
Abstract
Bacterial communities from freshwater lakes are shaped by various factors such as nutrients, pH value, temperature, etc. Their compositions and relative abundances would undergo changes to adapt the changing environments and in turn could affect the environments of freshwater lakes. Analyses of the freshwater lake’s bacterial communities under different environments would be of pivotal importance to monitor the condition of waterbody. In this study, we have collected freshwater samples from two lakes on Yun-Gui plateau of China, Lake Dianchi and Lake Haixihai and analyzed the bacterial community structures from these samples based on 16S rRNA sequencing. Results have shown that: Firstly, the bacterial community of these samples have very different taxonomical structures, not only between two lakes but also among the intra-groups for samples collected from Dianchi. Secondly, the differences between samples from two lakes are highly associated with the chemical-geographical properties of the two lakes. Thirdly, for samples of Dianchi and Haixihai, analytical results of physicochemical, taxonomical structure and relative abundance of community revealed that extreme physicochemical factors caused by human activities have strongly affected the bacterial ecosystem in Dianchi. These results have clearly indicated the importance of combining biological profiling and chemical-geographical properties for monitoring Chinese plateau freshwater bacterial ecosystem, which could provide clues for Chinese freshwater ecosystem remediation on plateau.
Similar content being viewed by others

Introduction
It is well-known that freshwater habitats, especially bacterial communities, play essential roles in global biogeochemical cycles. The taxonomical structure and relative abundance of bacterial community in freshwater would undergo changes to adapt the changing environments and they would in turn affect the environments of freshwater lakes. As reported, the quality of freshwater has already been seriously affected by salinization, eutrophication and microbiological pollution1. Particularly, eutrophication, the biological response to the excess input of nutrients mainly introduced by human activities2 into a water body, can arise under natural conditions. For example, cyanobacteria blooms, which occurred as a result of the rapid growth of Cyanobacteria, especially from genus Microcytis3,4 with complex communities of photosynthetic and heterotrophic microorganisms, are often associated with the production of a series of different secondary metabolites produced by members of the genera Anabaena, Microcystis, Planktothrix5, Aphanizomenon and Cylindrospermopsis6. The physicochemical characters of freshwater, such as temperature and nutrition, would affect the taxonomical structure and relative abundance of species within the bacterial communities7 in a water body.
Therefore, it would be of pivotal importance to investigate the taxonomical structure of freshwater communities and the driving forces that could shape the bacterial communities in freshwater, which would provide clues for freshwater ecosystem remediation. Previous studies have showed nitrogen, phosphorus8 and temperature9,10 as main driving forces that could alter the taxonomical structure of bacterial community in a surface water body. However, considering geographical regions, previous researches mainly studied oceans, rivers or plain lakes11,12, the precise information of the bacterial community in plateau lakes is still deficient. Furthermore, the subjects of these studies were mainly focused on the taxonomical composition of sediment community7. Therefore, there is a lack of studies on bacterial communities in Chinese plateau freshwater lakes.
It is generally known that lakes could exist in two alternative stable states of oligotrophy (clear with abundant submerged macrophytes) and eutrophy (turbid with abundant algae). These two types of lakes have different geobiochemical characteristics13. Comparing the structure of bacterial communities of these lakes not only will help us to have a better understanding of the environmental factors’ driving mechanisms of bacterial communities in the freshwater lakes, but also could provide valuable information to protect and remediate these lakes. Hence, it is essential to explore the bacterial communities in Chinese plateau freshwater lakes.
In this work, we have chosen two Chinese plateau freshwater lakes, Dianchi (DC) and Haixihai (HXH), from where community samples were collected and compared. DC is the largest plateau lake in China, located on the Yun-Gui plateau, Southwestern of China. Due to the aggravation of human activities, many parts of the lake have been seriously deteriorated by eutrophication and toxic cyanobacteria bloom. HXH is a relatively non-polluted, macrophytic and oligotrophic freshwater lake, also located on the Yun-Gui plateau, about 300 kilometers from DC. These two lakes represent two types of lakes: DC is eutrophic, turbid with abundant algae; while HXH is oligotrophic, clear with abundant submerged plants. By comparing the structure of bacterial communities of these lakes, we could be able to better understand the mechanisms through which environmental factors could alter bacterial communities in the freshwater lakes, which may provide clues for eutrophic lake remediation.
16S rRNA sequences of environmental DNA were commonly used to study the bacteria composition and diversity of natural bacterial communities7, decoding the taxonomical structure of communities with fairly high resolution. More importantly, uncultured bacteria in the community could be assessed qualitatively and quantitatively. In this study, we have used Illumina MiSeq platform for sequencing the V4-V5 regions of the 16S rRNA gene to profile the bacterial community’s patterns of samples collected from DC and HXH.
Based on 16S rRNA sequencing and data analyses for bacterial communities in two plateau lakes, we seek answers for the following questions: (i) Are taxonomical structures of bacterial communities of DC different from those of HXH? (ii) How do environmental factors affect the structure of bacterial communities? What are key environmental factors that could differentiate samples? (iii) What species could serve as biomarkers to differentiate samples from two lakes or those from intra-groups of DC?
Results and Discussions
General statistical information of environmental variables and sequencing data
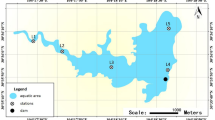
Environment characterization: 19 samples (14 samples from DC and 5 samples from HXH, each from a different site, Fig. 1) were collected and analyzed. These samples were collected at locations with different environmental characteristics.

The geographical locations of DC and HXH and the sampling sites for this study.
Red box indicates sampling location for DC samples, blue box indicates sampling location for HXH samples. The geographical locations were drawn using Adobe Photoshop and Adobe Illustrator.
Samples from DC and HXH have shown strong variations in dissolved oxygen (DO), total nitrogen (TN), ammonia nitrogen (NH4+-N), nitrate nitrogen (NO3−-N), nitrite nitrogen (NO2−-N), total phosphorus (TP), orthophosphate (PO43−-P), suspended solids (SS), potassium permanganate index (CODMn) and chlorophyll a (Chl-a) (t-test for per characterization, all P < 0.05, the value of each physicochemical character was shown in Supplementary Table S1). Phytoplankton biomass (proxied by Chl-a) of DC was higher than that of HXH. For intra-group samples from DC, TN (6.38–9.35 mg/L), TP (0.157–0.558 mg/L) and Chl-a (205.3–364.1 μg/L) in most of Caohai Lake (D-C) samples were all higher than what has been measured in Waihai Lake (D-W) (TN, 1.75–4.27 mg/L; TP, 0.096–0.205 mg/L; Chl-a, 57.2–148.6 μg/L), except DC7.
General statistics of sequencing results
For all of the 19 samples, 3,904,457 paired raw reads with read length of 301 bp were obtained. The number of reads per sample ranged from 222,894 to 428,888, with an average of 303,809 (Table 1). After trimming, screening, removing chimeras and singletons, 107,846 unique high quality reads per sample (2,047,735 reads in total for 19 samples) were obtained for downstream analysis.
Richness and diversity analyses based on Operational Taxonomic Units
A total of 3,836 OTU were obtained from 19 samples. Samples from DC contain more OTUs than HXH except sample DC7 (Table 1), probably because community diversity of DC7 has been damaged by the extreme environment (with very high TN and CODMn values). The GOOD’s coverage of these samples was estimated to be between 99.72% and 99.91% (Table 1). The rarefaction curves based on the OTUs (Fig. 2) have shown that all the samples tend to approach the saturation plateau, indicating that these two freshwater lakes have around 1,000~2,000 bacterial species. Among DC samples, the ACE and Chao1 values at 97% similarity levels were higher than those in HXH samples (Table 1), except sample DC7, meaning that the bacterial richness of DC samples was higher than those in HXH samples (Table 1). In general, the diversity of HXH was lower than that of DC, indicating that the ecological environment of HXH might be more fragile.

Rarefaction curves of OTUs for the bacterial communities’ samples from DC and HXH.
The rarefaction curves of determined tags tend to approach the saturation plateau.
Comparison of the composition of bacterial communities.
Bacterial community composition in Lake Dianchi and Lake Haixihai: Based on the OTUs’ information of each sample (except DC7), we found out that the top 20 OTUs represented the accumulative relative abundance of 35.24% and 58.25% in DC and HXH communities, respectively; and they have shared 7 OTUs in common (Table 2). Further analyses revealed that 163 abundant OTUs (in top 200 OTUs, except DC7) had significant differences between samples from DC and those from HXH (Supplementary file 1). Phylogenetic relationship analysis and relative abundance analysis on the top 200 OTUs (except DC7) have shown that these OTUs belong to the few most abundant phyla and their relative abundances varied between DC and HXH (Supplementary Figure S1).
Further analysis at phylum level showed that an average of 23.86 and 21.8 different phyla were identified for samples from DC and HXH. Dissimilarity tests based on the Bray-Curtis distance revealed that the community differences in phylum level between DC and HXH were significant (PERMANOVA, F = 8.354, P = 0.0004). Analyzed results revealed that the significant difference between DC and HXH could be owed to the existence of Cyanobacteria (Kruskal-Wallis test, except DC7, P = 0.003), Planctomycetes (Kruskal-Wallis test, except DC7, P = 0.002), Chlorobi (Kruskal-Wallis test, except DC7, P = 0.0008) and Verrucomicrobia (Kruskal-Wallis test, except DC7, P = 0.017).
More specifically, some taxa were more abundant in DC than in HXH at different taxonomical level. Firstly, as algal blooms related to eutrophication world-wide14, Cyanobacteria was characterized as the main causative organism15 (D-C, 0.65–2.33%, D-W, 5.31–15.83%). Secondly, in our study, Synechococcaceae (D-C, 0.12–1.52%, D-W, 4.38–15.67%) appeared as the main clade of Cyanobacteria (Fig. 3(b)) and dominated in DC samples compared to HXH. Thirdly, the Planctomycetales (0.35–1.35%) community amplified with Pirellulales (1.76–3.21%) can also be observed in large proportion in DC. Finally, the relative abundance of Saprospirae (a class of Bacteroidetes) had significant difference between DC and HXH (t-test, P < 0.05), which might be supported by a previous study, showing that the abundance of Bacteriodetes was associated with algae blooms16. The unclassified bacteria also took up a great proportion in samples from DC, indicating a much more complex taxonomical structure for samples in DC.

Taxonomical structure and relative abundance of each sample (a) at phylum level and (b) at family level.
At specific level, “others” represent those accounting for < 1% of the total OTUs in each sample and are shown in black at the top of each bar.
In contrast to samples of DC, Flavobacterium (a genus of Bacteroidetes, Fig. 3(b)) was the most abundant bacteria phylum in samples from HXH. In more details, HXH1-HXH5: 12.88–26.39%, HXH6 and HXH7: 4% and 8.68%. These value were small because of there were more than 49.9% OTU belonging to unclassified clades identified as the predominate group in the HXH6 and HXH7. Similarity phenomenon was found in various environments, such as soil and fresh water17. Studies showed that Flavobacterium, a heterotrophic bacteria that preferring an aerobic lifestyle18, was found to be abundant in freshwater and played a part in absorption and degradation of organic matter19. Moreover, Comamonadaceae (a family of Betaproteobacteria) and Verrucomicrobia, which have been discussed in a previous study showing that these taxa appear mostly in a specific degree of salinity20, account for a very large proportion of HXH (6.92% and 1.65%), but only a small percentage in DC (2.12% and 0.48%).
Although many environmental factors varied between DC and HXH, some dominant bacteria in samples from the two different lakes were similar. The main bacteria commonly existed in most freshwater lakes include Alphaproteobacteria (Table 2), which was abundant in both DC (7.30–19.86%) and HXH (12.47–18.37%). This indicated that Alphaproteobacteria was capable of living in both low nutrient environment and complex organic surroundings. Although the relative abundance of Alphaproteobacteria were moderate in many of the samples (Supplementary Figure S2), many studies have shown that they were ubiquitous and important for nitrogen cycle21. Additionally, Actinobacteria, especially ACK-M1 (a family of Actinobacteria, Fig. 3(b)), dominant in both DC and HXH (but there is no statistical difference between DC and HXH for Actinobacteria; Kruskal-Wallis test, except DC7, P = 0.144). Studies have shown that Actinobacteria dominated in all kinds of circumstances including soil and aquatic system21. The role of protistan predation may explain why they were common to the plankton22. Furthermore, Actinobacteria showed higher level of diversity in DC (Fig. 3(a)). It was rational to deduce that some specific bacteria of Actinobacteria could serve as saprophytic microbes that prefers eutrophic conditions23.
Furthermore, in order to examine the clustering patterns for samples from DC and HXH, a hierarchical clustering heatmap was generated based on all bacterial communities across all selected samples. The analyses have shown that DC7 was very distinct from other samples and samples of HXH were also different from samples of DC. Among samples from DC, samples other than the 5 samples (DC1, 3, 4, 5, 7) were clustered (Fig. 4). Samples from DC and HXH could also be distinguished by PCoA analysis (ANOSIM, P < 0.001), but group DC showed larger variance in one gradient direction (Fig. 5). Therefore, samples measured in DC and HXH could be clearly divided into three distinct clusters according to environmental factors: Cluster 1: samples in DC1, 3, 4, 5; Cluster 2: samples in DC6, 8, 9, 12, 14, 15, 16, 17, 20; Cluster 3: samples in HXH1, 2, 5, 6, 7. Additionally, the composition result, heatmap and PCoA result all revealed that DC7 was an exceptional sample. Therefore, comprehensive analytical results for DC7 would be detailed in a separate subsection.

Heatmap representing the differences among samples based on Bray-Crutis distance measures.
Bray-Curtis distances were calculated based on the formula:  . Computes the distance between μ and ν sample. i represents the OTU i in each sample. Based on the Bray-Crutis distance, a matrix was created among all samples and then for heatmap analysis.
. Computes the distance between μ and ν sample. i represents the OTU i in each sample. Based on the Bray-Crutis distance, a matrix was created among all samples and then for heatmap analysis.

PCoA of the dissimilarities among bacterial community taxonomical structures using thetaYC distances.
ThetaYC ( ) measures the dissimilarity between the structures of two communities, where ST is the total number of OTUs in communities μ and ν, μi and νi is the relative abundance of OTU i in community μ and ν, respectively. A matrix of pairwise thetaYC-based distances among all samples was calculated for PCoA analysis.
) measures the dissimilarity between the structures of two communities, where ST is the total number of OTUs in communities μ and ν, μi and νi is the relative abundance of OTU i in community μ and ν, respectively. A matrix of pairwise thetaYC-based distances among all samples was calculated for PCoA analysis.
Relationship between environmental variables and bacterial communities
Of the measured variables, we employed envfit24,25 as the analytical method in CCA package to identify a set of environmental factors that best explained the pattern of bacterial community clustering: TDS, Cond, TN, NH4+-N, NO3−-N, NO2−-N, TP, SS, pH, CODMn and Chl-a (Fig. 6). Results have shown that these factors: TN, TP, TDS, CODMn, SS and T, were correlated with the differences between bacterial communities from DC and HXH. The CCA results for distinguishing samples from HXH and DC (Fig. 6(a)) have shown a distinctively isolated point (DC7 sample), which was mostly influenced by Chl-a, TN and TP. And this result was consistent with the previous work8, which demonstrated that concentrations of both nitrogen and phosphorus could influence the growth of freshwater cyanobacteria in an eutrophic environment. In contrast, TDS, Cond, NH4+-N, SS and CODMn were the apparent dominant factors determining the bacterial community in HXH and DC when we excluded the influence of DC7 (Fig. 6(b)), in keeping with the result of physicochemical analysis. Meanwhile, Synechococcaceae was observed as the dominating clade of Cyanobacteria in DC samples. And it has been proven that Synechococcaceae was the resource of nitrogen and could use chlorophyll and assortment of phycobilins to perform oxygenic photosynthesis21. Planctomycetales was observed in large proportion of DC where there was a higher degree of TN, which has played a part in anaerobic ammonium oxidation26. This was consistent with the observation that Planctomycetales might play an important role in the ammonia cycle in the DC aquatic system27. Therefore, it was noteworthy that TP, TN and Chl-a were key factors for setting sample of DC7 as an outlier, which may shed light on the reason of the appearance of Exiguobacterium dominating in DC7. When it came to DC only (excluding DC7, Fig. 6(c)), it was apparent that pH was the most influential factor. Compared to D-W, the samples in D-C (DC1, 3, 4 and 5) located dispersedly on the graphical representation of CCA, showing fewer environmental factors with impact to D-C but larger differences among samples. These results conclusively demonstrated that bacterial communities in DC was different from those in HXH, supporting the hypothesis that bacterial community structure was affected by chemical geological factors7. The testing result might be explained by external factors, one of which was that the urban sewage and part of industrial effluent flew directly into D-C. The result has further illustrated that different environment factors, especially nutrient variations, were playing essential parts in forming the bacterial community of three clustered samples (Cluster 1, Cluster 2 and Cluster 3).

CCA plot showing the separation power of statistically significant environmental factors for differentiating samples.
(a) Samples from DC and HXH; (b) Samples from DC and HXH (except DC7); (c) Samples from DC (except DC7).
Although both DC and HXH were plateau lakes, the different nutrient conditions have made their community structures vary greatly. For DC, about 45% of the area’s treated/untreated wastewaters flowed into the small sublake D-C (DC1, 3, 4, 5), which was only 3% of the total areas of DC. Therefore, samples from D-C have shown a higher degree of variation compared with other DC samples. Physicochemical analysis as shown in Supplementary Table S1 also suggested that three clusters (Cluster 1, Cluster 2 and Cluster 3, representing D-C, D-W and HXH, respectively) could be clearly separated according to their ecological conditions provided with different nutrient composition for the bacteria to reside and proliferate (Figs 4 and 5). More specifically, Cluster 1: DC1, 3, 4, 5 showed lower pH values than other samples in DC, but a higher concentration of TN contributing to eutrophication. Chl-a, which acts as the origin of carbon for bacterial and influences pH by photosynthesis, was higher in DC than HXH, indicating the eutrophication in DC as well. Previous study has shown bacterial community groups associated with a relatively higher level of planktonic algae biomass28. Therefore, Chl-a might be selected as an important index for the breakout of cyanobacteria blooms.
We further examined the pattern of bacterial community’s taxonomical structures for samples from these plateau lakes and those from a plain lake. In this work, Lake Taihu (eutrophic lake) was selected as the representative plain lake for comparing the differences in taxonomical structures between bacterial communities from plain lake and those from plateau lakes (DC and HXH). The taxonomical structures for bacterial communities in Lake Taihu were obtained from a recent study for this lake29. On OTU level, the number of bacterial OTUs (about 5,295) for samples from Lake Taihu were higher than those from DC and HXH, which might suggest that the bacterial communities of plain lake were more complicated than those of plateau lakes. We also compared the bacterial communities of Lake Taihu, DC and HXH on phylum level. The proportion of top 4 phyla from samples in Lake Taihu (97.2%) was higher than those from DC and HXH (about 77.93% on average). And more remarkable, Actinobacteria was presented in Lake Taihu as dominant group (60.7 ± 10.2%)29 and its proportion was 15.68 ± 8.09% in DC and HXH, whereas dominant bacterial phyla in DC and HXH was Proteobacteria (average: 31.68%, ranging from 22.97% to 53.92%). Hence, the bacterial composition for plateau lakes’ bacterial communities were different from those of the plain lake.
Biomarker discovery and interpretation
To find out what species could be selected as biomarkers for such differences among DC (except DC7) and HXH, we chose relative abundance of top 200 OTUs, out of which 163 OTUs showed statistically significance (shown in Supplementary file 1) based on LEfSe method23, which may contribute to the differences. 163 OTUs could be classified into 10 phyla and 145 OTUs have statistically significant difference (P < 0.05) and 18 OTUs have highly significant variation (P < 0.01) between DC and HXH (shown in Supplementary file 1).
For intra-group of DC, although both D-C and D-W were of the same eutrophic environment in DC30, they were divided into two parts geographically by a man-made dike and biologically by the physicochemical conditions (Supplementary Table S1), which contributed to a result that discrepancy in bacterial community existed between samples in DC1, 3, 4 and 5 from other samples in DC (Figs 4 and 5). Except sample DC7, the community differences at phylum level between D-C samples and D-W samples were significant based on dissimilarity tests with Bray-Curtis distance (PERMANOVA, F = 6.584, P = 0.001). LEfSe was also applied to identify the difference among DC samples23. With the result from relative abundance of OTUs used as an input data, the 863 bacterial species could be divided into two distinct groups representing D-C and D-W, with a logarithmic LDA score (LEfSe uses LDA31 to estimate the effect size of each differentially abundant feature) no less than 3.8, demonstrating a high degree of difference in bacterial community between the two groups (Fig. 7(a)). Moreover, LEfSe highlighted several additional clades from class-level to genus-level showing differences between D-C and D-W. LEfSe reported 7 and 24 taxa to be more abundant with high logarithmic LDA score in D-W and D-C, respectively. In agreement with previous analyses, Proteobacteria were abundant in both D-W and D-C, but different clades in Proteobacteria possessed drastically different relative abundance: Rhodobacterials belonging to Alphaproteobacteria was the main clade of proteobacteria in D-W, while other clades like Betaproteobacteria and Gammaproteobacteria were more abundant in D-C. Other bacterial lineages enriched in D-C include Amoebophilaceae (a family within Bacteroidetes), GCA004 and SL56 within Chloroflexi and Verrucomicrobia. Other biomarkers in D-W include Firmus, Acidobacteria, Soilbactetres, Pseudanabaenales, Pseudanabaeba and Amaricoccus.

Comparison between bacterial community samples from D-W and D-C by indicator and cladogram.
Samples divided into two groups according to the logarithmic LDA score, which were all higher than 3.8. (a) Sorted by degree of difference and (b) overlaid on a complete cladogram. Cladogram showed the bacterial distribution of two types of freshwater and differences in abundance between them were presented as colors and circle’s diameters.
Bacterial communities for an extreme case at DC7
DC7 was an extreme case of our analysis of bacterial community structures. As we explained above, DC7 had the most special physicochemical condition among DC samples with an extremely high value of TN, TP, SS and Chl-a (Supplementary Table S1), which was a typical phenomenon in eutrophication environment. The result of heatmap analysis, PCoA analysis and CCA analysis (Figs 4, 5, 6) all separated DC7 from other samples, demonstrating the discrepancy and uniqueness of it. The sampling site of DC7 was close to two inlets of the most polluted rivers (Daqing River and Hai River), where a great deal of untreated urban effluent might be discharged into D-W through the two rivers32. Largely due to extreme water physicochemical characters, the bacterial community of DC7 with less richness was also clearly different from other samples in DC. The bacteria dominated in DC, such as Cyanobacteria (0.08%) and Planctomycetes (1.22%), were a small quantity of bacteria community in DC7 as a result of extreme harsh environment. The dominant clade of DC7, Firmicutes showed a relatively high proportion (69.5%, Fig. 3(a)). Additionally, the saprophytic microbes like Exiguobacterium, was the dominant genus (67.5%, Fig. 3(b)), Exiguobacterium preferred to live in eutrophic conditions23. Previous study revealed that Exiguobacterium was an alkaliphile and widely distributed in saline environment7. Therefore, we speculated that untreated urban effluent that might be discharged from two rivers (Fig. 1) into D-W (DC7) and changed the environment of DC7 into a more alkaline character than other samples, leading to prosperous clades of Firmicutes and especially Exiguobacterium genus under Firmicutes. Our study revealed the fact that extreme water physicochemical characters can deteriorate the native bacteria community and shape communities to be ones that could adopt the harsh environment.
Conclusion
Analyses of freshwater lake’s bacterial community have great importance for environment monitoring: Bacterial communities in freshwater lakes would undergo changes in composition and relative abundance to adapt the changing environment and they would in turn affect the freshwater lakes. Therefore, monitoring of bacterial communities and their surrounding physical-chemical-geographical properties would be important for the assessment of freshwater lake’s water quality.
In this study, we have collected 19 bacterial community samples from two plateau freshwater lakes DC and HXH of China and analyzed the bacterial communities from these samples. Results have shown that the community structures were different for those from DC and HXH. And intra-group of DC, the samples from D-C and D-W were also different. We have found Acidobacteria, Solibacteres, Pseudanabaenales, which were more abundant in D-W and Pneumophila, Xanthomonadaceae and SL56 abundant in D-C, as prominent biomarkers that could represent different sets of samples among DC samples. Moreover, we have examined DC7 with extreme water physicochemical characters and discovered that Exiguobacterium was dominant in DC7 as a saprophytic microbe preferring eutrophication.
The differences among these clusters of samples were associated with their physical-chemical-geographical environments. Canonical correspondence analyses have shown that the bacterial community of DC and HXH’s freshwater samples were shaped by multiple forces including TDS, Cond, TN, NH4+-N, NO3−-N, NO2−-N, TP, SS, pH, CODMn and Chl-a with different weights. And their concerted effects have shaped the bacterial communities as we have seen today.
These results have clearly indicated the importance of association of biological profiling and physical-chemical-geographical properties for examining Chinese plateau freshwater bacterial ecosystem: only in this way can we observe that genus Synechococcaceae has played an important role in the resource of nitrogen and oxygenic photosynthesis for samples in D-W and Pirellulales, which was abundant in DC, played a part in anaerobic ammonium oxidation.
As for future works, since the temperature would affect the bacterial community composition dramatically, it would be valuable to collect more samples on a regular basis in different seasons and more importantly only by collecting those samples could a predictive model be built that might be used to predict the environmental changes. Another direction of future research would be on collecting samples from more diverse lakes with more diverse physical-chemical-geographical properties, based on which a global picture could be drawn not only for more accurate predictive model, but also to establish a link between bacterial ecosystem for certain types of lakes or mid-sized ecosystems.
Materials and Methods
Study areas and sampling processes
DC is one of the most seriously polluted33, large (area 300 km2), shallow (average depth 4.2 m), freshwater lakes in China. The lake is located on the Yun-Gui plateau area of Southwestern China, near the southwestern outskirts of the capital of Yunnan Province, Kunming City. The water temperature ranges from 9.8 °C to 26.5 °C, with an annual average of about 16.0 °C and the lake water is alkaline and eutrophic. The lake serves for many social and economic purposes, such as flood control, irrigation, transportation, water supply, fishery and tourism. In recent decades, DC has gradually lost its function of water supply due to eutrophication and cyanobacteria blooms, which has resulted in the shortage of drinking water in Kunming City. The lake is divided into two parts: Caohai Lake (D-C) and Waihai Lake (D-W), by a man-made dike. D-C, lies at the north of DC, occupying about 3% of total surface of DC, receiving about 45% of the area’s treated/untreated wastewaters flowed into the lake30. The average depth is around 2.5 m. Although D-C is seriously eutrophication, almost no algae bloom occurred in the past decade. This may be due to the serious chemical pollution and relatively short water retention time (about 3 months). D-W is the main water body of DC and accounts for 97% of the whole area of the lake. The average depth of D-W is about 4.3 m. D-W was also seriously eutrophication and heavily cyanobacteria bloom had occurred in warm seasons. The water retention time in this sub-lake was about 2 years. The pollutants in this sub-lake mainly come from nearby 22 flowing rivers. HXH is a relatively non-polluted, macrophytic freshwater lake. The area is about 2.6 km2 and average depth is about 10 m with its maximum depth of about 16 m. The lake is surrounded by mountains on three sides and the annual average water temperature is about 13 °C.
In order to investigate and compare the bacterial communities from various environmental conditions in plateau lakes, we chose an oligotrophic lake (HXH) and a eutrophic lake (DC) for investigation. A total of 19 sampling sites were chosen and samples collected during 25–29, July, 2014. The geographical distributions of these samples were shown in Figs 1 and 5 sites (HXH1, 2, 5, 6 and 7) were located in HXH, which were almost distributed offshore throughout the lake, except HXH2, which was located near the lakeshore. 14 sites (DC1, 3–9, 12, 14–17 and 20) were located in DC. Among these sites, 4 sites (DC1, 3, 4 and 5) belong to D-C, which were distributed in this sublake; 10 sites (DC6-9, 12, 14–17 and 20) were located in D-W, which were distributed in this sublake. At each sampling site, 1 L mixed water sample was collected with 0.5 m depth of the water column by a cylinder sampler. The water samples were stored with portable cooler with ice bag and then carried to the laboratory for further microorganism and physicochemical characters analysis. The biological material (about 200 ml water and other parts were used for measuring the physicochemical character) of each station was size fractionated into individual samples by two kinds of filtration membranes: 20 μm tulle and 0.22 μm pore size filter membrane (Tianjin Jinteng Experiment Equipment Co., Ltd). Bacteria in the water samples were collected onto 0.22 μm pore size filter membrane. These filter membranes were sealed and stored at −20 °C until transported to the laboratory and then stored at −80 °C freezer before DNA extraction.
Physicochemical analysis
Environmental variables were collected for all freshwater samples (Supplementary Table S1). Water temperature (T), pH, dissolved oxygen (DO), conductivity (Cond) and Total Dissolved Solids (TDS) were measured in situ by portable meter (YSI ProPlus, USA) at the sampling sites. The other environmental parameters contained total phosphorus (TP), orthophosphate (PO43−-P), total nitrogen (TN), ammonia nitrogen (NH4+-N), nitrate nitrogen (NO3−-N), nitrite nitrogen (NO2−-N), suspended solids (SS), potassium permanganate index (CODMn) and chlorophyll a (Chl-a) were assayed according the Standard Methods (APHA, 1998).
DNA extraction
In the laboratory, the filter membrane was aseptically cut into quarters for DNA extraction. Unused quarters were restored at −80 °C freezer. Quarters used for extraction were isolated using the Water DNA Kit (Omega Bio-Tek, USA) as described by the manufacturer. Extracted DNA of each sample was stored at −20 °C.
Library Preparation and Illumina MiSeq Sequencing
Next generation sequencing library preparation and Illumina MiSeq sequencing were conducted as follows: DNA samples were quantified using a Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and DNA quality was checked on a 0.8% agarose gel. 5–50 ng metagenomic DNA was used to generate amplicons that cover small subunit ribosomal (16S rRNA) V4-V5 hypervariable regions of bacteria and archaea for each individual sample. The forward primer contained the sequence “GTGYCAGCMGCCGCGGTAA” and reverse primer contained the sequence “CTTGTGCGGKCCCCCGYCAATTC”. Sequencing library was constructed using a MetaVxTM Library Preparation kit (GENEWIZ, Inc., South Plainfield, NJ, USA). Indexed adapters were added to the ends of 16S rDNA amplicons by limited cycle PCR.
DNA libraries were validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and quantified by Qubit® 2.0 and real time PCR (Applied Biosystems, Carlsbad, CA, USA). DNA libraries were multiplexed and loaded on an Illumina MiSeq sequencer according to manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was performed using a 2 × 300 paired-end (PE) configuration; image analysis and base calling were conducted by the MiSeq Control Software (MCS) on the Illumina MiSeq sequencer.
Raw sequence process
The raw sequences of bacterial 16S rRNA gene amplicons were processed by MOTHUR34 version 1.33.3. To minimize the effects of random sequencing errors and avoid overestimates of phylogenetic relationship diversity35, the paired reads were processed by removing adapters, splicing using “make.contigs” command with default settings and removing the reads containing ambiguous base call (N), longer than 460 bp and shorter than 360 bp. We also performed the “chimera.uchime” script by MOTHUR34 to check putative chimeras and removed the chimeras with “remove.seqs” command. Putative contaminants (comparing candidate sequences against the Greengenes database36 August 2013 release) were removed from our datasets. Singletons were also removed. The MiSeq sequencing data for 19 freshwater lakes’ samples were deposited to NCBI SRA database with Bioproject number PRJNA299273.
Operational Taxonomical Units (OTU) assignment and taxonomy classification
The high-quality reads that passed MOTHUR quality control process were aligned using default parameters against the Greengenes database by MOTHUR34. Calculation of coverage percentage (GOOD’s coverage)37. Community richness diversity was calculated by estimating the numbers of OTUs based on the ACE38 and Chao139 values at the 97% similarity levels. Community diversity index: Shannon index (Shannon)40 and Simpson index (Simpson)41 were also conducted. Rarefaction analysis were also performed using MOTHUR34, based on which representative sequences for shared OTUs, as defined by 97% similarity, were obtained. Relative abundances of the bacterial taxa at phylum, family and genus level were calculated and compared.
Comparison of bacterial community structures
To probe into the causes for the differences among the bacterial communities, we compared the community structure at different taxonomical levels and analyzed the phylogenetic relationships of the top 200 OTUs qualitatively and quantitatively. A heatmap of Bray-Curtis distance42 from different samples was generated using the R package gplots. Pairwise ecological distances were also calculated for all samples using the thetaYC43 distance metric by MOTHUR34, which takes both community membership and relative abundance into account. These distances were then visualized by principal co-ordinate analysis (PCoA)44. Analysis of similarity (ANOSIM), which is a nonparametric test of differences between 2 or more groups based on any distance measure45, was used to statistically test the variation of bacterial community composition across sites. The significant values were computed based on permutation of group membership with 1,000 iterations. Permutational multivariate analysis of variance (PERMANOVA)46 with Bray-Curtis dissimilarity was used for comparing the community structure of each dataset and sub-dataset.
Relationship between environmental variables and bacterial communities
Canonical correspondence analysis (CCA), which was one of the best methods for direct gradient analysis in community ecology47, was used for revealing the correlations between bacterial communities and environmental factors. This procedure was conducted in R with the CCA function of Vegan package. The principle of choosing the significance of environmental factors was assessed with the “envfit” function24,25, which fitted environmental vectors onto ordination, determined r2 for environmental factors and defined the significance of each environmental variable (999 permutations) on all components conjointly with permutation procedure. Finally, we chose the environmental factors, which could show moderate correlation with both component-1 and component-2 in CCA analysis.
Biomarker analysis
Linear discriminate analysis (LDA) effect size (LEfSe)48, which is an algorithm for high dimensional biomarker discovery between two or more biological conditions or classes, was utilized to find biomarker in metagenomic analysis. In this work, LEfSe pipeline was used for discovering metagenomic biomarker48. For inter-group analyses, DC group (except DC7) and HXH were classified as two classes with subclasses at OTU level and for intra-group of DC (except DC7), two groups have been formed for comparison: D-C (DC1, 3, 4, 5) and D-W group (DC6–9, 12, 14–17 and 20). Alpha value for the factorial Kruskal-Wallis test49 among classes and the P-value of pairwise Wilcoxon test between subclasses were all smaller than 0.05. Threshold on the logarithmic LDA score for discriminative features was set to be 2.050.
Additional Information
How to cite this article: Han, M. et al. Comparison and Interpretation of Taxonomical Structure of Bacterial Communities in Two Types of Lakes on Yun-Gui plateau of China. Sci. Rep. 6, 30616; doi: 10.1038/srep30616 (2016).
References
Braga, E. S., Bonetti, C. V., Burone, L. & Bonetti Filho, J. Eutrophication and bacterial pollution caused by industrial and domestic wastes at the Baixada Santista Estuarine System–Brazil. Mar. Pollut. Bull. 40, 165–173 (2000).
Moss, B. R. Ecology of fresh waters: man and medium, past to future. (John Wiley & Sons, 2009).
Brittain, S. M. et al. Isolation and characterization of microcystins, cyclic heptapeptide hepatotoxins from a Lake Erie strain of Microcystis aeruginosa. J. Great Lakes Res. 26, 241–249 (2000).
Chen, Y., Qin, B., Teubner, K. & Dokulil, M. T. Long-term dynamics of phytoplankton assemblages: Microcystis-domination in Lake Taihu, a large shallow lake in China. J. Plankton Res. 25, 445–453 (2003).
Steffen, M. M. et al. Comparative metagenomics of toxic freshwater cyanobacteria bloom communities on two continents. PloS one 7, e44002–e44002 (2012).
Wiegand, C. & Pflugmacher, S. Ecotoxicological effects of selected cyanobacterial secondary metabolites a short review. Toxicol. Appl. Pharmacol. 203, 201–218 (2005).
Bai, Y. et al. Bacterial communities in the sediments of Dianchi Lake, a partitioned eutrophic waterbody in China. PLoS One 7, e37796 (2012).
Glibert, P. et al. Evidence for dissolved organic nitrogen and phosphorus uptake during a cyanobacterial bloom in Florida Bay. Mar. Ecol. Prog. Ser. 280, 73 (2004).
Lindström, E. S., Kamst-Van Agterveld, M. P. & Zwart, G. Distribution of typical freshwater bacterial groups is associated with pH, temperature and lake water retention time. Appl. Environ. Microbiol. 71, 8201–8206 (2005).
Murdoch, W. W., Scott, M. A. & Ebsworth, P. Effects of the general predator, Notonecta (Hemiptera) upon a freshwater community. J Anim Ecol, 791–808 (1984).
Lopez-Archilla, A. I., Marin, I. & Amils, R. Microbial Community Composition and Ecology of an Acidic Aquatic Environment: The Tinto River, Spain. Microb Ecol 41, 20–35 (2001).
DeLong, E. F. Microbial community genomics in the ocean. Nat Rev Microbiol 3, 459–469 (2005).
Scheffer, M., Hosper, S. H., Meijer, M. L., Moss, B. & Jeppesen, E. Alternative equilibria in shallow lakes. Trends Ecol Evol 8, 275–279 (1993).
du Plooy, S., Smit, A., Perissinotto, R. & Muir, D. Nitrogen uptake dynamics of a persistent cyanobacterium Cyanothece sp. bloom in Lake St Lucia, South Africa. African Journal of Marine Science 36, 155–161 (2014).
Muir, D. G. & Perissinotto, R. Persistent phytoplankton bloom in Lake St. Lucia (iSimangaliso Wetland Park, South Africa) caused by a cyanobacterium closely associated with the genus Cyanothece (Synechococcaceae, Chroococcales). Appl. Environ. Microbiol. 77, 5888–5896 (2011).
Wu, X., Xi, W., Ye, W. & Yang, H. Bacterial community composition of a shallow hypertrophic freshwater lake in China, revealed by 16S rRNA gene sequences. FEMS Microbiol Ecol 61, 85–96 (2007).
Cain, K. D. & Lafrentz, B. R. Laboratory maintenance of Flavobacterium psychrophilum and Flavobacterium columnare. Current protocols in microbiology Chapter 13, Unit 13B 11 (2007).
Tseng, C.-H. et al. Microbial and viral metagenomes of a subtropical freshwater reservoir subject to climatic disturbances. ISME J 7, 2374–2386 (2013).
Van Trappen, S., Mergaert, J. & Swings, J. Flavobacterium gelidilacus sp. nov., isolated from microbial mats in Antarctic lakes. Int J Syst Evol Microbiol 53, 1241–1245 (2003).
Campbell, B. J. & Kirchman, D. L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7, 210–220 (2013).
Newton, R. J., Jones, S. E., Eiler, A., McMahon, K. D. & Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75, 14–49 (2011).
Warnecke, F., Amann, R. & Pernthaler, J. Actinobacterial 16S rRNA genes from freshwater habitats cluster in four distinct lineages. Environ. Microbiol. 6, 242–253 (2004).
Wang, Y. et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 78, 8264–8271 (2012).
Dawson, K. S. et al. Quantitative fluorescence in situ hybridization analysis of microbial consortia from a biogenic gas field in Alaska’s Cook Inlet Basin. Appl. Environ. Microbiol. 78, 3599–3605 (2012).
Virtanen, R., Ilmonen, J., Paasivirta, L. & Muotka, T. Community concordance between bryophyte and insect assemblages in boreal springs: a broad‐scale study in isolated habitats. Freshwat. Biol. 54, 1651–1662 (2009).
Mishamandani, S., Gutierrez, T., Berry, D. & Aitken, M. D. Response of the bacterial community associated with a cosmopolitan marine diatom to crude oil shows a preference for the biodegradation of aromatic hydrocarbons. Environ. Microbiol. (2015).
Jiang, Y.-F. et al. Illumina-based analysis the microbial diversity associated with Thalassia hemprichii in Xincun Bay, South China Sea. Ecotoxicology 1–9 (2015).
Lü, M., Wang, J., Fan, Z. & Huang, Y. The spatial variations of bacterioplankton community composition in Lake Dianchi and their relation to environmental factors. Acta Sci. Circumstant 31, 299–305 (2011).
Li, J. et al. Annual periodicity in planktonic bacterial and archaeal community composition of eutrophic Lake Taihu. Sci Rep. 5 (2015).
Wang, Z. et al. Nitrogen removal from Lake Caohai, a typical ultra-eutrophic lake in China with large scale confined growth of Eichhornia crassipes. Chemosphere 92, 177–183 (2013).
Fisher, R. A. The use of multiple measurements in taxonomic problems. Annals of eugenics 7, 179–188 (1936).
Wang, Z. et al. Linear alkylbenzene sulfonate (LAS) in water of Lake Dianchi—spatial and seasonal variation and kinetics of biodegradation. Environmental monitoring and assessment 171, 501–512 (2010).
Luo, J., Yin, J., Cai, L., Zhang, K. & Hyde, K. D. Freshwater fungi in Lake Dianchi, a heavily polluted lake in Yunnan, China. Fungal Divers 16, 93–112 (2004).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 75, 7537–7541 (2009).
Kunin, V., Engelbrektson, A., Ochman, H. & Hugenholtz, P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Rea, M. C. et al. Effect of broad-and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 108, 4639–4644 (2011).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. science 308, 1635–1638 (2005).
Spellerberg, I. F. & Fedor, P. J. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon–Wiener’Index. Global Ecol. Biogeogr. 12, 177–179 (2003).
Magurran, A. E. Measuring biological diversity. (John Wiley & Sons, 2013).
Hunter, P. R. & Gaston, M. A. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466 (1988).
Clarke, K. R., Somerfield, P. J. & Chapman, M. G. On resemblance measures for ecological studies, including taxonomic dissimilarities and a zero-adjusted Bray–Curtis coefficient for denuded assemblages. J. Exp. Mar. Biol. Ecol. 330, 55–80 (2006).
Yue, J. C. & Clayton, M. K. A similarity measure based on species proportions. Commun. Stat. Theory. Methods. 34, 2123–2131 (2005).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Clarke, K. & Ainsworth, M. A method of linking multivariate community structure to environmental variables. Mar. Ecol. Prog. Ser. 92, 205–205 (1993).
Anderson, M. J. A new method for non‐parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Ter Braak, C. J. Canonical correspondence analysis: a new eigenvector technique for multivariate direct gradient analysis. Ecology 67, 1167–1179 (1986).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011).
Breslow, N. A generalized Kruskal-Wallis test for comparing K samples subject to unequal patterns of censorship. Biometrika 57, 579–594 (1970).
Aagaard, K. et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PloS one 7, e36466 (2012).
Acknowledgements
We are grateful to Chaoyun Chen from College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China and Andreas Harst from the Ruhr University Bochum, Bochum, Germany, Bochum, Germany, for their proofreading the manuscript. This work is partially supported by National Science Foundation of China grant 61103167 and 31271410, Ministry of Science and Technology’s high-tech (863) grant 2012AA023107 and 2014AA021502 and Sino-German Research Center grant GZ878.
Author information
Authors and Affiliations
Contributions
The whole study was designed by K.N. and Z.W. J.Z. and Z.W. collected samples, M.H. performed DNA extraction and sequencing, M.H. and Y.G. analyzed the data, M.H., C.Z., Z.W. and K.N. explained the results, M.H., Y.G., C.Z., Z.W. and K.N. wrote the initial draft of the manuscript, all revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Han, M., Gong, Y., Zhou, C. et al. Comparison and Interpretation of Taxonomical Structure of Bacterial Communities in Two Types of Lakes on Yun-Gui plateau of China. Sci Rep 6, 30616 (2016). https://doi.org/10.1038/srep30616
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep30616
This article is cited by
-
Corpse decay of wild animals leads to the divergent succession of nrfA-type microbial communities
Applied Microbiology and Biotechnology (2022)
-
Hidden in plain sight—highly abundant and diverse planktonic freshwater Chloroflexi
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
