addition, all samples showed more than 90% of properly pair reads. Three different RNA-seq metrics for quality control, GC content, duplicate distribution and the distribution of respective genetic coordinates, were performed (Supplementary Data 1). A normalization of the quantification data was also needed before all subsequent analysis to avoid statistical deviations due to differences in library and genetic sizes22 (Supplementary Data 1).
Differentially expressed genes (DEG) in each strain upon treatment with genistein in comparison to the wild-type strain grown in the absence of genistein were detected as described in Methods. Our first criterion was the selection of DEG showing [fold-changes] ≥3 (i.e. [log2] >1.6). Thus, 106 (1.52% of the genome), 14 (0.2%) and 71 (1.02%) DEG were detected in the wild-type, nodD1 and ttsI mutant strains, respectively, when compared to the wild-type strain grown in the absence of flavonoids (Fig. 1; Supplementary Data 2). The transcription of 18 of these genes was quantified by qPCR to validate the data set (Supplementary Table S1). In 17 out of these 18 genes, a linear correlation was obtained in the fold-change values obtained by both qPCR and RNA-seq (Supplementary Data 3).

Replicon distribution and number of differentially expressed genes in S. fredii HH103 upon induction with genistein.
Chr: chromosome, pl. a1: pSfHH103a1, pl. a2: pSfHH103a2, pl. b: pSfHH103b, pl. c: pSfHH103c, pl. d: pSfHH103d and pl. e: pSfHH103e. Red bars: wild-type strain, blue bars: nodD1 mutant; yellow bars: ttsI mutant.
In the wild-type strain, 98 genes were up-regulated in the presence of genistein and most of them (81) were located on the pSym. The rest of activated genes were found in the chromosome (11) and in plasmids c (5) and e (1). Only 8 genes were down-regulated in this condition (5 in the chromosome and 3 in plasmid e). Table 1 provides the list of the S. fredii HH103 genes showing the highest variations in expression (log2 fold-changes >3.3 or <−2) upon genistein treatment. The entire list of 106 DEG can be found as Supplementary Data 2. In addition to those related to the symbiotic T3SS and to the production of Nod factors, a large number of the genes over-expressed in these conditions coded for hypothetical proteins. These results indicate that a significant part of the genistein-induced regulon remains to be characterized.
The 14 DEG found in the nodD1 mutant in the presence of genistein were all down-regulated and distributed among the chromosome (4) and plasmids e (6), c (2), d (1) and a2 (1). In the ttsI mutant, 64 out of the 71 differentially expressed genes were up-regulated and most of them (53) were located on the symbiotic plasmid. The rest of the activated genes were found in plasmid c (6) and the chromosome (5). Down-regulated genes were located on the chromosome (4) and on plasmid e (3) (Fig. 1).
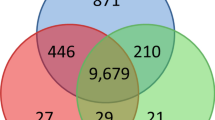
Figure 2 shows a comparative analysis of the sets of DEG upon treatment with genistein in HH103 and its nodD1 and ttsI derivatives. The 53 genes shared by HH103 and its ttsI mutant derivative are those regulated via NodD1 (directly or indirectly), with the exception of those genes regulated by both NodD1 and TtsI, which should be part of the 49 DEG identified only in the wild-type strain. We also found 4 genes that are affected by genistein in the three strains, indicating that they were regulated in a NodD1- and TtsI-independent manner.

Comparative analysis of the set of genes regulated by genistein in S. fredii HH103 (red circle) and its nodD1 (blue circle) and ttsI (yellow circle) mutant derivatives.
The differences among these strains are visualized by a Venn diagram. The number of genes that are either individual for a certain strain or that are affected in two or three strains are indicated.
Interestingly, we found a gene, SFHH103_00387 (annotated as fbpA), which was clearly repressed in the nodD1 and ttsI mutant backgrounds regardless the presence or absence of genistein. This pattern of expression was confirmed by qPCR (Supplementary Table S1, Supplementary Data 3). The product of this gene is predicted to be a periplasmic protein involved in the uptake of Fe3+. Whether this S. fredii HH103 protein is involved in Fe uptake and/or the reason why its expression is repressed in the absence of NodD1 or TtsI requires further investigation.
The S. fredii HH103 genes affected by genistein can be assigned to three different groups based on their promoter sequences
As mentioned above, RNA-seq analyses led to the identification of 106 S. fredii HH103 DEG upon treatment with genistein. However, we also considered genes that, showing lower changes of expression (as noeI or nolK), belonged to operons in which some of the genes exhibited clear genistein-mediated differential expression. The two copies of nopM present in the HH103 genome (expression fold-changes of 2.1 and 2.2) were also included because they were preceded by a well-conserved TB. Thus, the number of genes considered as affected by genistein in S. fredii HH103 raised to 117. Sixteen out of these 117 genes, however, were discarded by different reasons such as showing opposite direction to known nod boxes or being identical copies of a gene under the control of a NB (Supplementary Data 4). Therefore, only 101 genes, which could be assigned to three different groups based on the presence or absence of functional NB or TB in their promoter sequences, were subjected to further analyses.
S. fredii HH103 genes induced through nod boxes (NB)
Flavonoids exuded by legume roots activate the transcription of nod genes through their interaction with NodD. Although this protein interacts with specific promoter sequences (NB) even in the absence of inducers, binding of an appropriate flavonoid to NodD enhances the access of the RNA polymerase and increases the transcriptional level of the nod genes5,23.
The present study led to the functional study of the 15 NB present in the HH103 symbiotic plasmid20. Those NB located upstream of genes induced with genistein (>3 fold) in the wild-type and the ttsI mutant strains, but not in the nodD1 mutant, were considered active: NB1, NB2, NB8, NB9, NB10, NB13, NB14, NB15, NB17, NB18, NB19 (Supplementary Table S2, Supplementary Data 4). The genistein-induced expression of genes belonging to NB1, NB8, NB10, NB13, NB14, NB15, NB17 and NB19 has been validated by qPCR in this work (Supplementary Table S1) and that of noeL (NB2) and ttsI (NB18) has been previously demonstrated6,24. Quantitative PCR experiments confirmed that expression of psfHH103d_118 (NB9) was inducible by genistein in a NodD1-dependent manner (8.2-fold). Once the active NB were determined, 36 ORF located downstream these promoters were identified (Supplementary Table S2, Supplementary Data 4).
NB3 is situated upstream of nodD1 but opposed to this gene. In S. fredii, in contrast to other rhizobia, nodD1 is not located close to the common nodABC genes. Therefore, NB3 might be a reminiscence of a promoter controlling the transcription of nodABC in a S. fredii ancestor. The gene whose expression is controlled by NB3 (psfHH103d_384) codes for a hypothetical protein not present in other rhizobia and was induced 2.5-fold in the presence of genistein in HH103 and in its ttsI mutant, but not in the nodD1 derivative, suggesting that NB3 is active but shows a low efficiency.
S. fredii HH103 NB5 and NB16 lack downstream genes20. The downstream DNA regions showed very low number of reads regardless the presence of flavonoids and/or NodD1 (data not shown), which indicates that both NB are not functional. In addition, the absence of induction by genistein of the pseudogenes nodS and nodU of S. fredii HH103 indicated that the preceding NB12 was either not functional. These results are in agreement with those obtained in USDA25725 but in contrast to the situation described in NGR23421.
Figure 3A shows the alignments of the functional and non-functional NB of S. fredii HH103. Curiously, none of them, regardless their transcriptional activity, entirely follow the consensus NB sequence defined by Schlaman et al.3: ATCN9GATN7ATCN6ATCGATN6AAT. The three non-functional NB lack the initial A residue and fail in the last three nucleotides motif (AAT), suggesting that these residues could be very relevant for the functionality of this promoter region. Although the functional NB10 also fails in the initial A residue, it does contain the final AAT motif. We could not find a clear correlation between the sequences of the HH103 functional NB and their strength (fold induction of the gene located immediately downstream). Clearly, further research is required to clarify this issue.

Identified promoter sequences driving genistein-mediated expression in S. fredii HH103.
The fold-induction with genistein of the gene located immediately downstream is shown. (A) functional and non-functional nod boxes. (B) functional and non-functional tts boxes. (C) putative SyrM boxes located upstream of psfHH103d_306 and psfHH103d_322. (D) Imperfect tts and SyrM boxes located upstream of SFHH103_00346 (flgJ).
Some of the S. fredii HH103 functional NB control the expression of genes previously related to symbiosis (Supplementary Table S2, Supplementary Data 4). NB2 and NB8 are responsible for the expression of nodZnoeLnolK and nodABCIJnolO’noeI, respectively, which are dedicated to the synthesis and export of the different HH103 Nod factors20. NB14 controls the expression of the fixABCX genes, conserved in rhizobia and essential for nitrogen fixation in the S. meliloti-alfalfa symbiosis, since they probably code for a putative membrane complex participating in electron transfer to nitrogenase26,27. NB15 drives the expression of psfHH103d_257, which is involved in the flavonoid-induced synthesis of indole acetic acid (IAA)20, a phytohormone related to plant root growth promotion. Flavonoid-induced production of IAA has also been reported for S. fredii NGR234 and Rhizobium tropici CIAT 8999,28, suggesting that promotion of legume root growth could be widespread among rhizobia.
NB1 controls the expression of 5 genes (psfHH103d_373 to psfHH103d_370) possibly related to the synthesis of pentacyclic triterpenoids called hopanoids. It has been reported very recently that inactivation of the orthologous genes of Bradyrhizobium sp. and Bradyrhizobium diazoefficiens, whose expression is not driven by NB, negatively affects bacterial survival in stressful conditions and symbiotic performance with their host legumes29,30. The fact that in S. fredii HH103 hopanoid-related genes are induced by flavonoids in a NodD1 dependent manner suggests that hopanoids could have a bacterial protective role during symbiosis, although further research is required to elucidate this point.
On the other hand, RNA-seq analysis indicated that four NB (NB9, NB10, NB13 and NB17) control the expression of genes coding for conserved hypothetical proteins whose functions are currently unknown or poorly investigated (Supplementary Table S2, Supplementary Data 4). Future efforts must be focused on the study of these NodD1-flavonoid-activated genes, since they could shed light on molecular aspects of the symbiotic interaction between S. fredii HH103 and its legume hosts. As an example, S. fredii HH103 exopolysaccharide (EPS) production is inhibited by genistein11. However, among the DEG found upon a 24 h genistein treatment, there were not known genes (exo/exs) coding for enzymes directly involved in EPS production. Regarding EPS regulatory genes, in S. meliloti at least 8 proteins control succinoglucan (EPS I) production [reviewed by31]: MucR and SyrM act as positive regulators, whereas ExoX, ExoR, ExoS, ExsB, CbrA and EmmC repress this process. In S. fredii HH103, with the exception of syrM (fold-change +5.8, see below), the expression of the orthologous genes of these regulators was not greatly affected by the presence of genistein (fold-changes of −1.1 to +1.9). In addition to the putative involvement of syrM in the process, there is another HH103 gene, psfHH103d_161, that could be related to regulation of EPS production. This gene, which is under the control of NB10, is highly induced by genistein (fold-change +21.6) and codes for a hypothetical protein 58% identical to the R. leguminosarum biovar phaseoli PsiB. The fact that PsiB is involved in exopolysaccharide inhibition32 opens the possibility that the product encoded by psfHH103d_161 could be related to the genistein-mediated EPS repression exhibited by HH103.
The expression of two genes coding for transcriptional regulators, ttsI (NB18) and syrM (NB19), was also up-regulated via NodD1 and genistein (Supplementary Table S2, Supplementary Data 4). TtsI is the main activator of the S. fredii HH103 T3SS6 and SyrM is known to be a transcriptional regulator implied in the optimal performance of the S. meliloti-Medicago symbiosis33. This finding indicates that in S. fredii HH103, in addition to the expression of the symbiotic T3SS through TtsI, NodD1 and inducer flavonoids could activate the expression of a set of genes via SyrM. Moreover, NB19 controls the genistein-induced expression of two other genes: psfHH103d_368 and psfHH103d_369, which code for a putative transcriptional regulator and a 114-residue hypothetical protein, respectively.
S. fredii HH103 genes induced through tts boxes (TB)
Some Gram-negative bacterial strains possess a specialized apparatus for protein secretion termed T3SS. Pathogenic and symbiotic bacteria, including rhizobia, use this system to deliver effector proteins directly into the eukaryotic host cell34 in order to interfere with the host signal transduction cascades and promote pathogen or symbiotic infection by suppressing host defenses35,36. In rhizobia, these proteins are called Nops (Nodulation outer proteins).
S. fredii strains NGR234 and HH103 possess a T3SS that is induced by flavonoids and depends on NodD1 since expression of ttsI, the T3SS transcriptional regulator, is driven by a NB6. Thus, up-regulation of all T3SS genes is mediated by TtsI through binding to specific promoter sequences called tts boxes (TB). As previously mentioned, HH103 harbors 18 potential TB distributed among several replicons20. Our present study allowed the functional verification of these potential promoter regions, considering active those TB located upstream of genes transcriptionally activated with genistein in the wild-type strain but not in the nodD1 and ttsI mutants. RNA-seq analysis showed that only 11 tts boxes (TB1, 2, 3, 4, 5, 8, 9, 10, 11, 12 and 13) were active, controlling the expression of 35 ORFs (Supplementary Table S2, Supplementary Data 4). Although the genes located downstream of TB7 (psfHH103d_322-to psfHH103d_319) showed enhanced expression in the presence of genistein, this tts box was considered non-functional since this pattern of induction was not altered by the inactivation of ttsI. As discussed later, the upstream sequence of this genetic region (which includes the transcriptional regulator nodD2) contains a putative SyrM box which might explain why these genes are induced by genistein.
Most of the HH103 functional TB were located on the symbiotic plasmid, with the exception of TB1 and TB2, which were present in plasmid c. The differential expression of gunA (TB5), nopL (TB9), nopC (TB12) and nopI (TB2) was validated by qPCR (Supplementary Table S1; Supplementary Data 3) and previous works have shown that the expression of rhcJ (TB10), nopA (TB12), nopP (TB11) and nopX (TB8) was also regulated by flavonoids, NodD1 and TtsI6,37. All functional TB mainly followed the consensus GTCAGN5CGN2AGN10TA while the non-active TB failed to maintain the internal sequence CGN2AG (Fig. 3B). Four of these active TB (TB5, 8, 10 and 12) control the expression of operons coding for proteins that i) are components of the T3SS apparatus (such as nopBrhcJnolUrhcLNQRSTU and nopCAy4yQ, which depend on TB10 and TB12, respectively), ii) are implied in the translocation of proteins to the cytoplasm of the root cell (the nopX operon controlled by TB8), or iii) code for glycolitic enzymes (the gunA gene controlled by TB5) (Supplementary Table S3, Supplementary Data 4). The rest of the active tts promoter sequences (TB1, 2, 3, 4, 9, 11 and 13) drive the expression of genes that code for the putative effector proteins NopI, NopM/M2, NopD, NopL, NopP and NopT. These results are in agreement with previous reports in which the type 3-dependent secretion of at least nine S. fredii HH103 Nops (NopA, NopB, NopC, NopD, NopL, NopM/NopM2, NopP and NopX) has been demonstrated6,38. NopA, NopB and NopX are the main components of the T3SS extracellular appendages39,40,41. Secretion of NopP and NopC to the interior of Vigna unguiculata and Glycine max nodule cells, respectively, has been recently confirmed42,43 and the rest can be considered putative effectors. Results shown in this work allowed the identification, for the first time in a rhizobial strain, of the complete repertoire of T3SS-effectors. However, we cannot discard the future identification of other effectors that do not follow the up to date accepted signaling cascade that involves flavonoids, NodD1 and TtsI.
The confirmation that gunA and nopI were transcriptionally activated in a flavonoid-, NodD1- and TtsI-dependent manner was another interesting finding. GunA, previously identified only in B. japonicum, is a cellulase that could be implied on the breaking of the plant cell wall to insert the inyectisome in the plant cell membrane and allow protein translocation directly into the eukaryotic cytoplasm44. NopI, located on plasmid c downstream TB2, could be a new type of rhizobium-specific T3SS effector not previously described20 but further efforts are necessary to identify unequivocally this protein as a HH103 secreted effector. Interestingly, nopI appears to be the first gene of an operon that also contains three genes (SFHH103_04165 to SFHH103_04167) encoding the N-terminal, middle and C-terminal parts of a putative reverse transcriptase whose functionality and putative symbiotic involvement remain to be investigated.
S. fredii HH103 genes not controlled by NB or TB but regulated by genistein
The last set of HH103 DEG in the presence of genistein corresponds to those lacking defined NB or TB promoter regions. Our study revealed that 30 S. fredii HH103 ORFs can be included in this group (Supplementary Table S3, Supplementary Data 4). These genes were distributed among the chromosome and plasmids d and e, being 22 of them up-regulated and 8 down-regulated in the presence of genistein. The absence of known NB or TB promoter regions does not necessarily imply transcriptional independence of NodD1 and TtsI. In fact, the genistein-mediated differential expression of 24 of these 30 genes depends on the presence of a functional NodD1 protein, being 7 out of these 24 genes apparently also TtsI-dependent (Supplementary Table S3, Supplementary Data 4). This finding indicates that S. fredii HH103 NodD1 regulates a set of genes that do not depend on NB or TB. Regarding genes depending on NodD1, it could be possible that this additional level of regulation was mediated, at least partially, by the NB-controlled transcriptional regulator SyrM (NB19, see above). In this sense, well conserved SyrM boxes, as defined by Barnett and Long in the closely related S. meliloti Rm102133, were found upstream two operons that were highly induced by genistein and lacked functional NB or TB (Fig. 3C, Supplementary Table S3, Supplementary Data 4): psfHH103d_322-psfHH103d_319 (encoding hypothetical proteins and the regulatory protein NodD2) and psfHH103d_306-psfHH103d_311 (encoding hypothetical proteins putatively involved in electron transfer). As mentioned above, the tts box located upstream of psfHH103d_322- psfHH103d_319 has been considered as non-functional because this operon remains genistein-inducible in a ttsI mutant background. Putative SyrM boxes were located at positions −223 to −158 and −127 to −62 upstream the translation initial site of psfHH103d_322 and psfHH103d_306 respectively, but its functionality remains to be demonstrated.
The genistein-NodD1-induced expression of psfHH103d_306 was confirmed by qPCR (Supplementary Table S1, Supplementary Data 3). qPCR experiments also showed that HH103 nodD2 is positively regulated by NodD1 and genistein (±6.1 ± 1.1), suggesting that in S. fredii HH103 the expression of nodD2 (and the other genes of the operon) was most probably regulated through SyrM, as previously reported for the closely related strain NGR23445. Thus, in S. fredii HH103, regulation of symbiotic genes by flavonoids appeared to be rather complex since it involved NodD1, TtsI, NolR and possibly SyrM and NodD2. In fact, the latter has been reported to act as a repressor of nodD1 expression in S. fredii NGR234 and USDA19145,46. Further transcriptomic analyses of the role of NodD2, SyrM and NolR in gene regulation are required to determine the general regulatory circuit that controls the most basic aspects of the molecular dialogue involved in the symbiotic interaction between HH103 and its host legumes.
We have found several genes that are inducible by genistein and NodD1 and that do not contain NB, TB or SyrM boxes in their upstream regions, which indicates that unknown actors mediating the flavonoid-NodD1 regulatory cascade remain to be discovered. Among these genes, the expression pattern of psfHH103d_255 and SFHH103_02192 has been confirmed by qPCR (Supplementary Table S1, Supplementary Data 4). psfHH103d_255 is located on plasmid d and codes for a putative membrane protein belonging to the major facilitator superfamily MFS-1. SFHH103_02192 is situated in the chromosome and its predicted product is a hypothetical protein containing a complete COG2931 domain (Ca2+-binding protein, RTX toxin-related) as well as two incomplete Peptidase_M10_C domains (Peptidase M10 serralysin C terminal). Another chromosomal gene induced by NodD1 and genistein but not controlled by known regulatory promoters was ligE (SFHH103_00841), which codes for a β-aryl ether cleaving enzyme containing a GstA (gluthatione S-transferase, GST) domain. This kind of proteins is usually implied in detoxification processes. Plant GSTs are abundant in nodules and likely function to provide antioxidant defenses that are critical to support nitrogen fixation47. To our knowledge, this is the first time that a rhizobial (putative) GST is found to be inducible by NodD1 and flavonoids.
As mentioned above, our results indicated that the differential expression upon treatment with genistein of 7 CDSs with unknown promoter regions depended on the presence of both NodD1 and TtsI. Among these genes, we found a putative operon located on the chromosome that is composed of flgJ and two genes coding for conserved hypothetical proteins (Supplementary Table S3, Supplementary Data 4). The expression pattern of flgJ was confirmed by qPCR (Supplementary Table S1). flgJ codes for a flagellar protein containing a scaffolding domain required for polymerization of the distal rod of the flagellum48. Analysis of the upstream sequence of flgJ revealed the presence of imperfect tts and SyrM boxes (Fig. 3D), located at positions −152 to −126 and −817 to −752 upstream the translation initial site of flgJ, but to elucidate whether the differential expression of this operon depends on these putative promoter sequences requires further investigation. Nevertheless, the fact that flgJ was induced by genistein opens the possibility that flavonoids could affect HH103 motility. We plan to investigate this hypothesis in the next future. Interestingly, the bacterial flagellum shares a common ancestor with the T3SS49, which suggests that up-regulation of this gene could be related with the correct formation and assembly of T3SS machinery across the bacterial cell wall.
Finally, RNA-seq analysis showed that 4 genes were affected by genistein independently of the presence of NodD1 or TtsI. One of these genes, SFHH103_03875, located on the chromosome and induced with genistein (~6-fold), codes for a putative membrane permease containing two EamA domains. Database searching showed that this gene is well conserved among different rhizobia, but to our knowledge it has not been characterized. In addition, a set of three genes situated on plasmid e (SFHH103_05321 to SFHH103_05319) was repressed by the presence of genistein independently of NodD1 and TtsI, as validated by qPCR (Supplementary Table S1). This operon codes for conserved hypothetical proteins showing similarities with a TetR transcriptional regulator and two multidrug efflux transporter proteins (Supplementary Data 4). Recently, Rossbach and coworkers50 showed that three genes coding for a TetR regulator (EmrR) and two components (EmrAB) of an efflux system were induced by flavonoids in S. meliloti, most probably in a NodD-independent way. In addition to the different effect of flavonoids on their expression, emrAB are not orthologues of SFHH103_05320 and 05319. Thus, it is intriguing why two closely-related rhizobia harbor different efflux systems regulated by the presence of flavonoids but independently of NodD and why the efflux system of S. meliloti is induced whereas that of S. fredii is repressed.
Conclusion
A better understanding of the molecular mechanisms operating in the rhizobia-legume symbiosis is a requisite for the improvement of this interaction as well as for its putative extension to other families of plants of ecological and agronomical importance. The RNA-seq and qPCR analyses shown in this work define three groups of genes differentially expressed upon induction with genistein on S. fredii HH103: genes controlled by NB (e.g. nod and fix regions, ttsI and syrM genes), genes regulated by TB (e.g. rhc and nop regions) and genes not preceded by a NB or a TB (e.g. ligE and flgJ genes) (Fig. 4). A scheme summarizing the genistein stimulon (i.e., the set of genes whose expressions is affected by the presence of this flavonoid) of S. fredii HH103 is shown in Fig. 5. As can be observed, various regulatory elements remain to be identified. Our results are in agreement with previous reports indicating that nod gene inducing flavonoids possess a much broader transcriptional effect than the mere induction of genes implied in production of Nod factors and Nops12,13. Thus, there are many other DEG that remain to be characterized. Further efforts will be needed to describe the function of these genes, which would improve our understanding of the molecular dialogue established between both symbionts.

Linear representation of the complete RNA-seq-based transcriptomic data set of bacterial cultures induced with genistein for all replicons of S. fredii HH103.
Bars represent fold-change expression values. Each bar corresponds to one gene, being ordered according to their relative position in the replicon. Chr: Chromosome, pl. a1: pSfHH103a1, pl. a2: pSfHH103a2, pl. b: pSfHH103b, pl. c: pSfHH103c, pl. d (pSym): pSfHH103d and pl. e: pSfHH103e. Blue bars: ORFs regulated by genistein preceded by nod boxes and regulated by NodD1. Yellow bars: ORFs regulated by genistein preceded by tts boxes and regulated by TtsI. Red bars: ORFs regulated by genistein but not dependent on nod or tts boxes. Gene names shown correspond to those genes that have been annotated.

Model of the genistein stimulon of S. fredii HH103.
This scheme summarizes the information obtained in this work as well as results about the NolR repressor protein previously reported11. Induction and repression of genes (and processes) are indicated in red and in blue arrows, respectively. NB, nod box; TB, tts box; SB, putative SyrM box. Framed question marks indicate the possibility of the participation of unknown regulatory elements. The green circle surrounding SyrM, SB1 and SB2 denotes the putative involvement of these elements in the NodD1-genistein-mediated induction of psfHH103d_306-311 and psfHH103d_322-319.
Methods
Culture conditions and RNA extraction
Three Sinorhizobium fredii strains, namely HH103 RifR 51, HH103 RifRnodD1::Ω10 and HH103 RifRttsI::Ω6 were grown at 28 °C until stationary phase (OD600 ≈ 1,2) on yeast extract mannitol medium (YM)52, supplemented with genistein 3.7 μM when necessary. When required, the media were supplemented with the antibiotics rifampicin (50 μg ml−1) or spectinomycin (50 μg ml−1). Total RNA was isolated using a High Pure RNA Isolation Kit (Roche) according to the manufacturer’s instructions. Verification of the amount and quality of the resulting total RNA samples was carried out using a Nanodrop 1000 spectrophotometer (Thermo Scientific) and a Qubit 2.0 Fluorometer (Invitrogen). Two independent total RNA extractions were obtained for each condition.
Quantitative reverse transcription PCR
Results obtained in the RNA-seq analysis were validated by quantitative reverse transcription PCR (qRT-PCR) of 19 selected genes, which represented differentially and non-differentially expressed genes in the three strains in the presence of genistein. Total RNA was isolated using a High Pure RNA Isolation Kit (Roche) and RNAase Free DNAse (Qiagen) according to the manufacturer’s instructions. This (DNA free) RNA was reverse transcribed to cDNA using a QuantiTec Reverse Transcription Kit (Qiagen). Quantitative PCR was performed using a LightCycler 480 (Roche) with the following conditions: 95 °C, 10 min; 95 °C, 30 s; 50 °C, 30 s; 72 °C, 20 s; forty cycles, followed by the melting curve profile from 60 to 95 °C to verify the specificity of the reaction. The S. fredii HH103 RNA 16S gene was used as an internal control to normalize gene expression. The fold changes of two biological samples with three technical replicates in each condition were obtained using the ∆∆Ct method53. Selected genes and primers are listed in Supplementary Data 3.
RNA sequencing
Ribosomal RNA was depleted using a MICROB Express Bacterial mRNA Purification kit (Ambion), following the manufacturer’s protocol. Integrity and quality of the ribosomal depleted RNA was checked with Agilent Bioanalyzer 2100 (Agilent Technologies). RNA sequencing was carried out by Sistemas Genómicos (https://www.sistemasgenomicos.com/web_sg/) with the Next Generation Sequence (NGS) platform Illumina using the Illumina HiSeq 2000 sequencing instrument (Illumina). Ribosomal-depleted samples were used to generate whole transcriptome libraries following the manufacturer’s recommendations for sequencing on this NGS platform. Amplified cDNA quality was analyzed by the Bioanalyzer 2100 DNA 1000 kit (Agilent Technologies) and quantified using the Qubit 2.0 Fluorometer (Invitrogen).
Mapping of the RNA-seq data
The initial whole transcriptome paired-end reads obtained from sequencing were mapped against the latest version of the S. fredii HH103 genome (http://www.ncbi.nlm.nih.gov/assembly/GCF_000283895.1/) using the Life Technologies mapping algorithm version 1.3 (http://www.lifetechnologies.com/). Low-quality reads were eliminated using Picard Tools software version 1.83, remaining only high quality reads.
Assessment of differentially expressed genes
Gene prediction was estimated using the cufflinks method54. Quantification of gene expression levels was evaluated using the htseq-count method software, version 0.5.4p3 and the algorithm proposed by DESeq255,56. This method eliminates multimapped reads, considering only unique reads for the gene expression estimation. The edgeR method version 3.2.4 was applied for differential expression analysis among conditions57. This method uses a Poisson model to estimate the variance of the RNA-seq data for differential expressions and relies on different normalized processes based on depth global samples, CG composition and length of genes. RNA-seq data of each treatment were compared to those of the wild-type strain grown in the absence of genistein. Differentially expressed genes were defined as those genes with a fold-change lower or higher than −3 or 3, respectively, with a p value inferior to 0.05.
Consensus motifs
Functional nod and tts boxes of S. fredii HH103 identified by Vinardell et al.20 and validated by RNA-seq in this work were aligned using the ClustalW program and manipulated with Boxshade at EMBnet.
Search of conserved nucleotide motifs (such as SyrM boxes) was done by using the fuzznunc utility from the EMBOSS software package58.
Search of conserved protein motifs was performed at NCBI CD-search (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
RNA-seq data accession number
The RNA-seq data discussed in this publication have been deposited in the Sequence Read Archive of NCBI (BioProject database) under the BioProject ID PRJNA313151.
Additional Information
How to cite this article: Pérez-Montaño, F. et al. A transcriptomic analysis of the effect of flavonoids on Sinorhizobium fredii HH103 reveals novel rhizobial genes putatively involved in symbiosis. Sci. Rep. 6, 31592; doi: 10.1038/srep31592 (2016).
References
López-Baena, F. J., Ruiz-Sainz, J. E., Rodríguez-Carvajal, M. A. & Vinardell, J. M. Bacterial molecular signals in the Sinorhizobium fredii-soybean symbiosis. Int. J. Mol. Sci. 17, 755 (2016).
Suzaki, T. & Kawaguchi, M. Root nodulation: a developmental program involving cell fate conversion triggered by symbiotic bacterial infection. Curr. Opin. Plant. Biol. 21, 16–22 (2014).
Schlaman, H. R. M., Phillips, D. A. & Kondorosi E. Genetic organization and transcriptional regulation of rhizobial nodulation genes. In: The Rhizobiaceae (eds Spaink, H. P., Kondorosi, A. & Hooykaas, P. ) 361–386 (Kluwer Academic Publishers, 1998).
Spaink, H. P. Root nodulation and infection factors produced by rhizobial bacteria. Annu. Rev. Microbiol. 54, 257–288 (2000).
Peck, M. C., Fisher, R. F. & Long, S. R. Diverse flavonoids stimulate NodD1 binding to nod gene promoters in Sinorhizobium meliloti. J Bacteriol. 188, 5417–5427 (2006).
López-Baena, F. J. et al. Regulation and symbiotic significance of nodulation outer proteins secretion in Sinorhizobium fredii HH103. Microbiology 154, 1825–1836 (2008).
Pérez-Montaño, F. et al. Nodulation-gene-inducing flavonoids increase overall production of autoinducers and expression of N-acyl homoserine lactone synthesis genes in rhizobia. Res. Microbiol. 162, 715–723 (2011).
Pérez-Montaño, F. et al. The symbiotic biofilm of Sinorhizobium fredii SMH12, necessary for successful colonization and symbiosis of Glycine max cv Osumi, is regulated by Quorum Sensing systems and inducing flavonoids via NodD1. PLoS One 9, e105901 (2014).
Theunis, M., Kobayashi, H., Broughton, W. J. & Prinsen, E. Flavonoids, NodD1, NodD2 and nod-box NB15 modulate expression of the y4wEFG locus that is required for Indole-3-Acetic Acid Synthesis in Rhizobium sp. strain NGR234. Mol. Plant Microbe Interact. 17, 1153–1161 (2004).
Vinardell, J. M. et al. The effect of FITA mutations on the symbiotic properties of Sinorhizobium fredii varies in a chromosomal-background-dependent manner. Arch. Microbiol. 181, 144–154 (2004).
Vinardell, J. M. et al. NolR regulates diverse symbiotic signals of Sinorhizobium fredii HH103. Mol. Plant Microbe Interact. 17, 676–685 (2004).
Lang, K., Lindemann, A., Hauser, F. & Göttfert, M. The genistein stimulon of Bradyrhizobium japonicum. Mol. Genet. Genomics 279, 203–211 (2008).
Huyghe, A., Bakkou, N. & Perret, X. Profiling symbiotic responses of Sinorhizobium fredii strain NGR234 with RNA-seq. In: Biological Nitrogen Fixation 2 (ed. de Bruijn, F. J. ) 1153–1162 (John Wiley & Sons, 2015).
Pueppke, S. G. & Broughton, W. J. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant-Microbe Interact. 12, 293–318 (1999).
de Lyra, M. C. C. P. et al. Inactivation of the Sinorhizobium fredii HH103 rhcJ gene abolishes nodulation outer proteins (Nops) secretion and decreases the symbiotic capacity with soybean. Int. Microbiol. 9, 125–133 (2006).
Margaret, I. et al. Symbiotic properties and first analyses of the genomic sequence of the fast growing model strain Sinorhizobium fredii HH103 nodulating soybean. J. Biotechnol. 155, 11–19 (2011).
Schmeisser, C. et al. Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systems. Appl. Environ. Microbiol. 75, 4035–4045 (2009).
Schuldes, J. et al. Complete genome sequence of the broad-host-range strain Sinorhizobium fredii USDA257. J. Bacteriol. 194, 4483 (2012).
Weidner, S. et al. Genome sequence of the soybean symbiont Sinorhizobium fredii HH103. J. Bacteriol. 194, 1617–1618 (2012).
Vinardell, J. M. et al. The Sinorhizobium fredii HH103 genome: a comparative analysis with S. fredii strains differing in their symbiotic behaviour with soybean. Mol. Plant Microbe Interact. 28, 811–824 (2015).
Perret, X., Freiberg, C., Rosenthal, A., Broughton, W. J. & Fellay. R. High-resolution transcriptional analysis of the symbiotic plasmid of Rhizobium sp. NGR234. Mol. Microbiol. 32, 415–425 (1999).
Hansen, K. D., Irizarry, R. A. & Wu, Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 3, 204–216 (2012).
Li, F. Q., Hou, B. H., Chen, L., Yao, Z. J. & Hong, G. F. In vitro observation of the molecular interaction between NodD and its inducer naringenin as monitored by fluorescence resonance energy transfer. Acta. Biochim. Biophys. Sin. (Shanghai). 40, 783–789 (2008).
Lamrabet, Y. et al. Mutation in GDP-fucose synthesis genes of Sinorhizobium fredii alters Nod factors and significantly decreases competitiveness to nodulate soybeans. Mol. Plant Microbe Interact. 12, 207–217 (1999).
Krishnan, H. B., Lewin, A., Fellay, R., Broughton, W. J. & Pueppke, S. G. Differential expression of nodS accounts for the varied abilities of Rhizobium fredii USDA257 and Rhizobium sp. strain NGR234 to nodulate Leucaena spp. Mol. Microbiol. 6, 3321–3330 (1992).
Hirsch, A. M. & Smith, C. A. Effects of Rhizobium meliloti nif and fix mutants on alfalfa root nodule development. J. Bacteriol. 169, 1137–1146 (1987).
Edgren, T. & Nordlund, S. The fixABCX genes in Rhodospirillum rubrum encode a putative membrane complex participating in electron transfer to nitrogenase. J. Bacteriol. 186, 2052–2060 (2004).
del Cerro, P. et al. Regulatory nodD1 and nodD2 genes of Rhizobium tropici strain CIAT 899 and their roles in the early stages of molecular signaling and host-legume nodulation. BMC Genomics 16, 251 (2015).
Silipo, A. et al. Covalently linked hopanoid-lipid A improves outer-membrane resistance of a Bradyrhizobium symbiont of legumes. Nat. Commun. 5, 5106 (2014).
Kulkarni, G. et al. Specific hopanoid classes differentially affect free-living and symbiotic states of Bradyrhizobium diazoefficiens. MBio. 6, 5 (2015).
Janczarek, M. Environmental signals and regulatory pathways that influence exopolysaccharide production in rhizobia. Int J Mol Sci. 12, 7898–7933 (2011).
Mimmack, M. L., Borthakur, D., Jones, M. A., Downie, J. A. & Johnston, A. W. The psi operon of Rhizobium leguminosarum biovar phaseoli: identification of two genes whose products are located at the bacterial cell surface. Microbiology 140, 1223–1229 (1994).
Barnett, M. J. & Long, S. R. The Sinorhizobium meliloti SyrM regulon: effects on global gene expression are mediated by syrA and nodD3. J. Bacteriol. 197, 1792–1806 (2015).
Soto, M. J., Sanjuán, J. & Olivares, J. Rhizobia and plant-pathogenic bacteria: common infection weapons. Microbiology. 152, 3167–3174 (2006).
Alfano, J. R. & Collmer, A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu. Rev. Phytopathol. 42, 385–414 (2004).
Jiménez-Guerrero, I. et al. The Sinorhizobium (Ensifer) fredii HH103 Type 3 secretion system suppresses early defense responses to effectively nodulate soybean. Mol. Plant Microbe Interact. 28, 790–799 (2015).
López-Baena, F. J. et al. The absence of Nops secretion in Sinorhizobium fredii HH103 increases GmPR1 expression in Williams soybean. Mol. Plant Microbe Interact. 22, 1445–1454 (2009).
Rodrigues, J. A. et al. NopM and NopD are rhizobial nodulation outer proteins: identification using LC-MALDI and LC-ESI with a monolithic capillary column. J. Proteome Res. 6, 1029–1037 (2007).
Deakin, W. J., Marie, C., Saad, M. M., Krishnan, H. B. & Brougthon, W. J. NopA is associated with cell surface appendages produced by the type III secretion system of Rhizobium sp. strain NGR234. Mol. Plant Microbe Interact. 18, 499–507 (2005).
Lorio, J. C., Kim, W. S. & Krishnan, H. B. NopB, a soybean cultivar-specificity protein from Sinorhizobium fredii USDA257, is a type III secreted protein. Mol. Plant Microbe Interact. 17, 1259–1268 (2004).
Saad, M. M. et al. NopB, a type III secreted protein of Rhizobium sp. strain NGR234, is associated with pilus-like surface appendages. J. Bacteriol. 187, 1173–1181 (2005).
Schechter, L. M., Guenther, J., Olcay, E. A., Jang, S. & Krishnan. H. B. Translocation of NopP by Sinorhizobium fredii USDA257 into Vigna unguiculata root nodules. Appl. Environ. Microbiol. 76, 3758–3761 (2010).
Jiménez-Guerrero, I., Pérez-Montaño, F., Medina, C., Ollero, F. J. & López-Baena, F. J. NopC is a Rhizobium-specific type 3 secretion system effector secreted by Sinorhizobium (Ensifer) fredii HH103. PLoS One 10, e0142866 (2015).
Caldelari Baumberger, I., Fraefel, N., Göttfert, M. & Hennecke, H. New NodW- or NifA-regulated Bradyrhizobium japonicum genes. Mol. Plant Microbe Interact. 16, 342–351 (2003).
Kobayashi, H., Naciri-Graven, Y., Broughton, W. J. & Perret, X. Flavonoids induce temporal shifts in gene-expression of nod-box controlled loci in Rhizobium sp. NGR234. Mol. Microbiol. 51, 335–347 (2004).
Machado, D., Pueppke, S. G., Vinardell, J. M., Ruiz-Sainz, J. E. & Krishnan, H. B. Expression of nodD1 and nodD2 in Sinorhizobium fredii, a nitrogen-fixing symbiont of soybean and other legumes. Mol. Plant-Microbe Interact. 11, 375–382 (1998).
Dalton, D. A. et al. Physiological roles of glutathione s-transferases in soybean root nodules. Plant Physiol. 150, 521–530 (2009).
Cohen, E. J. & Hughes, K. T. Rod-to-hook transition for extracellular flagellum assembly is catalyzed by the L-ring-dependent rod scaffold removal. J. Bacteriol. 196, 2387–2395 (2014).
Blocker, A., Komoriya, K. & Aizawa, S. Type III secretion systems and bacterial flagella: insights into their function from structural similarities. Proc. Natl. Acad. Sci. USA 100, 3027–3030 (2003).
Rossbach, S., Kunze, K., Albert, S., Zehner, S. & Göttfert, M. The Sinorhizobium meliloti EmrAB efflux system is regulated by flavonoids through a TetR-like regulator (EmrR). Mol. Plant Microbe Interact. 27, 379–387 (2014).
Madinabeitia, N. et al. Sinorhizobium fredii HH103 has a truncated nolO gene due to a -1 frameshift mutation that is conserved among other geographically distant S. fredii strains. Mol. Plant-Microbe Interact. 15, 150–159 (2002).
Vincent, J. M. The modified Fahraeus slide technique. In A manual for the practical study of root nodule bacteria (ed. Vicent, J. M. ) (Blackwell Scientific Publications, 1970).
Pfaffl, M. W., Horgan, G. W. & Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36 (2002).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 31, 166–169 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140 (2010).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Acknowledgements
This work was supported by projects BIO2011-30229-C01 and AGL2012-38831 of the Ministerio de Economía y Competitividad of the Spanish government and projects P11-CVI-7050 and P11-CVI-7500 of the Junta de Andalucía. Dr. Pérez-Montaño (post-doctoral) and Mr. Acosta-Jurado and Mrs. Navarro-Gómez (PhD students) are recipients of grants of the V Plan Propio of the University of Seville (VPPI-US). We also would like to acknowledge Juan Carlos Treviño (Sistemas Genómicos S.L.) for his valuable help during all the bioinformatics analyses, Dr. Jonathan Trow (Sequence Read Archive from NCBI) for his technical assistance during the RNA-seq data submission and the Servicio General de Biología of the CITIUS of the University of Seville for their scientific equipment.
Author information
Authors and Affiliations
Contributions
F.J.L.-B. and J.M.V. conceived and designed the experiments. F.P.-M., I.J.-G., S.A.-J. and P.N.-G. performed the experiments. F.J.O., J.E.R.-S., J.M.V., F.P.-M., I.J.-G. and S.A.-J. analyzed the data. F.J.O., J.E.R.-S., F.J.L.-B. and J.M.V. contributed with reagents/materials/analysis tools. F.P.-M., F.J.L.-B. and J.M.V. wrote the paper. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pérez-Montaño, F., Jiménez-Guerrero, I., Acosta-Jurado, S. et al. A transcriptomic analysis of the effect of genistein on Sinorhizobium fredii HH103 reveals novel rhizobial genes putatively involved in symbiosis. Sci Rep 6, 31592 (2016). https://doi.org/10.1038/srep31592
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31592
This article is cited by
-
Fine-Tuned Immune Antagonism and Nodule-Specific Cysteine-Rich Peptides Govern the Symbiotic Specificity Between Alfalfa Cultivars and Ensifer meliloti
Journal of Plant Growth Regulation (2023)
-
Plant transcriptome analysis reveals specific molecular interactions between alfalfa and its rhizobial symbionts below the species level
BMC Plant Biology (2020)
-
Structural and enzymatic characterisation of the Type III effector NopAA (=GunA) from Sinorhizobium fredii USDA257 reveals a Xyloglucan hydrolase activity
Scientific Reports (2020)
-
GunA of Sinorhizobium (Ensifer) fredii HH103 is a T3SS-secreted cellulase that differentially affects symbiosis with cowpea and soybean
Plant and Soil (2019)
-
The non-flavonoid inducible nodA3 and the flavonoid regulated nodA1 genes of Rhizobium tropici CIAT 899 guarantee nod factor production and nodulation of different host legumes
Plant and Soil (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
