
Abstract
Novel biomarkers and non-invasive diagnostic methods are urgently needed for the screening of gastric cancer to reduce its high mortality. We employed quantitative proteomics approach to develop discriminatory biomarker signatures from human saliva for the detection of gastric cancer. Salivary proteins were analyzed and compared between gastric cancer patients and matched control subjects by using tandem mass tags (TMT) technology. More than 500 proteins were identified with quantification, and 48 of them showed significant difference expression (p < 0.05) between normal controls and gastric cancer patients, including 7 up-regulated proteins and 41 down-regulated proteins. Five proteins were selected for initial verification by ELISA and three were successfully verified, namely cystatin B (CSTB), triosephosphate isomerase (TPI1), and deleted in malignant brain tumors 1 protein (DMBT1). All three proteins could differentiate gastric cancer patients from normal control subjects, dramatically (p < 0.05). The combination of these three biomarkers could reach 85% sensitivity and 80% specificity for the detection of gastric cancer with accuracy of 0.93. This study provides the proof of concept of salivary biomarkers for the non-invasive detection of gastric cancer. It is highly encouraging to turn these biomarkers into an applicable clinical test after large scale validation.
Similar content being viewed by others

Introduction
Although the incidence and mortality have fallen dramatically over the past several decades, gastric cancer is still a major public health issue as the fourth most common cancer and the second leading cause of cancer related death worldwide1,2. Approximately 880,000 people succumb to this malignancy each year on the earth. Radical surgery is the only potential curative method for the localized disease. One of the main reasons for the high mortality is the delay in diagnosis due to the fact that early cancers are typically asymptomatic or causes only nonspecific symptoms3. About 90% of gastric cancers are adenocarcinomas. Non-Hodgkin’s lymphomas and leiomyosarcomas make up most of the remaining 10%4. Helicobacter pylori (H. pylori) infection is the strongest known risk factor for malignancies that arise within the stomach, and epidemiological studies have determined that the attributable risk for gastric cancer conferred by H. pylori is about 75%5,6,7,8. Usually the 5 year survival rates for gastric cancer are less than 30% in many countries, although it has been significantly improved in some Asian countries by up to 50% because of the early detection programs9. The corresponding 5 year survival rates for stage I and II patients are 67%, compared to 31% and 8% in stage III and IV patients, respectively10. Therefore, novel biomarkers and non-invasive diagnostic methods are urgently needed for the screening of gastric cancer to reduce the high mortality.
The detection of gastric cancer in the early stages is vitally important in ensuring an excellent prognosis8. However, as with other cancers, the challenges in early detection lie in the reality of non-specific symptoms and invasive physical procedures11. The symptoms of early stage cancer may be indistinguishable from those of benign dyspepsia, while the presence of alarm symptoms may imply an advanced and often inoperable disease. Dysphagia, weight loss, and a palpable abdominal mass appear to be major independent prognostic factors for gastric cancer. However, gastro-intestinal bleeding, vomiting and duration of symptoms, do not seem to have a relevant prognostic impact on the survival of gastric cancer3.
Biomarker discovery for gastric cancer has mainly focused on tissue12,13, blood14,15 or gastric juice samples16 for the identification of protein17, microRNA18, long non-coding RNA19, and DNA20 candidates. For example, serum TIMP-115 has been identified as prognostic biomarkers for gastric cancer. Clinical proteomic study has shown that IPO-38 protein is a promising biomarker both for diagnosis and for predicting prognosis of gastric cancer17. Besides, biomarker discovery has been carried out with the treatment of EGFR binding monoclonal antibody in advanced gastric cancer21,22. However, there are few reliable serum biomarkers for the diagnosis of gastric cancer so far. The available biomarkers of CEA, CA19-9 and CA72-4 are not sufficiently sensitive and specific for the detection of gastric cancer17.
Proteomics is a powerful approach for biomedical research. Mapping proteomes from tissues, cells, and organisms is being used to discover new disease biomarkers for clinical and diagnostic applications23. Cancer proteomics has been extensively used for the discovery of diagnostic biomarker for gastric cancer24,25. Different quantitative proteomics have been widely used for biomarker discovery in different types of samples, including surface-enhanced laser desorption/ionization26, two-dimensional gel electrophoresis-mass spectrometry27,28, and isobaric tag for relative and absolute quantitation (iTRAQ)29. Proteomics using mass spectrometry with Tandem Mass Tags (TMT) is a reliable technology for quantitative proteome analysis30. Each isobaric tagging reagent within a set has the same precursor mass and is composed of an amine-reactive NHS-ester group, a spacer arm and an MS/MS reporter. For each sample, a unique reporter mass results in the MS/MS spectrum (i.e., m/z 126–131 for TMT-6plex Isobaric Label Reagents). These reporter product ions are used to report relative protein expression levels31,32,33.
Endoscopy with biopsy sampling is the gold standard used in gastric cancer diagnosis34. However, the invasive and relentless character of this procedure makes it less suitable for fast screening35. Human saliva is a biological fluid with enormous diagnostic potentials. Because saliva can be non-invasively collected, it provides an attractive alternative approach for cancer diagnosis. Saliva harbors a wide array of components, especially proteins36,37, which can be very informative for the detection of oral diseases (e.g. oral cancer38 and Sjogren’s Syndrome39) and systemic diseases (e.g. breast cancer40 and lung cancer41). More specifically, saliva protein finger print has been preliminary analyzed for the early diagnosis of gastric cancer42. Besides, gram-negative bacterium Helicobacter pylori could secrete enzyme urease and convert urea into carbon dioxide and ammonia. Optoelectronic sensors have been developed to detect clinically relevant levels of carbon dioxide and ammonia in saliva that can potentially be used for early diagnosis of gastric cancer35.
In this study, we hypothesized that gastric cancer related proteins exist in human saliva, which could be clinically used to discriminate gastric cancer patients from healthy control subjects. Human saliva samples were collected from gastric cancer patients and matched healthy control subjects. Salivary proteins were analysed and compared between the two groups by using TMT technology for proteomic biomarker identification and quantification. Candidate proteomic biomarkers were selected and further verified by immunoassay. Their utility for the detection of gastric cancer has been evaluated. With the discovery and pre-validation of discriminatory proteomic markers from saliva, gastric cancer will be non-invasively detected with high specificity and sensitivity.
Results
The strategy for salivary proteomic biomarker discovery
The study design is briefly shown in Fig. 1A. The 40 gastric cancer saliva samples were collected from patients who have been diagnosed as gastric cancer by using biopsy at the Samsung Medical Center (Seoul, Korea); most of them were at their early stages, as a result of the National Cancer Screening Program for gastric cancer in Korea43. The saliva samples from 40 healthy control subjects were collected as controls by matching their age-, sex-, and ethnicity- with the cancer group. Their smoking and drinking history were matched generally by whether they are current or former smokers and their duration and intensity. Their H. pylori infection and intestinal metaplasia status was included. Patient demographics and clinical profiles are present in Table 1.

Study design (A) and experimental design (B) for proteomic biomarker development in human saliva.
Amylase depletion
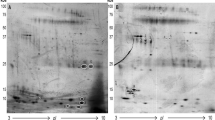
Alpha amylase is the most abundant protein in human saliva, accounts for about 50–60% of the total protein amount, which hurdled the detection and quantification of low abundant proteins. Depletion of these interfering proteins prior to definitive analyses should improve the resolution and sensitivity of salivary proteome analysis36. The one-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE) of salivary protein profiling before and after amylase depletion is shown in Fig. 2A. Obviously, the dominant band in the saliva without treatment disappeared after flow through the starch column. Figure 2B shows the two-dimensional difference gel electrophoresis (2D-DIGE) of two random saliva samples before and after amylase depletion, the dominant spots labeled in circle in Fig. 2B (a) significantly decreased when compare to that with amylase depletion in Fig. 2B (b).

(A) 1D SDS-PAGE of salivary proteins (a) before amylase depletion and (b) after amylase depletion; (B) 2D-DIGE of salivary proteins before amylase depletion (a) and after amylase depletion (b); Green: one saliva sample; Red: another saliva sample.
Cation exchange peptide fraction and peptides preparation
After amylase depletion, saliva proteins in each sample were reduced, alkylated and then digested by trypsin. According to the assignment in Fig. 1B, each sample in each group was labeled with corresponding tags of TMT-6plex. Combined peptides in group I and group II were fractionated by cation exchange chromatography column into 12 fractions, respectively, as shown in Supplementary Figure S2. All the fractions were dried under vacuum and were rehydrated in mobile phase B for further analysis.
Identification of differentially expressed proteins in saliva
Each fraction was loaded to liquid chromatography tandem mass spectrometry (LC-MS/MS) for protein identification and quantification. The raw data generated from the 12 fractions in each group was combined for protein database search and analysed in Proteome Discoverer with designed TMT workflow. Briefly, collision-induced dissociation (CID) spectrum was selected from total spectrum and used for protein identification. SEQUEST was interfaced with Proteome Discoverer for protein database search against IPI human database. Higher energy collisional dissociation (HCD) spectrum was extracted from the total spectrum and specifically used for reporter ion quantification. In this study, a global internal standard (GIS) was made and added to each group, specifically the GIS was labelled by TMT m/z at 126.1 in both groups. All the reading of other samples was compared with this GIS, which made the signal of all 10 samples in group I and group II comparable.
The database search results for group I and group II were exported to Microsoft Excel software, including the protein identification and quantification intensity ratios. In total, 519 proteins were identified from all the samples. The quantification data of cancer group and control group was extracted from the corresponding database search results. The distribution of individual proteins in cancer group and control group were systematically compared. 48 proteins showed significant difference (p < 0.05) between cancer group and control group (Table 2). For high throughput biomarker verification and validation, only these gastric cancer related candidates with available ELISA kits were selected for further evaluation.
Among these identified 519 proteins, their fold change ranged from 0.48 to 7.9 (Fig. 3A). 292 proteins were quantified with fold change in the range of 0.8 and 1.2. 80 proteins were up-regulated in the cancer group with fold change greater than 1.2. Meanwhile, 147 proteins were down-regulated in the cancer group (Fig. 3B) with fold change less than 0.8.

The fold change distribution (A) of these quantified proteins between cancer and control group. The regulation of discovered proteins (B) in cancer and control groups.
TMT based protein identification and quantification
The TMT quantification data of 6 candidates biomarker with p < 0.05, including CSTB, TPI1, DMBT1, Calmodulin-like protein 3 (CALML3), Immunoglobulin heavy chain (IGH), Interleukin-1 receptor antagonist (ILIRA), are shown in Fig. 4. The representative MS/MS spectrum of CSTB are shown in Fig. 5. Figure 5A,B are the CID spectrum and HCD spectrum for one peptide of CSTB, respectively. The rectangle labeled peaks in Fig. 5B are the TMT for quantification of this peptide. The reporter ion spectra for two different peptides of CSTB from group I, as shown in Fig. 5C,D, are very consistent, which represented the systematic down regulation of CSTB in cancer patients.

TMT quantification data for six candidate markers (A) CSTB, (B) TPI1, (C) DMBT1, (D) CALML3, (E) IGH and (F) IL1RA.

The MS/MS spectra of two peptides for CSTB with TMT labeling in group I: (A) CID spectrum for VHVGDEDFVHLR; (B) HCD spectrum for VHVGDEDFVHLR; (C) TMT reporter spectrum for VHVGDEDFVHLR; m/z = 127.1, 128.1 and 129.1 representing 3 normal samples, m/z = 130.1 and 131.1 representing 2 gastric cancer samples; (D) TMT reporter spectrum for peptide VFQSLPHENKPLTLSNYQTNK; m/z = 127.1, 128.1 and 129.1 representing 3 normal samples, m/z = 130.1 and 131.1 representing 2 gastric cancer samples.
Gene ontology analysis by PANTHER
Protein classification was finished by Panther Classification System based on their molecular function, related biological process, cellular component, protein class and related pathway. The Gene Oncology protein class analysis and pathway analysis of these proteins are shown in Fig. 6A,B, respectively.

Panther gene ontology pathway analysis of all proteins: (A) GO protein class analysis; (B) GO pathway analysis.
Candidate biomarker verification
Five proteins were selected for initial biomarker verification, including CSTB, TPI1, DMBT1, CALML3, and ILIRA. ELISA kits were extensively used for the target protein detection and quantification. Three of them were successfully verified in the discovery sample (n = 40) with p < 0.05, including CSTB, TPI1 and DMBT1. The dot plot of these biomarkers in the 20 cancer samples and 20 control samples are shown in Fig. 7.

Dot plot for biomarker verification in discovery sample set (n = 40): (A) CSTB; (B) TPI1; (C) DMBT1.
Candidate biomarker pre-validation
To further test the utility of these 3 candidates, a new sample set was utilized for pre-validation, which consisted of another 20 cancer samples and 20 control samples. The ELISA results (Supplementary Figure S1) demonstrated that all of them shown significant difference between gastric cancer patients and normal control (p < 0.05). To demonstrate the clinical utility of these salivary proteomic biomarker signatures for gastric cancer detection, logistic regression models were built based on different combinations of biomarkers. Figure 8A is the corresponding dot plot diagram of the three biomarker combination (CSTB, TPI1 and DMBT1) in the 40 pre-validation samples.

(A) dot plot diagram was based on the ELISA data of the gastric cancer group (n = 20) and healthy control group (n = 20). (B) the logistic regression model using 3 biomarkers (CSTB, TPI1 and DMBT1) in the pre-validation sample set results in AUC value of 0.93 with 85% sensitivity and 80% specificity (cutoff, 0.3721).
Biomarker performance and utility
Receiver operating characteristic (ROC) curve was built to evaluate the performance of these pre-validated biomarkers, yielding an area under ROC curve (AUC) value between 0.81 and 0.92. The combination of all there biomarkers could yield an AUC value of 0.93 with 85% sensitivity and 80% specificity (Fig. 8B).
Discussion
Biomarker discovery for gastric cancer detection
In total, we identified and quantified 519 proteins through the off-line two dimensional LC-MS/MS method (WCX-RPLC). The most abundant protein in saliva was selectively removed by affinity column, which greatly improved the resolution of biomarker discovery. When compared with ref. 37, about 20% of our identified proteins have been discovered by other approaches. Among these quantified proteins, there were 48 proteins shown significant difference between gastric cancer patients and normal subjects. Especially, about one third of these differentially altered proteins are gastric cancer related, either biologically or clinically, which demonstrated that human saliva could be a valuable medium for the detection of gastric cancer.
Of note it that most of these candidate biomarkers were down regulated in the saliva of gastric cancer patients. According to our preliminary work on salivary messenger RNA profiling and salivary microbial analysis from a similar saliva sample set, most identified candidates (which can differentiate gastric cancer patients from normal control subjects with p < 0.05) were also down regulated in cancer patients (data not shown). The consistency among protein, messenger RNA and microbial shown that there are some systematic changes occurred in human body that regulated by remote gastric cancer, which is fulfill the prospective of system biology.
Tumor-secreted exosomes have been found as a key player in determining cancer’s organotropic metastasis44. We proposed the role of cancer-derived exosomes in salivary biomarker development for systemic diseases and tested it in vitro45 and in vivo46. We found that suppression of exosome biogenesis result in the ablation of discriminatory salivary biomarker development, which might explain why saliva could be used for the detection of distal systemic disease, like gastric cancer.
The down-regulation of salivary biomarkers in cancer
Through initial verification in the discovery sample set and further confirmed in the pre-validation sample set, three proteins were consistently confirmed by ELISA. CSTB is an inhibitor of cathepsin proteases, which are increased in cancer. The protein levels of CSTB have been shown to correlate with tumor presence and stages. It has also been identified as a potential serum marker in hepatocellular carcinoma47. CSTB is a tissue and urinary biomarker for bladder cancer recurrence and disease progression48.
Through functional proteomics analysis, TPI1 has been identified in human gastric cancer cells as an anti-drug resistance agent49. It was also significantly regulated by H. Pylori in human gastric epithelial AGS cells7.
DMBT1 is a gene that is located at chromosome 10q 25.3–26.1, a possible tumor suppressor locus indicated by refinement of the losses of heterozygosity in various cancers50. The loss of DMBT1 expression may preferentially take place in well-differentiated gastric carcinoma. However, an upregulation of DMBT1 expression is more frequently found across all gastric cancer types51.
Human calmodulin-like protein (hCLP), is an epithelial-specific Ca2+-binding protein whose expression is strongly down regulated in cancers. Loss of immunoreactivity for human calmodulin-like protein is an early event in breast cancer development. The tumor-sensitive calmodulin-like protein is a specific light chain of human unconventional myosin X52. We also found that CALML3 down-regulated significantly in gastric cancer patients.
Diagnostic utility of salivary biomarkers
The diagnostic utility of these pre-validated biomarkers were evaluated by building the ROC curve and calculate their performance. By combining the three biomarkers through logistic regression, the biomarker panel could reach AUC value of 0.93 with 85% sensitivity and 80% specificity. The results collectively demonstrated that it is very promising to set up a saliva test for the detection of gastric cancer through using these developed biomarkers.
Conclusion
To the authors’ best knowledge, this is the first de novo proteomics biomarker discovery in human saliva for the detection of gastric cancer. New approaches and strategies were engaged for gastric cancer biomarker discovery. Through two phases biomarker development, 48 proteins were successfully discovered through amylase depletion and high throughput quantitative proteomic technology. ELISA further confirmed the presence of three candidates in the cancer saliva. Their performance for the detection of gastric cancer was evaluated, which is very encouraging for further definitive validation. Relay on the point of care technology, salivary diagnostic could be an ideal alternative way for the early detection and screening of gastric cancer.
Materials and Methods
Patients and samples
Our biomarker development consisted of two phases, including biomarker discovery phase and biomarker pre-validation phase (Fig. 1A). In total, 40 cancer patients and 40 normal control subjects were recruited for this study. All the saliva samples were collected under a protocol approved by institutional review board (IRB) of Samsung Medical Center and UCLA. All patients provided written informed consents. The methods were carried out in accordance with the approved guidelines. All experimental protocols were approved by Samsung Medical Center and UCLA Medical Centre Ethics Committee. Unstimulated saliva samples were consistently collected, processed, and stabilized as previously described40,41. All the samples were kept at −80 °C prior to assay. Identified proteomic biomarkers were first verified in the discovery sample set (20 gastric cancer samples and 20 healthy control samples) and then pre-validated in another sample set (20 gastric cancer samples and 20 healthy control samples).
Sample preparation
Saliva protein concentration was determined by BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). By taking 300 μg of proteins from each individual sample, every four samples were pooled into one sample in cancer group and healthy control group, respectively, thus 5 pool cancer samples and 5 pool healthy control samples were prepared. All the 10 pooled samples were subjected to potato starch affinity column for efficiently removal of alpha amylase as previously described53. Briefly, homemade affinity column packed with potato starch (Sigma Aldrich, Saint Louis, USA) was used to trap amylase and the flow through were collected for further analysis. Equal amount of protein from each pool sample was then used for the following experiment. Two pooled saliva samples were made from all the 10 pooled samples as a GIS for the comparison between two TMT-6plex experiments. 1D SDS-PAGE and 2D-DIGE were run as previously described41 to test the efficiency of amylase removal.
Reduction, alkylation, digestion, and labeling with TMT of the saliva samples
For each TMT-6plex experiment, 100 μg proteins in each pool saliva sample were dissolved in 45 μL of 200 mM TEAB. The sample was adjusted to a final volume of 100 uL with ultrapure water. With adding 5 μL 200 mM TCEP, the reaction was performed for 1 hour at 55 °C. Then, 5 μL of 375 mM IAA was added, and the mixture were reacted for 30 min in the dark at room temperature. 5 μL of freshly prepared trypsin (Promega, WI, USA) at 0.5 μg/uL concentration in TEAB (200 mM) was added. The digestion was performed overnight at 37 °C.
In group I, 1 GIS, 3 healthy control samples and two cancer samples were labelled by TMT with reporters at m/z = 126.1, 127.1, 128.1, 129.1, 130.1, 131.1, respectively (Fig. 1B). In group II, 1 GIS, the left 3 cancer samples and 2 healthy controls samples were labelled by another set of TMT with reporters at m/z = 126.1, 127.1, 128.1, 129.1, 130.1, 131.1, respectively. After 1 h of reaction at RT, 8 μL of 5% hydroxylamine (w/v) was added in each tube and mixed for 15 min. The six samples in each group were pooled into a new tube, respectively, and dried for storage at −80 °C.
Cation exchange fractionation of the pooled TMT-labeled saliva peptides
The pooled TMT-labelled saliva peptides were fractionated by cation-exchange chromatography using a flow rate at 0.8 mL/min on a 4.6 mm × 250 mm (5 μm, 125 Å) TSK gel CM-2SW column (Tosoh Bioscience, Stuttgart, Germany). The gradient was run as follows: 0–3 min 100% A (10 mM ammonium acetate, 25% acetonitrile, adjusted to pH = 3 with HAC), then to 100% B (200 mM ammonium acetate, 25% acetonitrile, adjusted to pH = 3 with HAC) at 15 min. As shown in Supplementary Figure S2, fractions were collected every minute and desalted via PepCleanTM C18 spin columns (Pierce, Rockford, IL, USA). The 12 fractions were dried under vacuum and stored at −80 °C for further LC-MS/MS analysis.
LC-MS/MS analysis
Peptides in each fraction were rehydrated in 2% (v/v) acetonitrile/0.1% (v/v) formic acid in water and injected with an autosampler (Eksigent NanoLC-2D, CA, USA). Peptides were first enriched on a reverse phase trap column (ProteoPep II, 100 μm × 2.5 cm, C18, 5 μm, 300 Å, New Objective, USA) and then eluted to analytical column (Magic C18AQ, 100 μm × 15 cm, 3 μm, 200 Å, Michrom Bioresources, USA). The mobile phase consisted of buffer (A) 2% acetonitrile and 0.1% formic acid in water, and buffer (B) 2% water and 0.1% formic acid in acetonitrile. A flow rate of 250 nL/min was applied for the separation of peptides for 140 mins. The gradient run was follows: 0–1 min, 2% B, then to 30% B at 90 min, 80% B at 110 min, and 2% B at 140 min. The mass spectrometer voltage was set to 1800 V and the heated capillary was kept at 180 °C. All mass spectra were acquired in the positive ionization mode with m/z scan range of 350–2000. The LTQ-Orbitrap XL (Thermo Fisher Scientific, San Jose, USA) was operated in a top 6 configuration at 60,000 resolving power (defined by m/Δm50%) for a full scan, with enabled charge state screening, monoisotopic precursor selection enabled, and + 1, and unassigned charge states rejected. After master scan, three most intense ions were subjected for collision-induced dissociation (CID) fragmentation using an isolation window of 3.0, collision energy of 30, default charge state of 2 and activation time of 30 ms. Fragmentation of three most intense TMT-reporter-labelled ions was achieved with HCD fragmentation at 7500 resolving power in the LTQ-Orbitrap using an isolation window of 2, collision energy of 40, default charge state of 2 and activation time of 30 ms.
Protein identification and quantification
LC-MS/MS data analysis was performed with Qual Brower (v2.0.7) and Proteome Discoverer (v1.3) interfaced SEQUEST (Human IPI database v3.78, 302626 entries). Up to two missed cleavage sites were allowed during the database search. Peptides and proteins identification were filtered with charge state dependent cross correlation (Xcorr) ≥2.0 and peptide rank No. 1 with requiring at least two peptides per protein. The filters allowed a 99% confidence level of protein identification with less than 1% false discovery rate. The Reporter Ions Quantitizer in the Proteome Discoverer was used to quantify the TMT reporter ion intensities at 126.13–131.14 m/z. Protein identification and quantification intensity ratios were exported to Microsoft Excel software. Reporter ion isotope correction factors were applied by subtracting the contribution of reporter ion isotopes to adjacent reporter ion intensities and adding these intensities back to the proper channel, after which data were normalized by median intensities for subsequent analyses.
ELISA
The ELISA tests for CSTB, TPI1 and DMBT1 (Antibodies-online, Atlanta, GA, USA) were performed according to the manufacturers’ instructions. All saliva samples were diluted 5 times with sample diluents for all three proteins.
Data analysis
The Graphpad Prism (Version 5.01) was used for all data analysis. For the number of proteins quantified in the 10 samples, p value was calculated based on t test and p < 0.05 was used as the cut-off for significance. The ROC curve and AUC value were constructed by numerical regression of the ROC curve. The confirmed gastric cancer related proteins were fitted for logistic regression models. Protein classification was finished by Panther Classification System (database version 6.1) based on their molecular function, related biological process, cellular component, protein class, and related pathway.
Additional Information
How to cite this article: Xiao, H. et al. Differential Proteomic Analysis of Human Saliva using Tandem Mass Tags Quantification for Gastric Cancer Detection. Sci. Rep. 6, 22165; doi: 10.1038/srep22165 (2016).
References
Crew, K. D. & Neugut, A. I. Epidemiology of gastric cancer. World J. Gastroenterol. 12, 354–362 (2006).
McLean, M. I. H. & El-Omar, E. M. Genetics of gastric cancer. Nat. Rev. Gastroenterol. Hepatol. 11, 664–674 (2014).
Maconi, G., Manes, G. & Porro, G. B. Role of symptoms in diagnosis and outcome of gastric cancer. World J. Gastroenterol. 14, 1149–1155 (2008).
Kelley, J. R. & Duggan, J. M. Gastric cancer epidemiology and risk factors. J. Clin. Epidemiol. 56, 1–9 (2003).
Plummer, M., Franceschi, S., Vignat, J., Forman, D. & de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 136, 487–490 (2015).
Polk, D. B. & Peek, R. M. Helicobacter pylori: gastric cancer and beyond. Nat. Rev. Cancer 10, 403–414 (2010).
Cho, S. O., Lim, J. W., Jun, J. H., Kim, K. H. & Kim, H. Helicobacter pylori in a Korean Isolate Expressed Proteins Differentially in Human Gastric Epithelial Cells. Dig. Dis. Sci. 55, 1550–1564 (2010).
Tan, Y. K. & Fielding, J. W. L. Early diagnosis of early gastric cancer. Eur. J. Gastroenterol. Hepatol. 18, 821–829 (2006).
Siegel, R., Miller, K. & Jemal, A. Cancer statistics, 2015. CA-Cancer J Clin. 65, 5–29 (2015).
Jung, J. J. et al. Chemokine growth-regulated oncogene 1 as a putative biomarker for gastric cancer progression. Cancer Sci. 101, 2200–2206 (2010).
Cui, J. A. et al. An integrated transcriptomic and computational analysis for biomarker identification in gastric cancer. Nucleic Acids Res. 39, 1197–1207 (2011).
Pan, S., Brentnall, T. A., Kelly, K. & Chen, R. Tissue proteomics in pancreatic cancer study: Discovery, emerging technologies, and challenges. Proteomics 13, 710–721 (2013).
Kang, X. D. et al. Prohibitin: a potential biomarker for tissue-based detection of gastric cancer. J. Gastroenterol. 43, 618–625 (2008).
Werner, S., Chen, H. D., Tao, S. & Brenner, H. Systematic review: Serum autoantibodies in the early detection of gastric cancer. Int. J. Cancer 136, 2243–2252 (2015).
Wang, C. S., Wu, T. L., Tsao, K. C. & Sun, C. F. Serum TIMP-1 in gastric cancer patients: A potential prognostic biomarker. Ann. Clin. Lab. Sci. 36, 23–30 (2006).
Hsu, P. I. et al. alpha 1-antitrypsin precursor in gastric juice is a novel biomarker for gastric cancer and ulcer. Clin. Cancer Res. 13, 876–883 (2007).
Hao, Y. et al. IPO-38 is identified as a novel serum biomarker of gastric cancer based on clinical proteomics technology. J. Proteome Res. 7, 3668–3677 (2008).
Liu, R. et al. A five-microRNA signature identified from genome-wide serum microRNA expression profiling serves as a fingerprint for gastric cancer diagnosis. Eur. J. Cancer 47, 784–791 (2011).
Zhou, X. Y., Yin, C. Q., Dang, Y. N., Ye, F. & Zhang, G. X. Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci Rep 5, 11516 (2015).
Watanabe, Y. et al. Sensitive and Specific Detection of Early Gastric Cancer with DNA Methylation Analysis of Gastric Washes. Gastroenterology 136, 2149–2158 (2009).
Shah, M. A. Update on Metastatic Gastric and Esophageal Cancers. J. Clin. Oncol. 33, 1760–1769 (2015).
Han, S. W. et al. Phase II study and biomarker analysis of cetuximab combined with modified FOLFOX6 in advanced gastric cancer. Br. J. Cancer 100, 298–304 (2009).
Hu, S. et al. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 5, 1714–1728 (2005).
Kawada, N. Cancer serum proteomics in gastroenterology. Gastroenterology 130, 1917–1919 (2006).
Lam, K. W. K. & Lo, S. C. L. Discovery of diagnostic serum biomarkers of gastric cancer using proteomics. Proteom. Clin. Appl. 2, 219–228 (2008).
Poon, T. C. W. et al. Diagnosis of gastric cancer by serum proteomic fingerprinting. Gastroenterology 130, 1858–1864 (2006).
Li, W. et al. Comparative proteomics analysis of human gastric cancer. World J. Gastroenterol. 14, 5657–5664 (2008).
Tsunemi, S. et al. Proteomics-based identification of a tumor-associated antigen and its corresponding autoantibody in gastric cancer. Oncol. Rep. 23, 949–956 (2010).
Chong, P. K. et al. ITIH3 is a Potential Biomarker for Early Detection of Gastric Cancer. J. Proteome Res. 9, 3671–3679 (2010).
Werner, T. et al. Ion Coalescence of Neutron Encoded TMT 10-Plex Reporter Ions. Anal. Chem. 86, 3594–3601 (2014).
Savitski, M. M. et al. Measuring and Managing Ratio Compression for Accurate iTRAQ/TMT Quantification. J. Proteome Res. 12, 3586–3598 (2013).
Nilsson, C. L. et al. Quantitative Phosphoproteomic Analysis of the STAT3/IL-6/HIF1 alpha Signaling Network: An Initial Study in GSC11 Glioblastoma Stem Cells. J. Proteome Res. 9, 430–443 (2010).
Dayon, L. et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 80, 2921–2931 (2008).
Ha, T. K. et al. Indication for endoscopic mucosal resection in early signet ring cell gastric cancer. Ann. Surg. Oncol. 15, 508–513 (2008).
Zilberman, Y. & Sonkusale, S. R. Microfluidic optoelectronic sensor for salivary diagnostics of stomach cancer. Biosens. Bioelectron. 67, 465–471 (2015).
Hu, S., Loo, J. A. & Wong, D. T. Human body fluid proteome analysis. Proteomics 6, 6326–6353 (2006).
Denny, P. et al. The proteomes of human parotid and submandibular/sublingual gland salivas collected as the ductal secretions. J. Proteome Res. 7, 1994–2006 (2008).
Hu, S. et al. Salivary Proteomics for Oral Cancer Biomarker Discovery. Clin. Cancer Res. 14, 6246–6252 (2008).
Hu, S. et al. Salivary proteomic and genomic biomarkers for primary Sjogren’s syndrome. Arthritis Rheum. 56, 3588–3600 (2007).
Zhang, L. et al. Discovery and Preclinical Validation of Salivary Transcriptomic and Proteomic Biomarkers for the Non-Invasive Detection of Breast Cancer. PLoS One 5, e15573 (2010).
Xiao, H. et al. Proteomic analysis of human saliva from lung cancer patients using two-dimensional difference gel electrophoresis and mass spectrometry. Mol. Cell. Proteomics, 10.1074/mcp.M1111.012112 (2012).
Wu, Z. Z., Wang, J. G. & Zhang, X. L. Diagnostic model of saliva protein finger print analysis of patients with gastric cancer. World J. Gastroenterol. 15, 865–870 (2009).
Lee, H. et al. Survival Outcome Associated with the Screening Interval for Gastric Cancer in Korea. Digestion 84, 142–148 (2011).
Hoshino, A. et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015).
Lau, C. S. & Wong, D. T. W. Breast Cancer Exosome-like Microvesicles and Salivary Gland Cells Interplay Alters Salivary Gland Cell-Derived Exosome-like Microvesicles In Vitro . PLoS One 7 (2012).
Lau, C. et al. Role of Pancreatic Cancer-derived Exosomes in Salivary Biomarker Development. J. Biol. Chem. 288, 26888–26897 (2013).
Lee, M. J. et al. Identification of cystatin B as a potential serum marker in hepatocellular carcinoma. Clin. Cancer Res. 14, 1080–1089 (2008).
Feldman, A. S., Banyard, J., Wu, C. L., McDougal, W. S. & Zetter, B. R. Cystatin B As a Tissue and Urinary Biomarker of Bladder Cancer Recurrence and Disease Progression. Clin. Cancer Res. 15, 1024–1031 (2009).
Wang, X. et al. Identification of triosephosphate isomerase as an anti-drug resistance agent in human gastric cancer cells using functional proteomic analysis. J. Cancer Res. Clin. Oncol. 134, 995–1003 (2008).
Kang, W. Q. et al. Induction of DMBT1 expression by reduced ERK activity during a gastric mucosa differentiation-like process and its association with human gastric cancer. Carcinogenesis 26, 1129–1137 (2005).
Conde, A. R. et al. DMBT1 is frequently downregulated in well-differentiated gastric carcinoma but more frequently upregulated across various gastric cancer types. Int. J. Oncol. 30, 1441–1446 (2007).
Rogers, M. S. & Strehler, E. E. The tumor-sensitive calmodulin-like protein is a specific light chain of human unconventional myosin X. J. Biol. Chem. 276, 12182–12189 (2001).
Deutsch, O. et al. An approach to remove alpha amylase for proteomic analysis of low abundance biomarkers in human saliva. Electrophoresis 29, 4150–4157 (2008).
Acknowledgements
This work was supported by NIH grants UH2 TR000923, LC110207 from Department of Defense, 20PT-0032 from TRDRP. The support from The Recruitment Program of Global Youth Experts of China, NSFC (No. 21305087), National High-tech R&D Program of China (863 Program, No. 2014AA020545), and Shanghai Jiao Tong University (No. YG2014QN21 and No. YG2015MS48) to H.X. are greatly acknowledged.
Author information
Authors and Affiliations
Contributions
D.T.W.W. and H.X. contributed to the study design. H.X. conducted the experiments, collected the data. H.X., Y.Z., Y.K., J.Y., L.Y.F. and C.X.C. contributed to the data analysis. S.K., J.J.K. and K.M.K. contributed to sample collection. The manuscript was written by H.X. All the authors reviewed the manuscript. All aspects of the study were supervised by H.X. and D.T.W.W.
Corresponding authors
Ethics declarations
Competing interests
David Wong is co-founder of RNAmeTRIX Inc., a molecular diagnostic company. He holds equity in RNAmeTRIX, and serves as a company Director and Scientific Advisor. The University of California also holds equity in RNAmeTRIX. Intellectual property that David Wong invented and which was patented by the University of California has been licensed to RNAmeTRIX. Additionally, he is a consultant to PeriRx. The other authors disclosed no potential conflicts of interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xiao, H., Zhang, Y., Kim, Y. et al. Differential Proteomic Analysis of Human Saliva using Tandem Mass Tags Quantification for Gastric Cancer Detection. Sci Rep 6, 22165 (2016). https://doi.org/10.1038/srep22165
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22165
This article is cited by
-
The salivary proteome in relation to oral mucositis in autologous hematopoietic stem cell transplantation recipients: a labelled and label-free proteomics approach
BMC Oral Health (2023)
-
The proteomic analysis of breast cell line exosomes reveals disease patterns and potential biomarkers
Scientific Reports (2020)
-
Analysis of Multimerin 1 (MMRN1) expression in ovarian cancer
Molecular Biology Reports (2020)
-
Saliva proteomics updates in biomedicine
Journal of Biological Research-Thessaloniki (2019)
-
Proteomic variation in metamorphosing Paralichthys olivaceus induced by exogenous thyroid hormone
Fish Physiology and Biochemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
