
Abstract
Cyclic di-GMP is a ubiquitous second messenger that regulates diverse cellular processes in bacteria by binding to various protein or riboswitch effectors. In Bacillus thuringiensis BMB171, a c-di-GMP riboswitch termed Bc2 RNA resides in the 5′-untranslated region (5′-UTR) of an mRNA that encodes a collagen adhesion protein (Cap). The expression of cap was strongly repressed in parent strain BMB171 because of the presence of Bc2 RNA but was significantly promoted in the Bc2 RNA markerless deletion mutant. Bc2 RNA acts as a genetic “on” switch, which forms an anti-terminator structure to promote cap read-through transcription upon c-di-GMP binding. As a result, cap transcription was de-repressed under high c-di-GMP levels. Therefore, Bc2 RNA regulates cap expression using a repression/de-repression model. Bc2 RNA-regulated Cap was also found to be tightly associated with motility, aggregation, exopolysaccharide secretion, biofilm formation, and virulence of B. thuringiensis BMB171 against its host insect Helicoverpa armigera.
Similar content being viewed by others


Introduction
Cyclic di-GMP is a universal and important bacterial second messenger1,2. Its intracellular concentration is controlled through the antagonistic activities of diguanylate cyclases (DGCs) and phosphodiesterases (PDEs)3. C-di-GMP was found to regulate various physiological processes in diverse bacteria4,5,6. It can also be recognized by the mammalian innate immune system, resulting in activation of type I IFN response7,8. C-di-GMP exerts its regulatory function by binding to a wide variety of effectors including kinases or phosphorylases6,9, transcription factors1,10,11,12, PilZ domain proteins1,13,14, degenerate DGCs or PDEs1,15,16, and riboswitches1,17.
Riboswitches are structured RNAs typically located in the 5′-untranslated regions (5′-UTRs) of mRNAs to regulate expression of downstream genes in response to changing concentrations of their cognate ligands18. A typical riboswitch usually consists of a highly conserved ligand-binding “aptamer domain” and a variable regulatory “expression platform”19. Two distinct classes of c-di-GMP riboswitches, termed c-di-GMP-I and c-di-GMP-II, were recently identified17,20. C-di-GMP-I consists of a highly conserved c-di-GMP binding aptamer domain and a variable expression platform domain that regulates downstream gene expression by forming a terminator/anti-terminator or sequester/anti-sequester structure17. Instead, c-di-GMP-II exhibits an allosteric self-splicing ribozyme activity triggered by c-di-GMP binding20.
Two c-di-GMP-I riboswitches, termed Bc1 RNA and Bc2 RNA, were experimentally identified in Bacillus thuringiensis BMB171 in our laboratory. Bc1 DNA resides upstream of the open reading frame (ORF) of a methyl-accepting chemotaxis protein (Mcp), while Bc2 DNA resides upstream of the ORF of a collagen adhesion protein (Cap). As a cell wall-anchored protein, Cap has been found to interact with collagen of animal tissues, affecting bacterial colonization, persistence, and virulence21,22. Adhesion to host cell surfaces and colonization onto host tissues are considered critical steps for microbial infection and survival23. In addition, Cap often acts directly as a virulence factor for many Gram-positive organisms22.
In the present manuscript, we found expression of complete cap gene (BMB171_C0936, new tag BMB171_RS05425) was ten times less than that of cap 5′-UTR in the parent strain BMB171, but was substantially promoted in the Bc2 RNA markerless deletion mutant ΔBc2, demonstrating that the transcription of cap was strongly repressed by the upstream Bc2 RNA riboswitch in BMB171. We provided solid evidence that Bc2 RNA acted as a genetic “on” switch, forming an anti-terminator structure upon c-di-GMP binding to enable read-through transcription of cap gene. Compared with BMB171, ΔBc2 exhibited motility deficiency, decreased exopolysaccharide (EPS) secretion, enhanced cell aggregation, and decreased biofilm formation. The cap deletion mutant Δcap exhibited the opposite trend in these physiological processes and showed enhanced virulence to the host insect larvae. To our knowledge, Bc2 RNA is the first experimentally identified c-di-GMP riboswitch in the B. cereus group strains. Our data clearly demonstrated that Bc2 RNA regulated the expression of cap by a repression/de-repression model.
Results
Bc2 RNA is located in the 5′-UTR of cap mRNA
Bc2 DNA contains an intrinsic transcription terminator sequence that is immediately adjacent to the ORF of Cap, suggesting that Bc2 RNA could be involved in regulating cap expression through a transcriptional termination strategy. Such an arrangement was found to be widely present in the B. thuringiensis and B. cereus genomes (Fig. 1a and Supplementary Fig. S1). Its potential function can be inferred from Vc2 RNA, a c-di-GMP-I riboswitch reported to regulate the expression of tfoX-like gene in Vibrio cholera17. Multiple sequence alignment revealed that the predicted aptamer region (10–40 nt) of Bc2 RNA was similar to that of Vc2 RNA, yet the predicted expression platform (40–90 nt) varied substantially in sequence. Thus, the conserved aptamer confers its binding specificity to c-di-GMP while the less conserved expression platform contributes to its diverse regulatory modes in bacteria. The c-di-GMP binding sites in Vc2 RNA are well conserved in Bc2 RNAs with some minor difference. Gα, one of the two guanine bases of c-di-GMP (termed Gα and Gβ, respectively), was predicted to recognize A11 of Bc2 RNA, while Gβ is predicted to form a canonical Watson-Crick base pair with C89 (Fig. 1b).

(a) Schematic representation of full length and terminated cap transcripts. TSSs (+1) are labeled by bent arrows. AUG is the start code. (b) Sequence alignments of Bc2 RNAs from B. thuringiensis and B. cereus with Vc2 RNA. White letters shaded in magenta, orange, green and black denote the conserved nucleotides of A, G, and C and U, respectively. The predicted c-di-GMP binding sites were highlighted by red stars, with the consensus sequences of Bc2 RNAs depicted below the alignments. The aptamer region and expression platform were underlined by blue and brown lines, respectively.
The transcription start site (TSS) of cap in BMB171 was clearly identified by 5′-RACE (Fig. 1a and Supplementary Fig. S2). The result indicated that there are 228 nucleotides (nt) from the TSS to cap and 76 nt to the aptamer region of Bc2 RNA, with a typical intrinsic transcription termination site for Bc2 RNA found in the region from +151 to +197 by bioinformatics analysis (Fig. 1a). The results suggested that the transcription of cap could be cis-regulated by Bc2 RNA, resulting in premature transcriptional termination or read-through via a riboswitch conformational change.
Bc2 RNA represses the cap gene expression
To explore the regulatory role of Bc2 RNA on cap expression, we first checked the transcription of the complete cap gene and its truncate 5′-UTR DNA that encompasses the Bc2 RNA sequence. In BMB171, the expression of 5′-UTR DNA was found to be almost 10 fold higher than that of cap gene (Fig. 2a). This demonstrated that most transcripts were terminated at the intrinsic terminator of Bc2 RNA, and the complete read-through of cap gene was at a relatively low level. However, the expression of complete cap was tremendously increased (about 20 fold higher) in the Bc2 RNA deletion mutant (ΔBc2) compared to that of the parent strain BMB171 (Fig. 2b), indicating that Bc2 RNA forms a terminator structure to strongly repress the transcription of cap gene.

(a) The relative expression levels of the 5′-UTR of cap in comparison with the complete cap gene by qPCR analyses in the BMB171 strain. (b) The differential expression levels of cap in the ΔBc2 and BMB171 strain by qPCR. Expression of gapdh gene was served as a negative control. (c) Products of in vitro transcription of DNA templates coding for the wild type (WT) and substitution mutant (A11T) riboswitches. The A11T mutant carries a single A to T mutation in the c-di-GMP binding pocket. FL and T denote full length and terminated transcripts, respectively. (d) The predicted secondary structures of Bc2 RNA in the absence and presence of c-di-GMP. Nucleotides predicted to bind to c-di-GMP are highlighted in red, and nucleotides predicted to form the terminator hairpin in the absence of c-di-GMP are highlighted in olive. The coding sequence of cap is boxed. Error bars depict SD of data from three independent experiments. ***P < 0.001.
Binding of c-di-GMP to Bc2 RNA enhances cap transcription in vitro
Second messenger-dependent transcription termination assays were carried out to determine whether Bc2 RNA functions as an aptamer for binding c-di-GMP, and to explore the potential regulatory mechanism of Bc2 RNA on cap expression in response to the changing concentrations of c-di-GMP.
From the Fig. 2c, one can clearly detect substantial transcriptional termination products in the absence of c-di-GMP (indicated by capital “T” letter). However, under increasing concentrations of c-di-GMP, a decreased transcriptional termination band and an increased read-through transcript band (indicated by capital: “FL”) were observed (Fig. 2c, left), demonstrating that c-di-GMP addition can change the Bc2 RNA riboswitch conformation, enabling RNA polymerase to read through the terminator sequence.
To test whether increased read-through transcription was triggered by the specific binding of c-di-GMP to the Bc2 RNA aptamer, the A11 nucleotide, one of conserved c-di-GMP binding nucleotides of the Bc2 RNA aptamer, was substituted to a thymidine. This substitution was supposed to reduce the binding affinity of Bc2 RNA with c-di-GMP. The results indeed showed that A11T substitution in Bc2 RNA abolished the effect of c-di-GMP on enhancing transcriptional read-through and the total transcription amount of the mutant was decreased (Fig. 3c, right). We further confirmed that Bc2 RNA is specific for c-di-GMP binding, as c-di-AMP, GTP and cGMP were not able to promote the transcriptional read-through of Bc2 RNA (Supplementary Fig. S3).

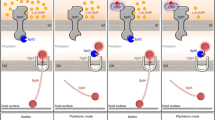
(a) Different intracellular c-di-GMP concentrations of the Δ2dgc, Δ3pde and BMB171 strains as determined by LC-MS/MS. (b) qPCR analyses of differential transcription profiles of cap in the Δ2dgc and Δ3pde mutants in comparison with the parent strain BMB171. (c) β-galactosidase activity analyses for B. thuringiensis strains carrying the Pcap-lacZ, PcapΔ-lacZ or promoterless Pnull-lacZ transcriptional fusion plasmids. (d) β-galactosidase activity analyses for B. thuringiensis strains carrying either Pcap-lacZ or Pcap-lacZ site-mutants or promoterless Pnull-lacZ transcriptional fusion plasmids. (e) Model showing proposed regulation of cap transcription by Bc2 RNA. Error bars depict SD of data from three independent experiments. ***P < 0.001; **P < 0.01; *P < 0.05.
These results support the hypothesis that the increasing transcriptional read-through is promoted by the specific interaction between c-di-GMP and Bc2 RNA. In the absence or at a low level of c-di-GMP, Bc2 RNA tends to form a stable terminator conformation that leads to transcriptional termination. While at a higher level of c-di-GMP, Bc2 RNA binds c-di-GMP and experiences a conformational change to form an anti-terminator structure, resulting in read-through and complete cap transcription (Fig. 2d).
C-di-GMP similarly enhances cap transcription through binding to Bc2 RNA in vivo
To explore the regulatory role of Bc2 RNA in vivo, we generated two BMB171 mutants (Δ2dgc and Δ3pde) that are capable of producing different amounts of intracellular c-di-GMP. Δ2dgc is a low intracellular c-di-GMP concentration mutant with two functional DGC genes (BMB171_C3708 (new tag BMB171_RS20080) and BMB171_C5008 (new tag BMB171_RS27040)) deleted (unpublished data), while Δ3pde is a high c-di-GMP concentration mutant with three c-di-GMP-specific PDE genes (BMB171_C0469 (new tag BMB171_RS02850), BMB171_C0550 (new tag BMB171_RS03240) and BMB171_C3420 (new tag BMB171_RS18570)) deleted (unpublished data). The intracellular c-di-GMP concentrations in Δ2dgc, Δ3pde and the parent strain BMB171 were then confirmed by LC-MS/MS (Fig. 3a and Supplementary Fig. S4). The relative expression levels of cap gene in these strains were finally measured by quantitative-PCR (qPCR) analyses. The results showed that the expression of cap gene was up-regulated in the high c-di-GMP concentration strain Δ3pde but down-regulated in the low c-di-GMP concentration strain Δ2dgc (Fig. 3b).
We also conducted the β-galactosidase activity assay to confirm the interaction between c-di-GMP and Bc2 RNA in vivo. Compared to the parent strain BMB171, expression of lacZ fused with the cap 5′-UTR DNA containing the entire wild type Bc2 RNA sequence, was significantly increased in the Δ3pde mutant but substantially decreased in the Δ2dgc mutant (Fig. 3c), which is in accordance with those measured by qPCR assay. In addition, the β-galactosidase activity was significantly promoted and showed little difference among these strains when lacZ was fused with the cap 5′-UTR DNA without the Bc2 RNA sequence (PcapΔ-lacZ) (Fig. 3c). Furthermore, the expression of lacZ fused with the cap 5′-UTR DNA in which the predicted c-di-GMP binding sites (A11, C37 and A38) in Bc2 RNA were substituted to thymidine also showed a significant decrease and was also insensitive to c-di-GMP concentration (Fig. 3d). As a negative control, the promoterless lacZ exhibited no β-galactosidase activity (Fig. 3c and d).
Taken collectively, these data indicated that the Bc2 RNA riboswitch repressed cap expression and deletion of Bc2 RNA enhanced cap transcription (Figs 2a,b and 3c); on the other hand, c-di-GMP binding to Bc2 RNA enhanced read-though, resulting in partial de-repression of cap expression (Figs 2c and 3b,c). Therefore, Bc2 RNA regulated the transcription of cap via a repression/de-repression model (Fig. 3e).
Enhanced Cap expression inhibits swimming motility of B. thuringiensis
C-di-GMP controls a variety of physiological processes, including switch between motile and sessile bacterial life styles2. It was also responsible for the motility regulation in B. thuringiensis, and increasing c-di-GMP resulted in decreased bacterial motility (unpublished data). We have also checked the effects of cap expression on B. thuringiensis motility by measuring the diameter of the swimming zone on the swimming plate containing 0.3% agar. The Δcap, BMB171 and ΔBc2 strains exhibited a similar growth rate on the LB plate (Fig. 4a). However, when grown in 0.3% agar swimming plate the Δcap mutant was observed to swim faster than the parent strain BMB171, whereas the swimming motility was nearly abolished for the ΔBc2 mutant (Fig. 4a). Because expression of the cap gene was particularly promoted in the ΔBc2 mutant, these results suggested that excessive Cap protein could inhibit the swimming motility of B. thuringiensis. Since the Bc2 RNA-cap locus arrangement is highly conserved in the B. thuringiensis and B. cereus genomes, the Bc2 RNA-dependent motility regulation by cap expression might represent a general regulatory mechanism for bacterial motility in the B. cereus group strains.

(a) Motility assay of ΔBc2, Δcap and BMB171 on LB plate containing 1.5% agar or swimming plate containing 0.3% agar. (b) Congo red binding assay. Cells were grown on LB agar supplemented with 1 mg/mL Congo red dye at 28 °C for 24 h. (c) Quantification of secreted EPS. (d) SEM imaging of bacteria mounted on microscope cover glasses. The extracellular matrix is indicated by arrows. Error bars depict SD of data from three independent experiments. **P < 0.01; *P < 0.05.
Excessive Cap expression blocks EPS secretion
Bacteria often grow as an associated multicellular community (biofilm) bound together by self-generated extracellular matrix comprising EPSs, secreted proteins, and extracellular DNA to adapt to the changing environment24. Since Congo red dye binding is positively correlated with the presence of EPSs in various bacteria25,26,27, we thus used the Congo red binding to assay the EPS production in the BMB171 and its mutants.
Based on this method, we found that both ΔBc2 and Δcap colonies showed a similar Congo red binding ability relative to BMB171, demonstrating that cell-bound EPS was present in these strains (Fig. 4b). However, different pink zones were observed around these colonies, possibly due to the different amounts of secreted EPS. The pink zone was thought to be secreted EPS that could bind Congo red dye. When comparing the pink zone area around each colony, the Δcap was shown to secrete more EPS than BMB171, while ΔBc2 strain secreted little EPS into the medium (Fig. 4b). To confirm these results, we further extracted and quantified secreted EPS from the mid-logarithmic cultures of these three strains grown in LB broth (Fig. 4c). The results indeed correlated well with those observed in the colony Congo red binding assays.
We also used scanning electron microscopy (SEM) to visualize the extracellular matrices. These strains were mounted on microscope cover glass to maintain the natural multicellular condition. A large amount of intercellular fiber-like connections for both BMB171 and Δcap cells were observed, whereas almost no such fiber was seen in the ΔBc2 mutant (Fig. 4d). Taken together, these results demonstrated that high levels of Cap led to decreased EPS secretion in B. thuringiensis.
Cap promotes aggregation of B. thuringiensis
Bacterial motility and EPS formation are correlated with aggregation, and elevated c-di-GMP levels usually lead to motility deficiency and increased aggregation in flagellated bacteria28,29.
The aggregation of ΔBc2, Δcap and BMB171 strains was tested in static broth cultures. We found both ΔBc2 and BMB171 aggregated quickly in the culture tube. Relatively, the aggregation was promoted in the ΔBc2 mutant. But the Δcap strain remained almost unchanged (Fig. 5a, left). In order to test whether the differences of the aggregation rates of these three strains were a result of their distinct swimming abilities or due to the potential influence from extracellular matrixes, the three strains were washed with phosphate buffered saline (PBS) to remove the extracellular matrix and re-suspended in the LB broth. The PBS-treated strains exhibited similar aggregation rates that were faster than untreated BMB171 (Fig. 5a, right). This result demonstrated that the extracellular matrix inhibited cell aggregation directly.

(a) Cell aggregation assay. Mid-logarithmic cultures grown in LB medium were divided into two groups. One group was directly applied for static incubation (Fig. 5a, left). The other was washed with PBS and re-suspended in the LB medium before static incubation (Fig. 5a, right). The interfaces of cell pellets and supernatant were indicated by white lines. (b) Representative photographs of biofilm formation assays for the ΔBc2, Δcap and BMB171 strains in glass bottles. Biofilm at the air-liquid-interface of the culture medium is indicated by arrows. (c) Quantification of biofilm formation by CV stain measured by UV spectrophotometer at 595 nm. Error bars depict SD of data from three independent experiments. *P < 0.05.
Excessive Cap expression leads to decreased biofilm formation
Bacterial biofilms are communities of cells bound together by self-produced extracellular matrix24, which provides mechanical stability of biofilms and mediates bacterial adhesion to surface30. Because the above experiments indicated that cap expression was highly correlated with the EPS production and cell aggregation, we hypothesized that biofilm formation might also be controlled by the Bc2 RNA/cap mediated signaling pathway. Compared with the parent strain BMB171, the Δcap mutant was found to form a thicker biofilm adhered to the tube at the air-liquid-interface of the culture medium (Fig. 5b). Instead, the ΔBc2 mutant did not lead to a dense biofilm formation under the similar culture condition. These data were also quantified by crystal violet stain and measured by a UV spectrophotometer (Fig. 5c). The results indicated that biofilm formation was clearly blocked in the ΔBc2 mutant but promoted in the Δcap mutant.
Cap and biofilm affect virulence of B. thuringiensis synergistically
B. thuringiensis produces intracellular insecticidal crystal proteins (ICPs) that kill a wide variety of insect larvae31,32. In addition to ICPs, B. thuringiensis also produces other types of virulence factors33,34. Since BMB171 was an acrystalliferous mutant strain35, therefore its virulence to insects must be due to other virulence factors such as Cap. In addition, biofilm formation is also positively correlated with virulence of pathogens36,37. Based on our data, we found that when cap is excessively expressed in the ΔBc2 mutant, its biofilm formation was abolished, whereas the Δcap mutant exhibited a reverse trend (Fig. 5b,c). We have further set up a virulence assay system using the Helicoverpa armigera larvae as the host and obtained some interesting results as shown in Fig. 6. Compared to the BMB171 and Δcap strains, ΔBc2 strain exhibited a stronger insecticidal activity (Fig. 6a). Moreover, both ΔBc2 and Δcap strains influenced the growth trend of insects. Compared to the insects fed with BMB171 and water, the growth of surviving larva fed with ΔBc2 and Δcap was strongly inhibited (Fig. 6b,c), possibly due to the combined virulence effects of Cap protein and biofilm formation.

(a) Survival rates of H. armigera larvae fed with ΔBc2 RNA, Δcap and BMB171 strains. (b) Biomass of survived H. armigera larvae fed with bacteria for seven days. (c) Photographs of survived H. armigera larvae. All experiments were repeated at least three times. ***P < 0.001.
Discussion
Bc2 RNA regulates downstream gene expression via an elaborate repression/de-repression model
Bacterial riboswitches can control biological processes at either transcriptional or translational level in a positive or negative way18. They can also regulate gene expression through controlling the stability of mRNA20. In B. thuringiensis, Bc2 RNA regulated the transcription of cap via a repression/de-repression model. When c-di-GMP concentration is low, Bc2 RNA forms an intrinsic transcription terminator structure that strongly repressed cap expression, but still allows some leaky cap expression to maintain basic physiological function. When c-di-GMP concentration is elevated, Bc RNA switches to an anti-terminator structure to promote read-through transcription of cap therefore, the expression of cap is de-repressed. B. thuringiensis harnesses this repression/de-repression model to control the expression of cap elaborately and strictly. When this regulatory pathway is disrupted, such as under abnormal c-di-GMP levels or excessive Cap, bacteria will exhibit many abnormal phenotype changes. As demonstrated in this manuscript, excessive Cap leads to decreased motility and EPS secretion, as well as to decreased biofilm formation and increased aggregation rate.
Bc2 RNA is the first experimentally validated c-di-GMP-binding riboswitch in the B. cereus group strains
C-di-GMP-mediated motility control is ubiquitous in bacteria, which is achieved mainly through inhibiting flagella and pili movements17. In Escherichia coli and Salmonella, c-di-GMP-binding protein YcgR inhibited cell motility through its interaction with flagellar motor proteins38,39. In B. subtilis, YpfA inhibited motility in response to rising levels of c-di-GMP, but the mechanism of YpfA-dependent motility regulation was currently unknown40,41. Interestingly, Both YcgR and YpfA homologies were absent in B. thuringiensis. In our study, we demonstrated that Bc2 RNA was a c-di-GMP-binding riboswitch, which was responsible for controlling the swimming motility of B. thuringiensis. As the Bc2 RNA-cap arrangement was conserved in B. cereus, B. thuringiensis and B. weihenstephanensis (Supplementary Fig. S1), it may represent a general mechanism of riboswitch-mediated motility regulation adopted by Bacillus.
C-di-GMP-I and c-di-GMP-II riboswitches are widely distributed in bacterial species, predominantly in the Clostridium, Bacillus, Vibrio and Geobacter genus (Supplementary Tables 2 and 3). The abundance of c-di-GMP riboswitches indicates its importance in bacteria. In Clostridium, four c-di-GMP-I riboswitches (Ct-E88, Cb-17B, Cb-E43 and Cd-630) were identified42,43. These c-di-GMP-I riboswitches use different recognition elements for c-di-GMP binding, resulting in different binding affinities42,43. As a result, c-di-GMP riboswitches might play different roles under various c-di-GMP levels. Moreover, the sequences of c-di-GMP-I have also been found in human gut metagenome DNA and blood disease bacterium R229 (Supplementary Table 2), indicating that c-di-GMP-I might control the physiological processes of human gut bacteria, or may be involved in the bacteria-human interaction. However, their potential regulatory mechanisms remain largely unknown to date.
Abundant functions of collagen binding protein
Collagen is an important component of animal tissue, and is also an important target for pathogens during their adhesion, colonization, and invasion of host cells44. Bacteria produce collagen-binding proteins to interact with collagens in host tissues, and these interactions are critical for their establishment and progression of infection22. B. thuringiensis Cap, a homolog to Staphylococcus aureus Cna45, contains an N-terminal signal peptide, followed by a binding domain A, a segment composed of repeated motifs often known as the B domains, and a C-terminal cell wall-anchoring region containing an LPXTG motif (Supplementary Fig. S5). A domain is responsible for the collagen binding, whereas the B repeat domain is thought to help expose the binding domain to the surface of bacterial cells. The C-terminal LPXTG motif is recognized by sortase A, a transpeptidase which cleaves the peptide bond between the Thr and Gly residues to covalently link the Thr residue to the peptidoglycan in the bacterial cell wall22. In BMB171, five different collagen adhesion proteins have been identified to date (Supplementary Fig. S5). Among them, only Cap possesses a typical collagen binding domain A and contains a long repeats of B domain.
B. thuringiensis Cap, a large 202 kDa cell wall-anchoring protein, is likely responsible for bacteria to cell interaction and adhesion. In the current manuscript, we first described that the B. thuringiensis Cap is an important mediator relating to many bacterial physiological processes such as motility, EPS formation, aggregation, biofilm formation, and virulence to insect larvae. However, the detailed mechanisms used by Cap to induce these diverse physiological changes in B. thuringiensis requires further investigation.
Methods
Bacterial strains and growth conditions
B. thuringiensis BMB17135 and its mutants were grown at 28 °C in lysogeny broth (LB) unless otherwise specified. E. coli DH5α used for cloning was grown in LB broth or LB agar plates at 37 °C. When necessary, relevant antibiotics were added to the cultures with the following final concentration: 50 μg/ml kanamycin, 25 μg/ml erythromycin and 100 μg/ml ampicillin. The bacterial strains and plasmids used in this study were listed in Table 1.
Construction of markerless gene deletion strains
Gene knockout based on homing endonuclease I-SceI mediated markerless gene deletion in B. thuringiensis was performed as previously reported with minor modifications46. Scheme of the gene deletion procedure was shown in Supplementary Fig. S6.
RNA isolation and qPCR
Logarithmic phase cells of B. thuringiensis strains grown in LB broth were prepared to isolate total RNA using the TRIzol reagent (life technologies, USA). First-strand cDNAs were synthesized using the PrimeScript RT reagent Kit with gDNA Eraser (Takara Biotechnology, Japan) according to the manufacturer’s instructions. The final cDNAs were served as template for PCR amplification using the specific primers listed in Supplementary Table 1. qPCR was performed as described previously with minor modification47. The gadph gene was served as internal reference and the degrees of expression change were calculated using comparative Ct method.
In vitro transcription termination assays
In vitro transcription termination assays were conducted using a method adapted from that described previously48. DNA templates harboring T7 promoter were amplified by PCR using primers listed in Supplementary Table 1. The template design was shown in Supplementary Fig. S7. Transcription reactions were expected to stop at the predicted intrinsic transcription terminator for terminated RNA transcripts (approximately 121 nt) or to stop at the terminus of the template for read-through transcripts (284 nt). Polymerization was initiated by adding 0.25 U of E. coli RNA polymerase holoenzyme (Biomics, China) into transcription mixtures containing 0.1 pmol of DNA template, 5 mM ATP, CTP and GTP, 0.5 mM UTP, 10 μCi [α-32P]-UTP, 20 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, 10% glycerol. Reaction mixtures were also supplemented with c-di-GMP (Biolog Life Science Institute, Germany) at final concentrations defined for each reaction. The reaction mixture was incubated at room temperature for 30 min and stopped by adding heparin at a final concentration of 0.5 mg/μl. The products were examined by 15% denaturing PAGE followed by analysis using a PhosphorImager (FUJIFILM, Japan).
β-galactosidase assays
The transcriptional fusion plasmids Pcap-lacZ, PcapΔ-lacZ, PcapA11T, PcapC37T, PcapA38T and Pnull-lacZ were constructed as shown in Supplementary Fig. S8. Vector Pcap-lacZ was transformed into the Δ2dgc, Δ3pde mutants and parent strain BMB171 respectively to create reporter strains Δ2dgc/Pcap-lacZ, Δ3pde/Pcap-lacZ and BMB171/Pcap-lacZ. Similarly, the reporter strains Δ2dgc/PcapΔ-lacZ, Δ3pde/PcapΔ-lacZ and BMB171/PcapΔ-lacZ without Bc2 RNA sequence in the promoter and the strains containing site-mutant Bc2 RNA sequence, as well as the negative control strains Δ2dgc/Pnull-lacZ, Δ3pde/Pnull-lacZ and BMB171/Pnull-lacZ were respectively constructed. All strains were grown in LB broth containing 25 μg/ml erythromycin at 28 °C for 6 h (Mid-logarithmic phase). The β-galactosidase activities were determined and converted to Miller units following the protocol reported previously49.
Measurements of intracellular c-di-GMP levels using LC-MS/MS
B. thuringiensis strains were grown to logarithmic phase (6 h) at 28 °C in LB broth. The cells were harvested at 4 °C and washed twice with distilled water. C-di-GMP was extracted from the cell pellets using the method reported by Burhenne and Kaever50. Detection of c-di-GMP was performed on a Finnigan Surveyor Plus liquid chromatography system followed by a Thermo Scientic TSQ Quantum Ultra EMR tandem mass spectrum system (San Jose, CA, USA). Intracellular c-di-GMP level was normalized by the corresponding wet cell weight.
Sliding motility assays
Overnight cultures of ΔBc2, Δcap and BMB171 strains were inoculated into LB broth at a final OD600 of 0.05 and then incubated at 28 °C for 6 h (mid-logarithmic phase) in shaking incubator. To score bacterial motility, 5 μl of the culture was spotted on LB plate containing 1.5% agar or swimming plate containing 0.3% agar and incubated at 28 °C for 6 h38,39.
Quantitative determination of EPS production
To determine EPS production of colonies, 10 μl of the mid-logarithmic cultures of ΔBc2, Δcap and BMB171 strains were respectively spotted on the LB plates containing 1.5% agarose and 1 mg/ml Congo red26. Plates were incubated at 28 °C for 24 h and imaged by a digital camera. To quantitatively determine secreted EPS, overnight cultures were inoculated into LB broth at a final OD600 of 0.05 and incubated at 28 °C for 6 h in shaking incubator. The supernatants were obtained by centrifuging at 14,000 ×g for 10 min. Potassium chloride was added to 200 ml of the supernatants at a final concentration of 1.0% (w/v). Two volumes of ethanol were then added to the mixtures and incubated at −20 °C overnight. The precipitated EPS were separated by centrifuging at 14,000 ×g for 20 min and dried at 60 °C overnight before determining their dry weight51. The EPS content was normalized by the corresponding wet cell weight.
Scanning electron microscopy
A 10-fold dilution of exponential cultures of various B. thuringiensis strains in fresh LB medium were individually dropped onto microscope cover glasses and incubated for 24 h at 28 °C. Cells were further fixed with 4% gluteraldehyde at room temperature for 2 h. The samples were then subjected to freezing drying and imaged with a scanning electron microscope (JSM-6390, JEOL, Japan).
Aggregation assays
Cells were cultured to exponential phase in LB medium and incubated at room temperature without any perturbation. To exclude the influence of extracellular matrix on aggregation, 4 ml exponentially growing cells in LB medium were harvested and washed for three times with PBS buffer. The cells were finally re-suspended in 4 ml fresh LB medium and incubated at room temperature statically.
Biofilm formation and quantification assays
Biofilm assays were performed as in previous study29. Exponential cultures grown in LB medium were transferred to the wells of polystyrene 24-well trays (2 ml per well) and incubated for 48 h at 28 °C. Liquid culture was gently removed and the wells were washed twice with distilled water. The adherent biofilm was quantified by staining with 1% crystal violet for 30 min at room temperature. The residual dye was washed away with distilled water and the 24-well trays were allowed to dry. The crystal violet-stained biofilms were then re-suspended in 2 ml 96% (v/v) ethanol, and the absorbance of biofilm-associated crystal violet was measured at 595 nm.
Virulence assays
H. armigera were reared on an artificial diet (70 g/l soybean meal, 40 g/l yeast extract, 125 ml/l acetic acid and 150 g/l agar) at 28 °C for 12 h photoperiod52. The ingredients were mixed and sterilized, and then cooled to 60 °C. Thereafter, 5 g/l vitamin C and 20 g/l penicillin were added and mixed completely. The artificial diet was immediately transferred to 24-well trays (1 ml/Well). 100 μl B. thuringiensis cultures in sporulation stage was added into the diet and allowed to dry. For every group, 72 larvae were transferred individually to three 24-well trays containing artificial diet to avoid cannibalism. The mortality of larvae was observed daily for 6 days. Each treatment was performed with at least three replicates.
Bioinformation analyses
The secondary structure prediction of Bc2 RNA was performed using the RNAfold webserver (http://nhjy.hzau.edu.cn/kech/swxxx/jakj/dianzi/Bioinf4/miRNA/miRNA1.htm). The multiple sequence alignments of Bc2 RNAs with Vc2 RNA were conducted using the ClustalW2 program (http://www.ebi.ac.uk/Tools/clustalw2/index.html), with final output processed by the ESPript 3.0 server (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Experimental data were statistically analyzed for significance using unpaired two-tailed Student’s t tests.
Additional Information
How to cite this article: Tang, Q. et al. Cyclic di-GMP contributes to adaption and virulence of Bacillus thuringiensis through a riboswitch-regulated collagen adhesion protein. Sci. Rep. 6, 28807; doi: 10.1038/srep28807 (2016).
References
Sondermann, H., Shikuma, N. J. & Yildiz, F. H. You’ve come a long way: c-di-GMP signaling. Curr. Opin. Microbiol. 15, 140–146, 10.1016/j.mib.2011.12.008 (2012).
Romling, U., Galperin, M. Y. & Gomelsky, M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 77, 1–52, 10.1128/MMBR.00043-12 (2013).
Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7, 263–273, 10.1038/nrmicro2109 (2009).
Caly, D. L., Bellini, D., Walsh, M. A., Dow, J. M. & Ryan, R. P. Targeting cyclic di-GMP signalling: a strategy to control biofilm formation? Curr. Pharm. Des. 21, 12–24 (2015).
Hengge, R., Grundling, A., Jenal, U., Ryan, R. & Yildiz, F. Bacterial signal transduction by c-di-GMP and other nucleotide second messengers. J. Bacteriol. 198, 15–26, 10.1128/JB.00331-15 (2015).
Lori, C. et al. Cyclic di-GMP acts as a cell cycle oscillator to drive chromosome replication. Nature 523, 236–239, 10.1038/nature14473 (2015).
Parvatiyar, K. et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 13, 1155–1161, 10.1038/ni.2460 (2012).
Shaw, N., Ouyang, S. & Liu, Z. J. Binding of bacterial secondary messenger molecule c di-GMP is a STING operation. Protein Cell 4, 117–129, 10.1007/s13238-012-2071-0 (2013).
Tuckerman, J. R., Gonzalez, G. & Gilles-Gonzalez, M. A. Cyclic di-GMP activation of polynucleotide phosphorylase signal-dependent RNA processing. J. Mol. Biol. 407, 633–639, 10.1016/j.jmb.2011.02.019 (2011).
Krasteva, P. V. et al. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327, 866–868, 10.1126/science.1181185 (2010).
Tao, F., He, Y. W., Wu, D. H., Swarup, S. & Zhang, L. H. The cyclic nucleotide monophosphate domain of Xanthomonas campestris global regulator Clp defines a new class of cyclic di-GMP effectors. J. Bacteriol. 192, 1020–1029, 10.1128/JB.01253-09 (2010).
Chin, K. H. et al. The cAMP receptor-like protein CLP is a novel c-di-GMP receptor linking cell-cell signaling to virulence gene expression in Xanthomonas campestris . J. Mol. Biol. 396, 646–662, 10.1016/j.jmb.2009.11.076 (2010).
Boehm, A. et al. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141, 107–116, 10.1016/j.cell.2010.01.018 (2010).
Pratt, J. T., Tamayo, R., Tischler, A. D. & Camilli, A. PilZ domain proteins bind cyclic diguanylate and regulate diverse processes in Vibrio cholerae . J. Biol. Chem. 282, 12860–12870, 10.1074/jbc.M611593200 (2007).
Abel, S. et al. Regulatory cohesion of cell cycle and cell differentiation through interlinked phosphorylation and second messenger networks. Mol. Cell. 43, 550–560, 10.1016/j.molcel.2011.07.018 (2011).
Lee, V. T. et al. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol. Microbiol. 65, 1474–1484, 10.1111/j.1365-2958.2007.05879.x (2007).
Sudarsan, N. et al. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321, 411–413, 10.1126/science.1159519 (2008).
Breaker, R. R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 4, 10.1101/cshperspect.a003566 (2012).
Breaker, R. R. Prospects for riboswitch discovery and analysis. Mol. Cell. 43, 867–879, 10.1016/j.molcel.2011.08.024 (2011).
Lee, E. R., Baker, J. L., Weinberg, Z., Sudarsan, N. & Breaker, R. R. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329, 845–848, 10.1126/science.1190713 (2010).
Miller, J. H. et al. The collagen binding protein Cnm contributes to oral colonization and cariogenicity of Streptococcus mutans OMZ175. Infect. Immun. 83, 2001–2010, 10.1128/IAI.03022-14 (2015).
Xu, Y., Liang, X., Chen, Y., Koehler, T. M. & Hook, M. Identification and biochemical characterization of two novel collagen binding MSCRAMMs of Bacillus anthracis . J. Biol. Chem. 279, 51760–51768, 10.1074/jbc.M406417200 (2004).
Vengadesan, K. & Narayana, S. V. Structural biology of Gram-positive bacterial adhesins. Protein Sci. 20, 759–772, 10.1002/pro.613 (2011).
Flemming, H. C. & Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633, 10.1038/nrmicro2415 (2010).
Koseoglu, V. K. et al. Listeria monocytogenes exopolysaccharide: origin, structure, biosynthetic machinery and c-di-GMP-dependent regulation. Mol. Microbiol. 96, 728–743, 10.1111/mmi.12966 (2015).
Spiers, A. J., Bohannon, J., Gehrig, S. M. & Rainey, P. B. Biofilm formation at the air-liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol. Microbiol. 50, 15–27 (2003).
Simm, R., Morr, M., Kader, A., Nimtz, M. & Romling, U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 53, 1123–1134, 10.1111/j.1365-2958.2004.04206.x (2004).
Bordeleau, E. et al. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile . J. Bacteriol. 197, 819–832, 10.1128/JB.02340-14 (2015).
Chua, S. L. et al. C-di-GMP regulates Pseudomonas aeruginosa stress response to tellurite during both planktonic and biofilm modes of growth. Sci. Rep. 5, 10052, 10.1038/srep10052 (2015).
Aguilar, C., Vlamakis, H., Guzman, A., Losick, R. & Kolter, R. KinD is a checkpoint protein linking spore formation to extracellular-matrix production in Bacillus subtilis biofilms. mBio 1, e00035–10, 10.1128/mBio.00035-10 (2010).
de Maagd, R. A., Bravo, A. & Crickmore, N. How Bacillus thuringiensis has evolved specific toxins to colonize the insect world. Trends Genet. 17, 193–199 (2001).
Raymond, B., Johnston, P. R., Nielsen-LeRoux, C., Lereclus, D. & Crickmore, N. Bacillus thuringiensis: an impotent pathogen? Trends Microbiol. 18, 189–194, 10.1016/j.tim.2010.02.006 (2010).
Wu, S. et al. Isolation and characterization of a novel native Bacillus thuringiensis strain BRC-HZM2 capable of degrading chlorpyrifos. J. Basic Microbiol. 55, 389–397, 10.1002/jobm.201300501 (2015).
de Maagd, R. A., Bravo, A., Berry, C., Crickmore, N. & Schnepf, H. E. Structure, diversity, and evolution of protein toxins from spore-forming entomopathogenic bacteria. Annu. Rev. Genet. 37, 409–433, 10.1146/annurev.genet.37.110801.143042 (2003).
He, J. et al. Complete genome sequence of Bacillus thuringiensis mutant strain BMB171. J. Bacteriol. 192, 4074–4075, 10.1128/JB.00562-10 (2010).
Al Safadi, R. et al. Correlation between in vivo biofilm formation and virulence gene expression in Escherichia coli O104:H4. PLoS One 7, e41628, 10.1371/journal.pone.0041628 (2012).
GIAOURIS, E. D. et al. Intra- and inter-species interactions within biofilms of important foodborne bacterial pathogens. Front. in Microbiol. 6, 10.3389/fmicb.2015.00841 (2015).
Paul, K., Nieto, V., Carlquist, W. C., Blair, D. F. & Harshey, R. M. The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol. Cell 38, 128–139, 10.1016/j.molcel.2010.03.001 (2010).
Fang, X. & Gomelsky, M. A post-translational, c-di-GMP-dependent mechanism regulating flagellar motility. Mol. Microbiol. 76, 1295–1305, 10.1111/j.1365-2958.2010.07179.x (2010).
Chen, Y., Chai, Y., Guo, J. H. & Losick, R. Evidence for cyclic di-GMP-mediated signaling in Bacillus subtilis . J. Bacteriol. 194, 5080–5090, 10.1128/JB.01092-12 (2012).
Gao, X. et al. Functional characterization of core components of the Bacillus subtilis cyclic-di-GMP signaling pathway. J. Bacteriol. 195, 4782–4792, 10.1128/JB.00373-13 (2013).
Luo, Y., Zhou, J., Wang, J., Dayie, T. K. & Sintim, H. O. Selective binding of 2′-F-c-di-GMP to Ct-E88 and Cb-E43, new class I riboswitches from Clostridium tetani and Clostridium botulinum respectively. Mol Biosyst. 9, 1535–9, 10.1039/c3mb25560c (2013).
Luo, Y., Chen, B., Zhou, J., Sintim, H. O. & Dayie, T. K. E88, a new cyclic-di- GMP class I riboswitch aptamer from Clostridium tetani, has a similar fold to theprototypical class I riboswitch, Vc2, but differentially binds to c-di-GMP analogs. Mol Biosyst. 10, 384–90, 10.1039/c3mb70467j (2014).
Salzillo, M. et al. Identification and characterization of enolase as a collagen-binding protein in Lactobacillus plantarum . J. Basic Microbiol. 55, 890–897, 10.1002/jobm.201400942 (2015).
Zong, Y. et al. A ‘Collagen Hug’ model for Staphylococcus aureus CNA binding to collagen. EMBO J. 24, 4224–4236, 10.1038/sj.emboj.7600888 (2005).
Zheng, C. et al. Functional analysis of the sporulation-specific diadenylate cyclase CdaS in Bacillus thuringiensis . Front. Microbiol. 6, 908, 10.3389/fmicb.2015.00908 (2015).
Tang, Q. et al. Mycobacterium smegmatis BioQ defines a new regulatory network for biotin metabolism. Mol. Microbiol. 94, 1006–1023, 10.1111/mmi.12817 (2014).
Artsimovitch, I. & Henkin, T. M. In vitro approaches to analysis of transcription termination. Methods 47, 37–43, 10.1016/j.ymeth.2008.10.006 (2009).
Wang, J. et al. High-throughput identification of promoters and screening of highly active promoter-5′-UTR DNA region with different characteristics from Bacillus thuringiensis . PLoS One 8, e62960, 10.1371/journal.pone.0062960 (2013).
Burhenne, H. & Kaever, V. Quantification of cyclic dinucleotides by reversed-phase LC-MS/MS. Methods Mol. Biol. 1016, 27–37, 10.1007/978-1-62703-441-8_3 (2013).
He, Y. W., Wu, J., Cha, J. S. & Zhang, L. H. Rice bacterial blight pathogen Xanthomonas oryzae pv. oryzae produces multiple DSF-family signals in regulation of virulence factor production. BMC Microbiol. 10, 187, 10.1186/1471-2180-10-187 (2010).
Paramasiva, I., Sharma, H. C. & Krishnayya, P. V. Antibiotics influence the toxicity of the delta endotoxins of Bacillus thuringiensis towards the cotton bollworm, Helicoverpa armigera . BMC Microbiol. 14, 200, 10.1186/1471-2180-14-200 (2014).
Janes, B. K. & S. Stibitz . Routine markerless gene replacement in Bacillus anthracis . Infection and immunity 74, 1949–1953, doi: 10.1128/IAI.74.3.1949-1953.2006 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant 31270105] and the Fundamental Research Funds for the Central Universities (grant 2662015PY175) to Jin He, and by the Ministry of Education, Taiwan, under the ATU plan, and by the National Science Council, Taiwan (grants 102-2113-M-005-006-MY3) to Shan-Ho Chou. We especially thank Dr. Jing Zhao from College of Plant Science, Huazhong Agricultural University for providing us H. armigera. And we thank Shu Liu and Bo Chen from College of Life Science and Technology, Huazhong Agricultural University for constructing Δ3pde and Δ2dgc mutant strain, respectively.
Author information
Authors and Affiliations
Contributions
J.H. and Q.T. designed the experiments. Q.T., K.Y., H.Q., Y.Z., W.W. and Y.F. did the experiments. J.H., S.H.C. and Q.T. wrote and revised the main manuscript text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tang, Q., Yin, K., Qian, H. et al. Cyclic di-GMP contributes to adaption and virulence of Bacillus thuringiensis through a riboswitch-regulated collagen adhesion protein. Sci Rep 6, 28807 (2016). https://doi.org/10.1038/srep28807
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28807
This article is cited by
-
Mutation of gdpS gene induces a viable but non-culturable state in Staphylococcus epidermidis and changes in the global transcriptional profile
BMC Microbiology (2022)
-
Genomic insights into the diversity of non-coding RNAs in Bacillus cereus sensu lato
Current Genetics (2022)
-
A c-di-AMP riboswitch controlling kdpFABC operon transcription regulates the potassium transporter system in Bacillus thuringiensis
Communications Biology (2019)
-
Cyclic di-GMP regulates Mycobacterium tuberculosis resistance to ethionamide
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
