
Abstract
Lymphatic filariasis (Lf) is one of the oldest and most debilitating tropical diseases. Millions of people are suffering from this prevalent disease. It is estimated to infect over 120 million people in at least 80 nations of the world through the tropical and subtropical regions. More than one billion people are in danger of getting affected with this life-threatening disease. Several studies were suggested its emerging limitations and resistance towards the available drugs and therapeutic targets for Lf. Therefore, better medicine and drug targets are in demand. We took an initiative to identify the essential proteins of Wolbachia endosymbiont of Brugia malayi, which are indispensable for their survival and non-homologous to human host proteins. In this current study, we have used proteome subtractive approach to screen the possible therapeutic targets for wBm. In addition, numerous literatures were mined in the hunt for potential drug targets, drugs, epitopes, crystal structures and expressed sequence tag (EST) sequences for filarial causing nematodes. Data obtained from our study were presented in a user friendly database named FiloBase. We hope that information stored in this database may be used for further research and drug development process against filariasis. URL: http://filobase.bicpu.edu.in.
Similar content being viewed by others

Introduction
Lymphatic filariasis (Lf) is one of the most neglected tropical diseases in many countries. It causes major public health problem1 such as physical disability, disfiguring and chronic morbidity2. It is predominantly caused by Wuchereria bancrofti (W. bancrofti) and Brugia malayi (B. malayi) and being transmitted in the host body through the bite of Anopheles mosquitoes (An farauti, An punctulatus, An koliensis and others) and Culex quinquefasciatus3.
Since 20 years, three classical drugs named Diethylcarbamazine (DEC), Ivermectin (IVM) and Albendazole (ALB)4 are in practice to cure this disease. It is noticeable that currently these drugs are being used in the Global Programme to Eliminate Lymphatic Filariasis (GPELF) program, which has targeted to eliminate this disease by 20205. Several studies have shown that Mass Drug Administration (MDA) does not cure filarial infections completely; it only reduces new infections by clearing larvae from the human blood2,6,7,8. DEC and IVM kill microfilaria (mf) but it remains ineffective against adult worms4,9. Moreover, in African countries, DEC causes severe side-effects in the Onchocerca infected patient and IVM cannot be prescribed to the Loa-loa patient10,11,12. Thus, hunting of widespread macrofilaricides through the most essential proteins of pathogens are still remains a big challenge to wipe out this disease13. To overcome with this limited drug and drug targets for Lf, we initiated our research project to recognize the novel therapeutic targets for Lf, which should be essential for their survival and does not show any similarity with the human host proteins.
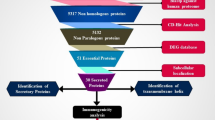
In the past few years, several investigators have made extensive efforts to identify better drugs and therapeutic targets to enhance the treatment of Lf14,15,16,17,18,19,20,21,22. A tremendous job was done by the division of Parasitology, New England Biolabs, by mining the essential genes of B. malayi23. It provides a new gateway to develop and design new therapeutics for brugian filariasis. Unfortunately, Wolbachia (Wol) which is an excellent drug target for Lf was untouched. It is an obligate intracellular symbiotic bacterium of filarial nematodes. It plays very crucial role in the development, vitality and fertility of the filarial nematodes24,25. Targeting Wol would be very effective approach to monitor the human filariasis infections. In 2009, the first computational effort was made to identify the essential genes of the unculturable Wolbachia endosymbiotic bacterium26 in the lack of complete proteome of Wol. At present, we have complete genome of Wolbachia from Brugia malayi (wBm)27, which could be use to define the most suitable therapeutic targets in wBm through a hierarchical proteome subtractive approach. Several researchers have used this approach to identify the possible therapeutic targets28,29,30,31,32 in various pathogens. Therefore, we have adopted this approach with slight addition and modification to come up with possible therapeutic targets. The complete work flow of our approach has been demonstrated in Fig. 1. In this current approach, we have added estimation of the identified therapeutic targets through the druggability test and ensured that all identified drug targets are non-homologous to human proteins. Identified potential therapeutic targets were further modelled to get insight into their molecular organization and protein conformations.

Schematic representation of the various steps involved to identify the potential therapeutic targets and vaccine targets of wBm through extensive proteome subtractive approached.
All identified therapeutic targets in this study was modelled and stored in an open access database named FiloBase. In addition, we have modelled forty drug targets of B. malayi and incorporated in our database which was earlier identified by Kumar et al. from the England Biolabs23. In order to supplement and make it more informative, other significant filarial information such as ESTs sequences, potential epitopes, experimental drugs, experimental structures of nematode proteins was incorporated in FiloBase by extensive literature survey which was in demand to facilitate the drug discovery process of Lf33. At present, FiloBase contains 119 potential drug targets for Lf; we hope that FiloBase will be worthwhile to expedite the process of drug discovery for the better treatment of Lf.
Materials and Methods
Identification of pathogens metabolic pathways
Kyoto Encyclopedia of Genes and Genomes (KEGG) is a manually curated database which helps to understand the biological systems of the organisms. Metabolic pathway of wBm and human was retrieved from the KEGG database and manual comparison was performed to find out the unique and common pathways in pathogens. Metabolic pathways which were present in both (human and wBm) were considered as common pathways and those which were present only in wBm not in human host were identified as a unique pathway. Protein sequence of both the pathways were retrieved from the UniProt database and passed through the further proteome subtractive channel.
Mining of non-homologous to human proteins of wBm
Retrieved protein sequences of wBm from both the pathways were subjected to BLASTp and sequence similarity search was performed against the human proteome database. The main objective of this step was to define the non-homologous to human proteins in wBm. It is likely to prevent the cross-reactivity of drug compounds with the human host proteins34. Here, we have used ‘Expect’ value (e-value) <0.005 and a minimum bit score of >100 to exclude the homologous sequences. Proteins which showed “HITS” with the above mentioned cut-off values were considered as non-homologous proteins35,36,37 and carried for further screening process, while remaining sequences were excluded from the list.
Essentiality assessment of wBm proteins
In order to identify the essential proteins of wBm, resultant non-homologous to human protein sequences of wBm were subjected to the protein BLAST tool and similarity search was performed against the essential protein sequence of bacteria from Database of Essential Gene (DEG)38 with an e-value <0.0001 and bit score >10039. DEG is a database which contains indispensable genes from bacteria, archaea and eukaryote organisms which support their cellular life. Currently, it holds 12,926 essential genes of bacteria. Based on the assumptions that similar proteins which are essential in one bacteria may be essential for another bacteria, hits found with DEG database with the above mentioned cut-off values were expected to represent the crucial proteins of wBm while remaining proteins were not, therefore excluded from the list of probable drug target.
Drug prioritization
All these non-homologous to human and essential proteins of wBm can be treated as potential therapeutic targets. However, being non-homologous to human and indispensable for the pathogen survival is not only the criteria to identify the most suitable drug targets for any pathogens, other vital parameters such as low molecular weight, sub-cellular localizations and their ability to interact with potential drugs (druggability) are also playing very significant role to identify the potential drug targets. Therefore, screening of potential therapeutic targets of wBm were carried out by identifying their biological significance and sub-cellular localization using Gneg-mPLoc40. Gneg-mPLoc is a powerful tool to identify the sub-cellular location of gram negative bacteria proteins. It identifies the gram negative protein localization in the following eight locations (1) cytoplasm, (2) extracellular, (3) fimbrium, (4) flagellum, (5) inner membrane, (6) nucleoid, (7) outer membrane and (8) periplasm40. Resultant data sets of Gneg-mPLoc were further cross-checked with CELLO v.2.541 and PSORTb II42 which are another web based tool for the prediction of sub-cellular localization of protein. The main objective of this study was to classify the proteins as potential drugs and vaccine targets. The cytoplasmic and inner membrane proteins were considered as potential drug targets, whereas the surface membrane, peri-plasmic and extracellular proteins were considered as potential vaccine targets. Prevention from the Lf can be accomplished through the vaccine development which involves antigenic surfaces to trigger the humoral immune responses. Therefore, membrane associated protein candidates are likely to represents as potential therapeutic targets to develop new vaccine, epitopes and as well as powerful drug candidates.
Druggability analysis
The most reliable way to identify the druggability of a protein is to identify the similar protein which binds to the drug-like compound43,44. Therefore, all identified potential drug targets were further evaluated based on the druggability test. In this step, all identified potential drug targets were searched against the DrugBank database45 and TTD database46. For this study, we have downloaded all FDA approved drugs (1,478), FDA approved Biotech drugs (183), experimental drugs (2,718), approved enzymes (174) and approved transporters (79) from DrugBank and TTD database (2134). Hits found with DrugBank and TTD database was considered as druggable targets while remaining were considered as novel drug targets which further need to be validated experimentally.
Tertiary structure identification
The tertiary structures of identified possible therapeutic and vaccine targets were searched in the PDB47 database for experimental structure. Available structures were incorporated in our database, while, un-available structures were searched for suitable templates for building their tertiary structure using NCBI protein BLAST tool. Template structures which have shown sequence identity more than 40% and query coverage more than 90% were modelled using Modeller9v1048 and sequence which exhibits lower sequence identities with template proteins were modelled using I-TASSER (Iterative Threading ASSEmbly Refinement) server49 or Phyre2 (Protein Homology/analogY Recognition Engine) server50. I-TASSER builds a 3D model of the user given protein sequences based on the multiple threading approach through LOMETS (Local Meta-Threading Server) and iterative template fragment assembly51 while Phyre2 predicts protein structure based on the remote homology recognition techniques. All the modelled structures were verified using Structural Analysis and Verification Server (SAVeS) for PROCHECK52 to evaluate their stereo-chemical quality by analyzing residue-by-residue geometry and ERRAT53 was used to evaluate their overall structural quality. PROVE program was also used to verify our modelled structure. It verifies the structure based on the Z-score deviation.
Database development and organization
All identified potential drug targets were stored in a user friendly open access database named FiloBase. All data were stored in a My-SQL and hosted using an Apache server. The complete flow-chart of the database development was illustrated in Fig. 2. The user interface of the database was prepared in PHP and JavaScript whereas; back-end was supported by PHP and Bio-Perl. The entire database contents was classified in five different search categories, (1) Drug targets of B. malayi and W. bancrofti, (2) EST sequences of B. Malayi, B. timori, B. Pahangi and Wol, (3) Epitope sequences from extensive literature survey, (4) Available and potential drug compounds with literature support and (5) Potential vaccine targets for wBm.

Architecture of the FiloBase database.
User can easily fetch desired information by selecting the query field and enter their query keywords in the given text box from the home page of the database. To make our retrieval system user friendly and more convenient, we have classified all the information and made available by a click on the table menu on the home page of the database (Fig. 3a).

The snapshot of the “Home Page” and “Result Page” of the FiloBase.
(a) Navigation window: It includes direct link to the various classified data in a single click, (b) Query search through text query, (c) List of potential drugs for Wolbachia endosymbiont, (d) Summary tab of the potential drug targets: It consists brief information about the therapeutic target, (e) Sequence tab: it contains the sequence and chain information of target protein, (f) Model verification tab: It contains model verification results obtained from PROCHECK, ERRAT and PROVE, (g) 3D structure tab: It contains the 3D modelled structure of the target protein with their active site pockets and (h) KEGG tab: It contains the graphical output of KEGG pathways if available.
Results and Discussion
Identification of metabolic pathways for wBm
Here, we report first computational approach to identify potential therapeutic targets of wBm by the protein subtractive approach. It was proved as a successful approach to identify the potential drug target proteins which are involved in various metabolic pathways of pathogen, but absent in the host organism and essential for their survival54. Overall summary of the project is depicted in the Table 1. In this current study, we have considered several vital parameters and systematic approach with a drug prioritization method to come with superior drug and vaccine targets. Thus, identified drug targets supposed to exhibits less side-effect and may signify as a supreme drug or a vaccine targets for the treatment of Lf.
Initially, metabolic pathway information of wBm and human host was collected from the KEGG database. At present, KEGG contains 281 metabolic pathways of human and 65 metabolic pathways of wBm. Following our protocol, we identified 5 unique metabolic pathways and 44 common metabolic pathways of wBm (Supplementary Table 1). Protein sequences from unique (50) and common metabolic pathways (460) of wBm were retrieved from the UniProt database and submitted to NCBI Protein BLAST, against the human. It results 156 proteins that showed “NO HITS” against the human proteome, remaining 311 proteins were excluded from the lists.
Essential proteins of wBm
In order to identify the essentiality of 156 non-homologous protein sequence of wBm, a sequence similarity searched was performed against the DEG database using protein BLAST tool. It resulted, 115 proteins, among these, 24 wBm proteins were belonging to the unique metabolic pathways and 91 proteins were from common metabolic pathways (Supplementary Table 2). These 115 proteins were likely to represents the significant role in metabolic pathways of wBm, non-homologous to human and indispensable for their survival. Further we identified the length of the essential proteins. These initial target protein lists can be used as a potential drug target; however, being non-homologous to human, essential and their involvement in various metabolic pathways is not an only criterion for the selection of potential drug targets. Therefore, to minimize the drug side-effects and for the identification of most potential drug targets of wBm, we further investigated these drug targets for low molecular weight, sub-cellular localization, experimental structure and druggability parameters. Through low molecular weight analysis we identified 10 protein sequences which have sequence length less than 100 kD. As it has been reported earlier by Dutta et. al. that short length sequences have less chance to represent an attractive drug target55, we screened this short length protein sequences from the potential drug target list. It results 105 protein sequences.
Prioritization of essential non-homologous proteins of wBm
Since, sub-cellular localization of protein plays a major role to understand the protein functions which could be essential for drug discovery and development process56. To investigate the sub-cellular localization of these 105 protein sequences; Gneg-mPLoc was used and resultant data was crosschecked with CELLO server. The main objective of this step is to identify the locations of the proteins in the cell which helps to differentiate between the drug and vaccine targets32. Protein founds in the cytoplasmic, periplasmic or inner-membrane region was considered as potential drug targets while extracellular proteins were recognized as potential vaccine targets. We identified 101 potential drug targets (Supplementary Table 3) and 4 potential vaccine targets (Table 2).
Druggability of essential non-homologous proteins of wBm
The druggability of essential wBm proteins were evaluated based on the assumptions that a druggable protein targets should interact with drug-like compound57,58. For this reason, each essential, non-homologous protein of wBm which plays a key role in metabolic pathway were accessed in the standalone BLASTp tool to find the similar drug target homology against the DrugBank and TTD database with an e-value less than 0.005. Proteins which showed hits with the defined cut-off values were recognized as significant homologs59 whereas, remaining proteins were not therefore excluded from the future study (Supplementary Table 4). Out of 101, we identified 61 drug targets which have shown similarity with the drug targets available in DrugBank and TTD database. Interestingly, 56 drug targets from DrugBank and 25 drug targets from TTD database have shown similarity, among these 20 drug targets were common in both the database. Ultimately, we identified 61 highly potential druggable targets (Table 3) and 4 potential vaccine targets (Table 2). These proposed therapeutic targets can be utilized for the further experimental studies to develop the most anti wBm therapeutics to treat the Lf.
Data compilation and database development
Identified drug targets and vaccine targets were stored in a user friendly database and we named it as FiloBase-a comprehensive drug target database for filariasis. The snapshot of the home page and the result page of FiloBase have been shown in Fig. 3.
The inaugural release of FiloBase contains a total no. of 119 potential drug targets of filarial nematodes. We have collected 58 drug targets of B. malayi from the literature survey23,33 and 61 drug targets from our current study through subtractive proteome approach for wBm. Since, structural information plays an important role for the drug and vaccine development60. These drug targets were stored in our database which can be fetched through the classified tab options present in the left-side of the database page (Fig. 3a). User can also enter their keywords to the query box available at the home page of the database and retrieve desired information. It will fetch the drug target information, related to the user given query (Fig. 3b).
All identified drug targets were modelled for their structural information using Modeller9v11, Phyre2.0 and I-TASSER server. Reliability of modelled drug targets was further evaluated using PROCHECK, ERRAT and PROVE server. The verification data of the modelled structure are stored in the model verification tab of the result page of the potential drug targets. User can fetch the desire information from the given link and can further improve the quality of the modelled structure if needed. All verified structures were incorporated in our database with additional information about potential drug targets to render it more enlightening and useful. Possible binding site residues were identified based on the template proteins and structure was shown three dimensionally using JSmol visualize.
In sequence tab, solvent accessibility and secondary structure were shown for each potential drug targets. User can get the graphical view of the secondary structure through the link given at the result page of the database. Alignment file of target and template proteins is also given in the result page to database to crosscheck the quality of the modelled structure (Fig. 3). This information may be useful for further development and to get insight into the structural level of the potential therapeutic targets.
In 3D Structure tab, a user friendly JSmol visualizer tool was used to visualize the modelled structure of therapeutics targets. In addition, based on the template, we have identified possible binding sites and listed in the result page. User can click on the active site pockets to explore the binding pockets and get insight into their positions and contributing residues using JSmol button (Fig. 3f). In KEGG tab, we have shown the role of the corresponding drug target in the metabolic pathway (Fig. 3g).
To make our database more informative, we have cross-linked our database with various related databases such as KEGG database for Pathways analysis, sequence similarity database in KEGG (SSDB) for motif search, UniProt and EMBL database for sequence information.
Various literatures33,61,62,63, databases and various websites [World Health Organization (WHO), Centre for Disease Control and Preventions (CDC), National Vector Borne Disease Control Programme-India (NVBDCP), DrugBank and Drugs.com (http://www.drugs.com/), Medscape (http://emedicine.medscape.com/), GAELF (http://www.filariasis.org/), The carter center (http://www.cartercenter.org/health/lf/)] were mined in the hunt of available drugs for the treatment of Lf and incorporated in our database with their 3D structure information. Drugs available in various brand names have been also collected with their general descriptions, clinical pharmacology, pharmacokinetics, contraindications, warnings, precautions and dosage and administration information data. Entire drug candidates are linked to DrugBank, KEGG and PubChem database for further information.
In addition, we have collected 71,009 EST sequences from various filariasis causing nematodes [B. malayi (65,536), B. pahangi (318), B. timori (26), W. bancrofti (5103) and wBm (26)] and stored in our database. These EST sequences of filariasis causing nematodes and wBm bacteria with add-on information will be useful for various groups of researchers.
Due to the advancements in biological sciences, an enormous number of sequence data were generated. Therefore, BLAST has become essential tool for biologists to compare these proteins and nucleotide sequences with largest set of datasets to investigate the similarities between them. For that reason, we have developed a standalone BLAST tool for filariasis and named it FiloBLAST. Query sequences can be entered in the single letter amino acid or in nucleotide code and based on the input sequence type algorithm can be selected. At present, user can BLAST their query sequences against the local copy of EST databases, Protein databases and drug targets from DrugBank databases. For EST databases, we have collected EST sequences from all the filariasis causing nematodes and made a local copy for BLAST search. We have also downloaded EST sequences from NEMBASE464 and given a link to the NEMBASE4 database for further information. Likewise, we have also collected data from Protein Data Bank65, Swiss-Prot66 and DrugBank67 database and made a local copy so that user can query their nucleotide and protein sequences against these databases to fetch the desired information in a single platform.
For potential and experimentally proved epitope sequences of filariasis causing nematodes were identified through the enormous literature survey and databases such as PubMed and Google Scholar. Various epitope databases were queried (IEDB68, BCIpep69 and SEDB58) to recognize the experimental and potential epitopes. At present, FiloBase contains 62 epitopes; 27 from B. malayi and 35 from W. bancrofti. A quick link has been provided in the home page of the database to fetch this information in a single click. We will timely update our database as earliest filarial data will be reported in the literature or authentic web resources. We have further provided an online data submission tool for users to upload their data in our database. Submitted data will be verified and may incorporate into our database. User can send their suggestions for further improvement of FiloBase.
Conclusions
In summary, owing to the emergence of resistant and limitations of access drugs for Lf, current research interest is focused on the identification of novel drug and vaccine targets to enhance the treatment of Lf. Using comparative proteome subtractive approach, we have pruned those drug targets which may be less effective or may cause severe side-effects. Therefore, proteins which are indispensable to the survival of pathogens and non-homologous to host proteins were considered as potential therapeutic targets. Druggability approach was further applied to the potential drug target data sets to expose the more probable drug targets. Our investigation reveals 61 potential drug targets and 4 potential vaccine target for wBm which could be further validated experimentally through the drug and vaccine design pipelines. Identified drug and vaccine targets were further modelled and then verified for their structural quality. Sequentially, we have enriched these data with essential information which may enlighten and support further research on wBm. All information was stored in a single stop web based platform. We named it FiloBase. We have also collected filariasis related information and records from various literatures and authenticated website to make accessible for the filarial researcher community through a user friendly database. In future, our database can be hereby extended by identifying potential drug targets from other filarial causing nematodes.
Additional Information
How to cite this article: Sharma, O. P. and Kumar, M. S. Essential proteins and possible therapeutic targets of Wolbachia endosymbiont and development of FiloBase-a comprehensive drug target database for Lymphatic filariasis. Sci. Rep. 6, 19842; doi: 10.1038/srep19842 (2016).
References
Rebollo, M. P. & Bockarie, M. J. Toward the elimination of lymphatic filariasis by 2020: treatment update and impact assessment for the endgame. Expert review of anti-infective therapy 11, 723–731, 10.1586/14787210.2013.811841 (2013).
Krentel, A., Fischer, P. U. & Weil, G. J. A review of factors that influence individual compliance with mass drug administration for elimination of lymphatic filariasis. PLoS neglected tropical diseases 7, e2447, 10.1371/journal.pntd.0002447 (2013).
Graves, P. M. et al. Lymphatic filariasis in Papua New Guinea: distribution at district level and impact of mass drug administration, 1980 to 2011. Parasites & vectors 6, 7, 10.1186/1756-3305-6-7 (2013).
Hoerauf, A. Filariasis: new drugs and new opportunities for lymphatic filariasis and onchocerciasis. Current opinion in infectious diseases 21, 673–681, 10.1097/QCO.0b013e328315cde7 (2008).
Addiss, D. G. Global elimination of lymphatic filariasis: a “mass uprising of compassion”. PLoS neglected tropical diseases 7, e2264, 10.1371/journal.pntd.0002264 (2013).
Sharma, O. P. et al. Modeling, docking, simulation and inhibitory activity of the benzimidazole analogue against beta-tubulin protein from Brugia malayi for treating lymphatic filariasis. Med Chem Res 21, 2415–2427, 10.1007/s00044-011-9763-5 (2012).
Osei-Atweneboano, M. Y., Eng, J. K. L., Boakye, D. A., Gyapong, J. O. & Prichard, R. K. Prevalence and intensity of Onchocerca volvulus infection and efficacy of ivermectin in endemic communities in Ghana: a two-phase epidemiological study. Lancet 369, 2021–2029, 10.1016/S0140-6736(07)60942-8 (2007).
Schwab, A. E., Boakye, D. A., Kyelem, D. & Prichard, R. K. Detection of benzimidazole resistance-associated mutations in the filarial nematode Wuchereria bancrofti and evidence for selection by albendazole and ivermectin combination treatment. American Journal of Tropical Medicine and Hygiene 73, 234–238 (2005).
Gillan, V., O’Neill, K., Maitland, K., Sverdrup, F. M. & Devaney, E. A Repurposing Strategy for Hsp90 Inhibitors Demonstrates Their Potency against Filarial Nematodes. PLoS neglected tropical diseases 8, e2699, 10.1371/journal.pntd.0002699 (2014).
Brady, M. & Global Alliance to Eliminate Lymphatic, F. Seventh meeting of the Global Alliance to Eliminate Lymphatic Filariasis: reaching the vision by scaling up, scaling down and reaching out. Parasites & vectors 7, 46, 10.1186/1756-3305-7-46 (2014).
Albiez, E. J. et al. Chemotherapy of onchocerciasis with high doses of diethylcarbamazine or a single dose of ivermectin: microfilaria levels and side effects. Tropical medicine and parasitology : official organ of Deutsche Tropenmedizinische Gesellschaft and of Deutsche Gesellschaft fur Technische Zusammenarbeit 39, 19–24 (1988).
Boussinesq, M., Gardon, J., Gardon-Wendel, N. & Chippaux, J. P. Clinical picture, epidemiology and outcome of Loa-associated serious adverse events related to mass ivermectin treatment of onchocerciasis in Cameroon. Filaria journal 2 Suppl 1, S4, 10.1186/1475-2883-2-S1-S4 (2003).
Towards a strategic plan for research to support the global program to eliminate lymphatic filariasis. Summary of Immediate needs and opportunities for research on lymphatic filariasis. Philadelphia, Pennsylvania, USA, December 9-10, 2003. The American journal of tropical medicine and hygiene 71, iii, 1–46 (2004).
Sharma, O. P., Vadlamudi, Y., Liao, Q., Strodel, B. & Suresh Kumar, M. Molecular modeling, dynamics and an insight into the structural inhibition of cofactor independent phosphoglycerate mutase isoform 1 from Wuchereria bancrofti using cheminformatics and mutational studies. Journal of biomolecular structure & dynamics 31, 765–778, 10.1080/07391102.2012.709460 (2013).
Pham, J. S. et al. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. International journal for parasitology. Drugs and drug resistance 4, 1–13, 10.1016/j.ijpddr.2013.10.001 (2014).
Govindarajan, M. & Sivakumar, R. Ovicidal, larvicidal and adulticidal properties of Asparagus racemosus (Willd.) (Family: Asparagaceae) root extracts against filariasis (Culex quinquefasciatus), dengue (Aedes aegypti) and malaria (Anopheles stephensi) vector mosquitoes (Diptera: Culicidae). Parasitology research 113, 1435–1449, 10.1007/s00436-014-3784-1 (2014).
Sharma, O. P., Agrawal, S. & Kumar, M. S. Physicochemical properties of the modeled structure of astacin metalloprotease moulting enzyme NAS-36 and mapping the druggable allosteric space of Heamonchus contortus, Brugia malayi and Ceanorhabditis elegans via molecular dynamics simulation. Interdisciplinary sciences, computational life sciences 5, 312–323, 10.1007/s12539-013-0182-9 (2013).
Sahare, K. N. & Singh, V. Antifilarial activity of ethyl acetate extract of Vitex negundo leaves in vitro. Asian Pacific journal of tropical medicine 6, 689–692, 10.1016/S1995-7645(13)60119-4 (2013).
Joseph, S. K. & Ramaswamy, K. Single multivalent vaccination boosted by trickle larval infection confers protection against experimental lymphatic filariasis. Vaccine 31, 3320–3326, 10.1016/j.vaccine.2013.05.077 (2013).
Galvin, B. D. et al. A target repurposing approach identifies N-myristoyltransferase as a new candidate drug target in filarial nematodes. PLoS neglected tropical diseases 8, e3145, 10.1371/journal.pntd.0003145 (2014).
Saini, P. et al. Effect of ferulic acid from Hibiscus mutabilis on filarial parasite Setaria cervi: molecular and biochemical approaches. Parasitology international 61, 520–531, 10.1016/j.parint.2012.04.002 (2012).
Yadav, M., Singh, A., Rathaur, S. & Liebau, E. Structural modeling and simulation studies of Brugia malayi glutathione-S-transferase with compounds exhibiting antifilarial activity: implications in drug targeting and designing. Journal of molecular graphics & modelling 28, 435–445, 10.1016/j.jmgm.2009.10.003 (2010).
Kumar, S. et al. Mining predicted essential genes of Brugia malayi for nematode drug targets. PloS one 2, e1189, 10.1371/journal.pone.0001189 (2007).
Taylor, M. J. Wolbachia endosymbiotic bacteria of filarial nematodes. A new insight into disease pathogenesis and control. Archives of medical research 33, 422–424 (2002).
Taylor, M. J. & Hoerauf, A. A new approach to the treatment of filariasis. Current opinion in infectious diseases 14, 727–731 (2001).
Holman, A. G., Davis, P. J., Foster, J. M., Carlow, C. K. & Kumar, S. Computational prediction of essential genes in an unculturable endosymbiotic bacterium, Wolbachia of Brugia malayi. BMC microbiology 9, 243, 10.1186/1471-2180-9-243 (2009).
Foster, J. et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS biology 3, e121, 10.1371/journal.pbio.0030121 (2005).
Hosen, M. I. et al. Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdisciplinary sciences, computational life sciences 6, 48–56, 10.1007/s12539-014-0188-y (2014).
Arvind, A., Jain, V., Saravanan, P. & Mohan, C. G. Uridine monophosphate kinase as potential target for tuberculosis: from target to lead identification. Interdisciplinary sciences, computational life sciences 5, 296–311, 10.1007/s12539-013-0180-y (2013).
Madagi, S. et al. Identification of membrane associated drug targets in Borrelia burgdorferi ZS7- subtractive genomics approach. Bioinformation 6, 356–359 (2011).
Butt, A. M. et al. Mycoplasma genitalium: a comparative genomics study of metabolic pathways for the identification of drug and vaccine targets. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 12, 53–62, 10.1016/j.meegid.2011.10.017 (2012).
Barh, D. et al. In Silico Subtractive Genomics for Target Identification in Human Bacterial Pathogens. Drug Develop Res 72, 162–177, 10.1002/Ddr.20413 (2011).
Sharma, O. P., Vadlamudi, Y., Kota, A. G., Sinha, V. K. & Kumar, M. S. Drug targets for lymphatic filariasis: a bioinformatics approach. Journal of vector borne diseases 50, 155–162 (2013).
Sarkar, M., Maganti, L., Ghoshal, N. & Dutta, C. In silico quest for putative drug targets in Helicobacter pylori HPAG1: molecular modeling of candidate enzymes from lipopolysaccharide biosynthesis pathway. Journal of molecular modeling 18, 1855–1866, 10.1007/s00894-011-1204-3 (2012).
Collins, J. F., Coulson, A. F. & Lyall, A. The significance of protein sequence similarities. Computer applications in the biosciences : CABIOS 4, 67–71 (1988).
Pearson, W. R. Effective protein sequence comparison. Methods in enzymology 266, 227–258 (1996).
Pearson, W. R. Comparison of methods for searching protein sequence databases. Protein science : a publication of the Protein Society 4, 1145–1160, 10.1002/pro.5560040613 (1995).
Luo, H., Lin, Y., Gao, F., Zhang, C. T. & Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic acids research 42, D574–580, 10.1093/nar/gkt1131 (2014).
Jadhav, A., Ezhilarasan, V., Prakash Sharma, O. & Pan, A. Clostridium-DT(DB): a comprehensive database for potential drug targets of Clostridium difficile. Computers in biology and medicine 43, 362–367, 10.1016/j.compbiomed.2013.01.009 (2013).
Shen, H. B. & Chou, K. C. Gneg-mPLoc: a top-down strategy to enhance the quality of predicting subcellular localization of Gram-negative bacterial proteins. Journal of theoretical biology 264, 326–333, 10.1016/j.jtbi.2010.01.018 (2010).
Yu, C. S., Lin, C. J. & Hwang, J. K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein science : a publication of the Protein Society 13, 1402–1406, 10.1110/ps.03479604 (2004).
Nakai, K. & Horton, P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends in biochemical sciences 24, 34–36 (1999).
Hajduk, P. J., Huth, J. R. & Tse, C. Predicting protein druggability. Drug discovery today 10, 1675–1682, 10.1016/S1359-6446(05)03624-X (2005).
Keller, T. H., Pichota, A. & Yin, Z. A practical view of ‘druggability’. Current opinion in chemical biology 10, 357–361, 10.1016/j.cbpa.2006.06.014 (2006).
Knox, C. et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic acids research 39, D1035–1041, 10.1093/nar/gkq1126 (2011).
Chen, X., Ji, Z. L. & Chen, Y. Z. TTD: Therapeutic Target Database. Nucleic acids research 30, 412–415 (2002).
Bernstein, F. C. et al. The Protein Data Bank. A computer-based archival file for macromolecular structures. European journal of biochemistry/FEBS 80, 319–324 (1977).
Eswar, N. et al. Comparative protein structure modeling using MODELLER. Current protocols in protein science/editorial board, John E. Coligan … [et al.] Chapter 2, Unit 2 9, 10.1002/0471140864.ps0209s50 (2007).
Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics 9, 40, 10.1186/1471-2105-9-40 (2008).
Kelley, L. A. & Sternberg, M. J. Protein structure prediction on the Web: a case study using the Phyre server. Nature protocols 4, 363–371, 10.1038/nprot.2009.2 (2009).
Wu, S. & Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic acids research 35, 3375–3382, 10.1093/nar/gkm251 (2007).
Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. Journal of biomolecular NMR 8, 477–486 (1996).
Colovos, C. & Yeates, T. O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein science : a publication of the Protein Society 2, 1511–1519, 10.1002/pro.5560020916 (1993).
Barh, D. et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chemical biology & drug design 78, 73–84, 10.1111/j.1747-0285.2011.01118.x (2011).
Dutta, A. et al. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In silico biology 6, 43–47 (2006).
Caragea, C., Caragea, D., Silvescu, A. & Honavar, V. Semi-supervised prediction of protein subcellular localization using abstraction augmented Markov models. BMC bioinformatics 11 Suppl 8, S6, 10.1186/1471-2105-11-S8-S6 (2010).
Damte, D. et al. Putative drug and vaccine target protein identification using comparative genomic analysis of KEGG annotated metabolic pathways of Mycoplasma hyopneumoniae. Genomics 102, 47–56, 10.1016/j.ygeno.2013.04.011 (2013).
Shanmugham, B. & Pan, A. Identification and characterization of potential therapeutic candidates in emerging human pathogen Mycobacterium abscessus: a novel hierarchical in silico approach. PloS one 8, e59126, 10.1371/journal.pone.0059126 (2013).
Barh, D. et al. Exoproteome and secretome derived broad spectrum novel drug and vaccine candidates in Vibrio cholerae targeted by Piper betel derived compounds. PloS one 8, e52773, 10.1371/journal.pone.0052773 (2013).
Sharma, O. P., Jadhav, A., Hussain, A. & Kumar, M. S. VPDB: Viral Protein Structural Database. Bioinformation 6, 324–326 (2011).
Ottesen, E. A. Lymphatic filariasis: Treatment, control and elimination. Advances in parasitology 61, 395–441, 10.1016/S0065-308X(05)61010-X (2006).
Simonsen, P. E. et al. Lymphatic filariasis control in Tanzania: effect of six rounds of mass drug administration with ivermectin and albendazole on infection and transmission. BMC infectious diseases 13, 335, 10.1186/1471-2334-13-335 (2013).
Sodahlon, Y. K. et al. A success story: Togo is moving toward becoming the first sub-Saharan African nation to eliminate lymphatic filariasis through mass drug administration and countrywide morbidity alleviation. PLoS neglected tropical diseases 7, e2080, 10.1371/journal.pntd.0002080 (2013).
Elsworth, B., Wasmuth, J. & Blaxter, M. NEMBASE4: the nematode transcriptome resource. International journal for parasitology 41, 881–894, 10.1016/j.ijpara.2011.03.009 (2011).
Sussman, J. L. et al. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta crystallographica. Section D, Biological crystallography 54, 1078–1084 (1998).
Watanabe, K. & Harayama, S. [SWISS-PROT: the curated protein sequence database on Internet]. Tanpakushitsu kakusan koso. Protein, nucleic acid, enzyme 46, 80–86 (2001).
Wishart, D. S. et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic acids research 36, D901–906, 10.1093/nar/gkm958 (2008).
Kim, Y. et al. Immune epitope database analysis resource. Nucleic acids research 40, W525–530, 10.1093/nar/gks438 (2012).
Saha, S., Bhasin, M. & Raghava, G. P. Bcipep: a database of B-cell epitopes. BMC genomics 6, 79, 10.1186/1471-2164-6-79 (2005).
Acknowledgements
Om Prakash Sharma is grateful to the Council of Scientific & Industrial Research (CSIR), India for the Senior Research Fellowship. Research carried out in the laboratory of the Centre for Excellence in Bioinformatics, Pondicherry University-India.
Author information
Authors and Affiliations
Contributions
O.P. and M.S.K. conceived the study. O.P. and M.S.K. performed the model output analysis and wrote the initial draft of the paper. O.P. and M.S.K. contributed to interpreting results, discussion of the associated results and improvement of this paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sharma, O., Kumar, M. Essential proteins and possible therapeutic targets of Wolbachia endosymbiont and development of FiloBase-a comprehensive drug target database for Lymphatic filariasis. Sci Rep 6, 19842 (2016). https://doi.org/10.1038/srep19842
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19842
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
