
Abstract
In this work, we present a facile and low-cost approach to synthesize heteroatom doped porous carbon via hydrothermal treatment of stem bark of broussonetia papyrifera (BP) as the biomass precursor in diluted sulfuric acid, and following thermal activation by KOH at 800 °C. The morphology, structure and textural property of the prepared porous carbon (PC) are investigated by scanning electron microscopy, transmission electron microscopy, N2 sorption isotherms, and X-ray photoelectron spectroscopy. The porous carbon possesses a high BET surface area of 1759 m2 g−1 and an average pore size of 3.11 nm as well as hetero-oxygen (9.09%) and nitrogen (1.7%) doping. Such porous carbon shows outstanding capacitive performances of 416 F g−1 and 300 F g−1 in three and two-electrode systems, respectively. As a solid-state adsorbent, the obtained porous carbon has an excellent CO2 adsorption capacity at ambient pressures of up to 6.71 and 4.45 mmol g−1 at 0 and 25 °C, respectively. The results present one novel precursor-synthesis route for facile large-scale production of high performance porous carbon for a variety of great applications including energy storage and CO2 capture.
Similar content being viewed by others

Introduction
Activated carbons (ACs) have been attracting a lot of investigation in previous decades. The reason for this attention is due to it is cheap, abundant, environmentally safe, commercially available and a sustainable biological resource1. Generally, ACs are usually prepared by a two-step process, in which carbon precursors are pyrolyzed at temperatures in the range of 600 ~ 900 °C in an inert atmosphere such as Ar or N2, followed by physical activation at elevated temperature in the presence of suitable oxidizing atmosphere chemical activation process using chemical agents such as KOH, NaOH, H3PO4, and ZnCl21,2,3,4. It has been demonstrated that the incorporation of heteroatoms, such as sulfur, boron, nitrogen, and oxygen, into the carbon lattice can significantly enhance mechanical, semi-conducting, field-emission, and electrical properties of carbon materials. For example, nitrogen doping is considered to be the most promising method for enhancing surface polarity, electric conductivity and electron-donor tendency of the ACs, thereby improving their performances in CO2 capture, electric double-layer capacitors, fuel cells, and catalysis5. To prepare these materials, one approach is to use nitrogen-containing original precursors6 or ionic liquids7 for pyrolysis, and another approach is to post-treat carbon with N-containing dopants such as ammonia, amine, or urea8. Biomass is an attractive original precursor, which is cheap, environment friendly and readily available in high quality and quantity. In addition to its carbon and water content, biomass is rich in elements such as sulfur, nitrogen, and phosphorus, as well as metals such as Fe, Cu, and others9. Up to now, however, only a small portion of biomass materials such as pig bones, egg white protein, amaranth, hemp10, bean dregs11,12, seaweeds13 or seaweed biopolymers14,15, etc., are used as carbon precursors for supercapacitor electrodes and CO2 capture. Therefore, the search for new precursors, which are cheap, accessible and with potential to create significant economic valorisation, is highly desired. Herein we employ stem bark with complex multilayered structure from broussonetia papyrifera (BP) as the carbon precursor to develop a simple and facile method for the large-scale production of nitrogen doped ACs.
It is well known that BP is a species of flowering plant in the family moraceae. Importantly, the initial oxygen and nitrogen contents of the stem bark from BP are around 50 wt% and 1.5 wt%, respectively16,17,18,19. Therefore, the BP stem bark should be a promising carbon precursor for the production of heteroatom-doped ACs due to its cheap, abundant, and a sustainable biological resource. In the present study, we prepared hetero-oxygen (9.09%) and -nitrogen (1.7%) co-doped porous carbon with a high BET surface area via hydrothermal treatment of stem bark with complex multilayered structure from BP as the biomass precursor, and following thermal activation by KOH at 800 °C. The applications of this novel heteroatom doped porous carbon in high-power supercapacitors and CO2 capture are also demonstrated.
Results and Discussion
Synthesis and Characterization of Nitrogen Doped Porous Carbon
As mentioned previously, heteroatom doped porous carbon with high BET surface area was prepared via a two-step progress, in which the BP stem bark was first hydrothermal treated in 1 M diluted sulfuric acid and then thermal activated by KOH at 700~900 °C. The resultant carbon materials are denoted as PC-700, PC-800, and PC–900, respectively. In the first step of hydrothermal treatment, the BP stem barks undergo acid-catalyzed dehydration, fragmentation, rearrangement and polymerization reactions to produce acid-insoluble intermediate biochar including spherical conjugated aromatic carbonaceous materials20,21,22 and pseudo-lignin, which consists of carbonyl, carboxylic, aromatic and aliphatic structures. Usually, spherical structure with diameter of 2~5 μm and flake from residual BP stem bark can be observed, which is consistent with our SEM observations (see Fig. 1a). Then during thermal activation at high temperature, the KOH activation involves primary reaction of 6KOH + 2 C = 2 K + 3 H2 + 2 K2CO3, decomposition of K2CO3 to K2O, and reactions of K/K2O/K2CO3/CO2 with carbon23,24. In addition, the abundant oxygen functional groups in biochar can react with KOH during heating, and generate H2O and CO2 gases because of dehydration and decarboxylation24. Particularly, upon heating to high temperature, the biochar undergo much stronger dehydrogenation and intermolecular dehydration reactions. As a result, serious structural changes are induced, leading to a much more porous network in final products. Considering the fact that the synthesis parameters, such as the hydrothermal temperature, H2SO4 concentration, biochar/KOH mass ratio, and activation temperature, play important roles in the regulation of the capacitance performance of the final porous carbons, a series of experiments were performed in order to screen the optimal synthesis conditions. Our experimental results confirm that the optimal synthesis conditions are: hydrothermal temperature = 170 °C, H2SO4 concentration = 1 M, biochar/KOH mass ratio = 1/2, and activation temperature = 700 ~ 900 °C (see Supporting Information, Figs S2 and S3 and Tables S1 and S2). Figure 1b shows the SEM image of the resultant PC–800, demonstrating that the prepared porous carbon sample possesses a continuous branched porous framework consisted of carbon nanosheets and fully interconnected pores ranges from several to hundreds of nanometers (Fig. 1b and the inset). The porous sheet-like structure is further studied by TEM observation. The typical TEM image (see Fig. 1c) reveals that the PC–800 has obvious porous texture. In addition, the localized graphitic structure which leads to an enhanced conductivity, can be seen from high-resolution TEM image in Fig. 1d.

SEM images of biochar (a) and the PC-800 (b). (c,d) TEM images of the PC-800 at different magnifications.
The textural structure of the obtained porous carbons is investigated by N2 adsorption at 77.3 K. As shown in Fig. 2a, the isotherms of PCs exhibit a typical type-IV curve with an obvious type-H4 hysteresis loop at p/po = 0.45. The existence of type-H4 hysteresis loop is indicative of the presence of large mesopores embedded in a matrix with interconnected channels of much smaller size25,26. Figure 2b shows the pore size distribution (PSD) of the samples at different activation temperatures calculated according to NLDFT model. The meso and macro-pores increase with the temperature increment. The textural and electrochemical properties of these samples are summarized in Table 1 in detail. The PC–800 exhibits the BET surface of 1759 m2 g−1, which is much higher than that of PC–700 (1393 m2 g−1) and PC–900 (1229 m2 g−1) as well as the highest pore volume of 0.92 m3 g−1.

N2 adsorption-desorption isotherms (a) and pore size distributions (b) of the PC-700, PC-800, and PC-900.
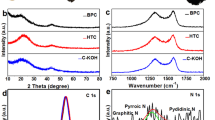
The XRD patterns of the obtained PC samples at 700, 800 and 900 °C are shown in Fig. 3a. All samples show two weak diffraction peaks centered at 2θ = 23.2° and 43°, respectively. The value of d002 is about 0.38 nm, larger than that of graphite (0.335 nm), implying a random combination of graphitic and turbostratic stacking27,28. In addition, with increasing calcination temperature, the intensities of the 2θ peaks at approximately 43 °C increases, due to the formation of higher degree of graphitic structure at higher carbonization temperature, which will greatly improve the electrical conductivity29,30,31. Figure 3b is the corresponding Raman spectra, exhibiting two bands at around 1339 cm−1 (D band) and 1582 cm−1 (G band) for all samples. The D band is related to the presence of disordered carbon structures, while the G band is associated with the vibration of sp2-hybridized carbon atoms in a graphitic layer32,33. The intensity ratio of D and G bands (ID/IG) is known to be an indicator of the degree of graphitization, and the smaller the ID/IG ratio, the higher the degree of graphitization. Here, the ID / IG peak ratio of PC–700, PC–800, and PC–900 are determined to be 0.93, 0.90, and 0.86, respectively. The results are consistent with those from XRD patterns.

XRD patterns (a) and Raman spectra (b) of the PC-700, PC-800 and PC-900.
XPS measurements were further used to analyze the components and chemical bonding at the surface of the BP-derived porous carbons. As shown in Fig. 4a, all samples exhibit a predominant C 1 s peak at 285 eV, a weak N 1 s peak at ca. 401 eV, and an O 1 s peak at ca. 533 eV. Table S3 provides the C, O, and N contents obtained from XPS analysis. The N contents are found to be 1.66, 1.43, and 0.99 atom% for the PC-700, PC-800 and PC-900, respectively, while the O contents are 10.91, 9.09, and 6.42 atom%. Clearly, the N and O contents of the prepared samples are dependent on the activation temperature. The high resolution XPS spectra of N 1 s for PC–700, PC–800 and PC–900 (see Fig. 4b–d) reveal the presence of four types of N configurations. The peaks at 398.5, 400.1, and 401.1 eV indicate the N doping in the form of pyridinic-N, pyrrolic-N, and quaternary-N30,34, respectively. As reported previously, electrochemical performance of carbons can be further enhanced by surface functionalities of O and N) due to additional pseudocapacitance35,36,37.

XPS survey spectra (a) and High-resolution XPS spectrum of N1s peaks (b–d) of the PC-700, PC-800, and PC-900.
Capacitance of the BP-Derived PCs
Electrochemical performance of BP-derived PCs as electrode materials for supercapacitors was initially estimated using a standard three-electrode system in 6 M KOH solution. The typical CV curves of the BP-derived PCs at a scan rate of 5 mV s−1 and 100 mV s−1 in the potential range between −1 and 0 V (vs. Hg/HgO) are shown in Fig. 5a,b. No matter at a lower scan rate of 5 mV s−1 (see Fig. 5a) or a higher scan rate of 100 mV s−1(see Fig. 5b) all PCs electrodes show a quasi-rectangular shape in the CV curves, suggesting an ideal electric double-layer capacitance behavior based on ionic adsorption and exchange. On the other hand, at a higher scan rate of 100 mV s−1, no dramatic change of the CV curves is observed. The GCD curves of these PCs electrodes are linear and display an isosceles triangle shape (Fig. 5c), indicating that the BP-derived PCs in KOH solution demonstrate ideal charge and discharge characteristics for electric double-layer capacitor. Figure 5d summarizes the gravimetric specific capacitance of three samples according to the GCD testing. It is found that the PC–800 electrode has the highest capacitance at the same current density. For instance, it possesses a specific capacitance of about 416 F g−1 at 0.5 A g−1, which is higher than those from the PC–700 and PC–900 electrodes. Moreover, the capacitance of the PC–800 electrode shows only a slight decrease to 333 F g−1 when the current density increased to 50 A g−1, suggesting good rate capability. Notably, the specific capacitance value of 416 F g−1, is higher than the reported values in aqueous electrolyte38,39. In addition, the PC–800 electrode shows better supercapacitor performance compared to electrodes made from various other carbon-based materials using other biomass as reported in the literatures38,40,41. The superior specific capacitance of the PC–800 electrode is ascribed to its high specific surface area, hierarchical porous structures, good conductivity, and heteroatom-doping. Considering that the rate capability is an important factor for the use of supercapacitors in power applications. The CV tests at different scan rates and GCD tests at different current densities were performed. As shown in Fig. 6c, with the scan rate increasing from 5 to 400 mV s−1, the CV curves of the PC–800 electrode become somewhat distorted but still retain a rectangular-like shape, suggesting an excellent high-rate capacitive behavior. Similarly, the GCD curves of the PC-800 electrode display a nearly isosceles triangle shape at different current densities (see Fig. 6a,b). More importantly, the GCD curves still maintain triangle shapes at a high current density of 50 A g−1, implying good coulombic efficiency (93%) and ideal capacitive behavior. The rate capability and cycling performance of the BP-derived PC electrodes based supercapacitor were further tested with a two-electrode system. The CV tested at different scan rates of 10 ~ 1000 mV s−1 and GCD tested at 0.05~2 A g−1 were utilized to examine the rate capability of the PC–800 based supercapacitor. As shown in Fig. 7a,b, the CV curves possess a typical rectangular shape at relative low scan rates. With increasing scan rate up to 1000 mV s−1, the CV curve still exhibits rectangular shape with only a little distortion, indicating an ideal electrochemical capacitive behavior with rapid diffusion and easy transportation of electrolyte ions to the interface of the electrode42. Figure 7c shows the GCD curves of the PC-800 supercapacitor. Clearly, the curve of 2 A g−1 still maintains linear and no obvious IR drop appears, suggesting characteristics of a little internal resistance and high rate capability of the PC–800 based supercapacitor. The cycling stability of the PCs based supercapacitor during the charge-discharge process is considered as another crucial factor in practical applications. As shown in Fig. 7d, the PC–800 based supercapacitor exhibits high capacitance retention of about 87.2% after 20000 charge-discharge cycles in a two-electrode configuration at 2 A g−1, indicating excellent electrochemical cycling stability.

CV curves tested at a scan rate of 5 mV s−1 (a) and 100 mV s−1 (b), the GC curves tested at a current density of 0.5 A g−1 (c), the correlation of specific capacitances with current densities (d) of the PC-700, PC-800, and PC-900.

GC curves tested at 0.5–20 A g−1 (a,b) and the CV curves tested at scan rates of 5–400 mV s−1 (c) of the PC-800 electrode.

CV curves at 10–1000 mV s−1 (a,b), GC discharge curves of 0.05–2 A g−1 (c) and cycling performance (d) of the PC-800 based supercapacitor.
CO2 adsorption properties of the BP-Derived PCs
The CO2 adsorption on BP-Derived PCs was investigated at 25 and 0 °C under atmospheric pressure (1000 mbar). The CO2 adsorption isotherms measured at ambient conditions for all BP-Derived PCs are presented in Fig. 8. The CO2 adsorption capacities for the PC-700, PC-800 and PC-900 are about 1.06, 1.08 and 0.85 mmolg-1 at 100 mbar, respectively (Fig. 8a). The results suggest that at very low pressure, the CO2 adsorption capacity of the porous carbons are highly depend on their nitrogen content (1.61%, 1.43% and 0.99% shown in Table S3)11,19. When the pressure increases to 1000 mbar, the CO2 adsorption capacities increase to 3.91, 4.45 and 3.76 mmol g-1, respectively, for the PC-700, PC-800 and PC-900, which is consistent with the BET results in Table 1. Particularly, the PC-800 has the unprecedented CO2 adsorption capacities of 4.45 and 6.71 mmol g−1 at 25 and 0 °C, respectively, which are higher than those of the reported carbons and comparable to the nitrogen containing carbons19,31,43. For example, the activated polypyrrole showed the CO2 uptake only of 6.2 mmol g−1 at 0 °C44,45 and active poplar anthers had the CO2 uptake only of 4.18 mmol g−1 at 25 °C19. It should be pointed that the N2 adsorption capacities for the PC–800 under ambient pressure at 0 °C is only 0.48 mmol g−1 (7.1% of CO2 adsorption capacities). Therefore, the PC–800 with even minor nitrogen doping, is still a potential selective adsorbent for CO2 and N2 separation. Figure 8d shows the CO2 adsorption capacities at 0 °C for the PC–800 from 0 to 1000 mbar with seven repeated runs. Clearly, the adsorption amounts for the PC-800 keep almost unchanged at 0 °C in the measured pressure ranges within seven runs, demonstrating its high recyclable stability for capture of CO2.

CO2 adsorption isotherms (a,b) on different porous carbons at 25 °C and 0–1000 mbar, (c) CO2 absorption isotherms and N2 adsorption isotherms on the PC-800 and (d) CO2 adsorption isotherms on the PC-800 at 0 °C and 0–1000 mbar within seven repeated cycles with regeneration.
Conclusions
In summary, hetero-oxygen and nitrogen doped porous carbons have been produced via hydrothermal treatment of stem bark with complex multilayered structure from BP as the biomass precursor in diluted sulfuric acid, and following thermal activation by KOH at 800 °C. The as-synthesized hetero-atom porous carbon possesses a partial graphitic structure with a very high specific surface area of 1759 m2 g−1 and a high pore volume of 0.92 cm3 g−1. The hetero-atom doped PCs based electrode for supercapacitor displays a high specific capacitance of 416 F g−1 at 0.5 A g−1, together with superior rate capability and cycling stability. As a solid-state adsorbent, the hetero-atom doped PCs has an excellent CO2 adsorption capacity at ambient pressures of up to 6.71 and 4.45 mmol g−1 at 0 and 25 °C, respectively. Considering the worldwide abundance and recyclability of BP, its stem bark can be acted as a novel biomass source for large-scale production of the PCs for high-performance supercapacitors and CO2 adsorbent.
Experimental
Materials
BP stem bark was collected from a local plantation and was prior washed, cut into pieces (1 cm × 1 cm) and dried. All of the other chemicals were analytical grade and were purchased from Aladdin Reagent (Shanghai) Co., Ltd. without further treatment.
Preparation of Nitrogen Doped Porous Carbon
Nitrogen doped porous carbon were prepared by carbonization and activation of the hydrothermal product of BP stem bark. Detailed procedures are as follows: 10.0 g of BP stem bark and 200 mL of 1 M diluted sulfuric acid were placed in a 250 mL stainless steel autoclave. The autoclave was sealed and heated at 170 °C for 10 h and then allowed to cool to room temperature. The resulting carbonaceous solid, denoted as biochar, was recovered by filtration, washed with distilled water, and dried. The biochar material was chemically activated using potassium hydroxide. The biochar and KOH were thoroughly ground in an agate mortar in a 1:2 mass ratio, and then the mixture was heated at 700 ~ 900 °C (5 °C min−1) for 1 h under argon flow. After that, the activated samples were thoroughly washed with 10 wt % HCl and distilled water. Finally, the carbons were dried in an oven at 100 °C for 12 h.
Structural Characterization
The morphology of the obtained porous carbons was characterized by scanning electron microscopy (SEM, JEOL JSM-6610LV and JEOL S-4800) operated at an acceleration voltage of 10 kV. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-1011 microscope operating at 200 kV. High-resolution TEM (HRTEM) was performed using a JEM-2100 F microscope operating at an accelerating voltage of 200 kV. The crystallographic information of porous carbons was investigated by powder X-ray diffraction (XRD, Rigaku D/Max 2500PC). Raman spectra were collected on a Renishaw inVia Raman spectrometer. X-ray photoelectron spectroscopy (XPS) was performed on a 1063 photoelectron spectrometer (Thermo Fisher Scientific, England) with Al-Kα X-ray radiation as the X-ray source for excitation. The textural properties were characterized by N2 sorption measurements at 77.3 K (Micromeritics TriStar II 3020). The specific surface area was obtained by Brunauer-Emmett-Teller (BET) method. The pore size distribution (PSD) was calculated by the nonlocal density functional theory (NLDFT) method. The total pore volume (Vtotal) was estimated from the adsorbed amount at a relative pressure p/p° of 0.99. Micropore volume (Vmic) was calculated using the t-plot method.
Electrode Preparation and Electrochemical Measurements
The working electrodes were typically fabricated by mixing the porous carbon as the active material (80 wt%), acetylene black (62 m2/g, 10 wt%), and polytetrafluoroethylene (PTFE, 10 wt%) in ethanol and then coated onto the nickel foam current collectors (1 cm × 1 cm) with a spatula. The premade electrodes were pressed under a pressure of 10 MPa for 5 min and finally dried at 130 °C for 12 h in an oven. The symmetric two-electrode supercapacitor was assembled into a total of 2032 stainless steel coin cells in air with nearly identical (both weight and size) electrodes, the electrolyte (6 M KOH) and a glassy fibrous separator.
The capacitive performance of single electrode was studied on a CHI760D electrochemical workstation (CH Instruments Inc., Shanghai, China) using a standard three-electrode system with platinum wire and Hg/HgO electrode as counter and reference electrodes, respectively, in 6 M KOH electrolyte at 25 °C. Cyclic voltammetry (CV), galvanostatic charge-discharge (GCD) technique and alternating current impedance were employed in the electrochemical investigations. CV tests of individual electrode were carried out between −1.0 and 0 V (vs. Hg/HgO). GC tests were performed at different current density varying from 0.5 to 50 A g−1 in the same potential range as the CV test. The specific capacitance based on GC was calculated using the equation17,18: Cg = I/(mdV/dt), where I is the constant current and m the mass of active materials, and dV/dt is calculated from the slope obtained by fitting a straight line to the discharge curve from the end of the voltage drop to the end of the discharge process. Gravimetric capacitance from GC of the two two-electrode configuration was calculated by using the formula Cg = 4I/(mdV/dt).
CO2 Capture Measurements
The CO2 adsorption isotherms of the samples were measured using a Micromeritics TriStar II 3020 static volumetric analyzer at 0 and 25 °C. The N2 adsorption isotherms of the BP–800 were measured using a Micromeritics TriStar II 3020 static volumetric analyzer at 0 °C. Prior to each adsorption experiment, the sample was degassed for 10 h at 150 °C to ensure that the residual pressure was below 1 × 10−3 mbar. After the samples were cooled down to 0 or 25 °C, CO2 was introduced into the system. The CO2 adsorption capacity in terms of the adsorbed volume under standard temperature and pressure was then recorded. The recycling adsorption test of CO2 was performed with a simple regeneration by evacuating at 150 °C for 10 h under a pressure of 1 × 10−3 mbar19.
Additional Information
How to cite this article: Wei, T. et al. A Facile and Low-Cost Route to Heteroatom Doped Porous Carbon Derived from Broussonetia Papyrifera Bark with Excellent Supercapacitance and CO2 Capture Performance. Sci. Rep. 6, 22646; doi: 10.1038/srep22646 (2016).
References
Sevilla, M. & Mokaya, R. Energy storage applications of activated carbons: supercapacitors and hydrogen storage. Energy Environ. Sci. 7, 1250–1280 (2014).
Lillo-Ródenas, M., Cazorla-Amorós, D. & Linares-Solano, A. Understanding chemical reactions between carbons and NaOH and KOH: an insight into the chemical activation mechanism. Carbon 41, 267–275 (2003).
Molina-Sabio, M., Rodriguez-Reinoso, F., Caturla, F. & Selles, M. Porosity in granular carbons activated with phosphoric acid. Carbon 33, 1105–1113 (1995).
Tsai, W., Chang, C. & Lee, S. A low cost adsorbent from agricultural waste corn cob by zinc chloride activation. Bioresour. technol. 64, 211–217 (1998).
Wang, R. et al. Nitrogen-rich mesoporous carbon derived from melamine with high electrocatalytic performance for oxygen reduction reaction. J. Power Sources 261, 238–244 (2014).
Hao, P. et al. Graphene-based nitrogen self-doped hierarchical porous carbon aerogels derived from chitosan for high performance supercapacitors. Nano Energy 15, 9–23 (2015).
Guo, D.-C. et al. Ionic liquid C16mimBF4 assisted synthesis of poly (benzoxazine-co-resol)-based hierarchically porous carbons with superior performance in supercapacitors. Energy Environ. Sci. 6, 652–659 (2013).
Wei, J. et al. Controllable synthesis of rich ritrogen-doped ordered mesoporous carbon for CO2 capture and supercapacitors. Adv. Funct. Mater. 23, 2322–2328 (2013).
Liu, X. et al. Biomass-derived nitrogen self-doped porous carbon as effective metal-free catalysts for oxygen reduction reaction. Nanoscale 7, 6136–6142 (2015).
Wang, H. et al. Interconnected carbon nanosheets derived from hemp for ultrafast supercapacitors with high energy. ACS nano 7, 5131–5141 (2013).
Xing, W. et al. Superior CO2 uptake of N-doped activated carbon through hydrogen-bonding interaction. Energy Environ. Sci. 5, 7323–7327 (2012).
Rufford, T. E., Hulicova-Jurcakova, D., Zhu, Z. & Lu, G. Q. Nanoporous carbon electrode from waste coffee beans for high performance supercapacitors. Electrochem. Commun. 10, 1594–1597 (2008).
Pintor, M.-J. et al. Preparation of activated carbon from Turbinaria turbinata seaweeds and its use as supercapacitor electrode materials. C. R. Chim. 16, 73–79 (2013).
Wei, L. et al. Hydrothermal carbonization of abundant renewable natural organic chemicals for high-performance supercapacitor electrodes. Adv. Energy Mater. 1, 356–361 (2011).
Yan, J., Wang, Q., Wei, T. & Fan, Z. Recent Advances in Design and Fabrication of Electrochemical Supercapacitors with High Energy Densities. Adv. Energy Mater. 4, 1–41 (2013).
Xi, D., Li, J., Kuang, Y.-w., Xu, Y.-m. & Zhu, X.-m. Influence of traffic exhausts on elements and polycyclic aromatic hydrocarbons in leaves of medicinal plant Broussonetia papyrifera. Atmos. Pollut. Res. 4, 370–376 (2013).
Zhu, Y. et al. Carbon-based supercapacitors produced by activation of graphene. Science 332, 1537–1541 (2011).
Wang, R. et al. Promising porous carbon derived from celtuce leaves with outstanding supercapacitance and CO2 capture performance. ACS appl. mater. interfaces 4, 5800–5806 (2012).
Song, J., Shen, W., Wang, J. & Fan, W. Superior carbon-based CO2 adsorbents prepared from poplar anthers. Carbon 69, 255–263 (2014).
Alatalo, S.-M. et al. Soy protein directed hydrothermal synthesis of porous carbon aerogels for electrocatalytic oxygen reduction. Carbon 96, 622–630 (2016).
White, R. J., Brun, N., Budarin, V. L., Clark, J. H. & Titirici, M. M. Always look on the “light” side of life: sustainable carbon aerogels. ChemSusChem 7, 670–689 (2014).
Antonietti, M., Fechler, N. & Fellinger, T.-P. Carbon aerogels and monoliths: control of porosity and nanoarchitecture via Sol–Gel routes. Chem. Mater. 26, 196–210 (2014).
He, X. et al. Synthesis of hierarchical porous carbons for supercapacitors from coal tar pitch with nano-Fe2O3 as template and activation agent coupled with KOH activation. J. Mater. Chem. A 1, 9440–9448 (2013).
Zheng, X. et al. Oriented and Interlinked Porous Carbon Nanosheets with an Extraordinary Capacitive Performance. Chem. of Mater. 26, 6896–6903 (2014).
Sing, K. S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure and appl. chem. 57, 603–619 (1985).
Groen, J. C., Peffer, L. A. A. & Pérez-Ramı́rez, J. Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis. Microporous and Mesoporous Mater. 60, 1–17 (2003).
Sevilla, M. & Fuertes, A. B. Fabrication of porous carbon monoliths with a graphitic framework. Carbon 56, 155–166 (2013).
Inagaki, M. New carbons-control of structure and functions. (Elsevier, 2000).
Ma, Y. et al. The production of carbon microtubes by the carbonization of catkins and their use in the oxygen reduction reaction. Carbon 49, 5292–5297 (2011).
Qian, W. et al. Human hair-derived carbon flakes for electrochemical supercapacitors. Energy Environ. Sci. 7, 379–386 (2014).
Yu, J. et al. One-pot synthesis of highly ordered nitrogen-containing mesoporous carbon with resorcinol–urea–formaldehyde resin for CO2 capture. Carbon 69, 502–514 (2014).
Ferrari, A. & Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev.B. 64, 1–13 (2001).
Kaufman, J., Metin, S. & Saperstein, D. Symmetry breaking in nitrogen-doped amorphous carbon: Infrared observation of the Raman-active G and D bands. Phy. Rev. B 39, 13053–13060 (1989).
Kapteijn, F., Moulijn, J., Matzner, S. & Boehm, H.-P. The development of nitrogen functionality in model chars during gasification in CO2 and O2 . Carbon 37, 1143–1150 (1999).
Li, Z. et al. Mesoporous nitrogen-rich carbons derived from protein for ultra-high capacity battery anodes and supercapacitors. Energy Environ. Sci. 6, 871–878 (2013).
Chandra, V. et al. Highly selective CO2 capture on N-doped carbon produced by chemical activation of polypyrrole functionalized graphene sheets. Chem. Commun. 48, 735–737 (2012).
Bagreev, A., Angel Menendez, J., Dukhno, I., Tarasenko, Y. & Bandosz, T. J. Bituminous coal-based activated carbons modified with nitrogen as adsorbents of hydrogen sulfide. Carbon 42, 469–476 (2004).
Peng, C. et al. Promising activated carbons derived from waste tea-leaves and their application in high performance supercapacitors electrodes. Electrochim. Acta 87, 401–408 (2013).
Jiang, L. et al. High rate performance activated carbons prepared from ginkgo shells for electrochemical supercapacitors. Carbon 56, 146–154 (2013).
Li, X. et al. Preparation of capacitor’s electrode from sunflower seed shell. Bioresour. technol. 102, 1118–1123 (2011).
Zhu, H., Wang, X., Yang, F. & Yang, X. Promising carbons for supercapacitors derived from fungi. Adv. mater. 23, 2745–2748 (2011).
Wang, J. et al. Fabrication of porous carbon spheres for high-performance electrochemical capacitors. RSC Adv. 4, 7538–7544 (2014).
Sha, Y. et al. Facile preparation of nitrogen-doped porous carbon from waste tobacco by a simple pre-treatment process and their application in electrochemical capacitor and CO2 capture. Mater. Res. Bull. 64, 327–332 (2015).
Sevilla, M., Valle-Vigón, P. & Fuertes, A. B. N-doped polypyrrole-based porous carbons for CO2 capture. Adv. Funct.Mater. 21, 2781–2787 (2011).
Xu, L. et al. Nitrogen-doped porous carbon spheres derived fromd-glucose as highly-efficient CO2 sorbents. RSC Adv. 5, 37964–37969 (2015).
Acknowledgements
Financial support from Program for NSFC (51472209, 51272219, U1401241), RFDP (20124301110006) the Research Foundation of Education Bureau of Hunan Province, China (Grant No. 15B237) and the Construct Program of the Key Discipline in Hunan Province is greatly acknowledged.
Author information
Authors and Affiliations
Contributions
T.W., X.W. and H.L. designed the experiments and wrote the manuscript. T.W. worked on materials synthesis and characterization of morphology and composition. Q.Z. and Y.G. performed supercapacitor fabrication, galvanostatic charge-discharge measurements, and CV measurements. T.W. contributed to data analysis and analytical characterization. X.W. and H.L. managed the research team. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wei, T., Zhang, Q., Wei, X. et al. A Facile and Low-Cost Route to Heteroatom Doped Porous Carbon Derived from Broussonetia Papyrifera Bark with Excellent Supercapacitance and CO2 Capture Performance. Sci Rep 6, 22646 (2016). https://doi.org/10.1038/srep22646
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22646
This article is cited by
-
In situ formation of Co3O4 nanoparticles embedded N-doped porous carbon nanocomposite: a robust material for electrocatalytic detection of anticancer drug flutamide and supercapacitor application
Microchimica Acta (2021)
-
Fabrication and Characterization of Silica Based Ceramic Composite for Filtration Applications
Silicon (2021)
-
Natural biomass derived hard carbon and activated carbons as electrochemical supercapacitor electrodes
Scientific Reports (2019)
-
Biomass-derived carbon materials with structural diversities and their applications in energy storage
Science China Materials (2018)
-
A novel hierarchical porous nitrogen-doped carbon derived from bamboo shoot for high performance supercapacitor
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
