
Abstract
The application of next generation sequencing (NGS) technology in the diagnosis of human pathogens is hindered by the fact that pathogenic sequences, especially viral, are often scarce in human clinical specimens. This known disproportion leads to the requirement of subsequent deep sequencing and extensive bioinformatics analysis. Here we report a method we called “Preferential Amplification of Pathogenic Sequences (PATHseq)” that can be used to greatly enrich pathogenic sequences. Using a computer program, we developed 8-, 9- and 10-mer oligonucleotides called “non-human primers” that do not match the most abundant human transcripts, but instead selectively match transcripts of human pathogens. Instead of using random primers in the construction of cDNA libraries, the PATHseq method recruits these short non-human primers, which in turn, preferentially amplifies non-human, presumably pathogenic sequences. Using this method, we were able to enrich pathogenic sequences up to 200-fold in the final sequencing library. This method does not require prior knowledge of the pathogen or assumption of the infection; therefore, it provides a fast and sequence-independent approach for detection and identification of human viruses and other pathogens. The PATHseq method, coupled with NGS technology, can be broadly used in identification of known human pathogens and discovery of new pathogens.
Similar content being viewed by others

Introduction
Next generation sequencing (NGS) technologies1,2, including 2nd and 3rd generation DNA sequencing platforms, have started a revolution in genomics and provided opportunities for its broad application in many other fields3,4,5, including the diagnosis of human pathogens6,7,8,9,10. Examples of NGS application in the fields of virology and infectious diseases include: 1) epidemiology investigation of infectious disease outbreaks11,12; 2) etiologic diagnosis of viral infections using a meta-genomic approach13,14; 3) discovery of new human viruses4; and 4) discovery of other new pathogenic viruses15. Detailed reviews offer an introduction to NGS technology applications in virus discovery and clinical/diagnostic virology7,8,10. However, NGS technology is still a research tool, rather than a diagnostic tool and cannot be used in current infectious disease diagnostic laboratories due to 1) the scarcity of pathogen sequences in human clinical samples; 2) the necessary subsequent requirement of extensive deep sequencing; and 3) the complexity of bioinformatics analysis required in order to identify the pathogenic sequences. For example, the average viral genome in a human clinical sample is about 1-100 per 10 million human genome sequence reads. Many laboratories have developed various strategies, from consensus PCR assays that use degenerate primers to computational subtraction of large sequence data in order to find possible unknown pathogens, with little success. These “search for a needle in a haystack” strategies have proven to be a very difficult task.
To make NGS technology a practical tool for detecting human pathogens, the key is to greatly increase the presence of pathogenic sequences in a clinical sample. To address this challenge, we developed a method we called “Preferential Amplification of Pathogenic Sequences (PATHseq)” which can be used to preferentially amplify non-human sequences in a clinical sample. This method is based on the following facts: 1) active infection is the result of pathogenic gene expression, which produces RNAs, or pathogenic transcripts; 2) only about 3% of the human genome generates transcripts. Among these, the top 1,000 and 2,000 most abundant human transcripts comprise more than 65% and 72% of all human transcripts, respectively16; 3) by selectively excluding the amplification of these abundant human transcripts, we can preferentially amplify pathogenic transcripts in human clinical samples; 4) pathogenic transcripts can be further enriched through subtractive hybridization against a reference (normal) human transcription library (human transcriptome). The PATHseq technology, in combination with NGS technology, has the potential to provide comprehensive and unbiased detection of human pathogens responsible for any infectious disease.
Results
The most abundant human transcripts
The recent completion of the Encyclopedia of DNA Elements (ENCODE) project17 provides a genome-wide “landscape of transcription in human cells” in 14 different cell lines. Although the size of the human genome is huge (containing over 3 billion base pairs (bp)), it encodes only about 20,000 protein-coding genes, accounting for a very small fraction (approximately 2%) of the genome. Based on the publicly available ENCODE database16, the total human large transcripts (>200 bp RNAs) in GM12878 (a cell line that contributed most to the ENCODE database) are 161,999. Among these, 86,248 transcripts are reproducible (in a duplicated experiment). These 86,248 transcripts are defined as human transcriptome (Table 1). A recent report found that most protein-coding genes have one major transcript expressed at significantly higher level than others and in human tissues these major transcripts contribute almost 85 percent to the total mRNA18. Given that the average length of human mRNAs is 1.3 kb19, the complexity can be reduced by 26.8 times (3,000,000/(86,248 × 1.3)), if we sequence cDNA instead of genomic DNA. This strategy has been successfully used by several laboratories in search of human pathogens4,5,20. However, this strategy is still impractical for diagnostic laboratories, because the number of human transcripts is still too large compared to the relative scarcity of pathogenic transcripts.
In order to solve this problem, we developed an alternative strategy using the most abundant human transcripts (Table 1). Our strategy relies on the removal of the most abundant human transcripts from clinical samples to selectively enrich the pathogenic sequences and further reduce the sequencing complexity. As shown in Table 1, we found the most abundant 1,000 and 2,000 transcripts comprised about 65% and 72% of all human transcripts, respectively, based on ENCODE data17.
Non-human oligonucleotides
We further developed a computer program to look for specific patterns in the human transcriptome database. We used the following steps to find the shortest unmatched k-mers of nucleotides in the human transcriptome. First, we collated a set of the Ensembl transcript names (e.g., ENST00000387347) and retrieved DNA sequences corresponding to the transcript names from the human cDNA sequences of the Ensembl release 73 available at ftp://ftp.ensembl.org/pub/release-73/fasta/homo_sapiens/cdna. Secondly, we searched for the shortest unmatched k-mers in the collated set of DNA sequences21,22. The computer program counted k-mers for a given k or the size of substring in a DNA sequence. It started from k = 1 and checked if all of the possible k-mers occurred at least once. It stopped when it reached a k value where there was at least one k-mer that was not found as a substring in the set of transcripts. As predicted, we found that human transcript sequences are not randomly distributed. Using this computer program, we were able to generate a set of 88 8-mer oligonucleotides (Table 2) that do not match the sequences of the 2,000 most abundant human transcripts. This set of oligonucleotides is, therefore, named as “non-human oligonucleotides”. In other words, by using this set of oligos as primers in the construction of cDNA library, we can get rid of 72% of human transcripts from clinical samples, greatly increasing the chance of selectively targeting pathogenic sequences. Theoretically, this set of primers has the probability to amplify any sequences larger than 5,958 bp (48 × 8 /88), which should include almost all human pathogens (both viruses and bacteria). To test how likely this set of short oligonucleotides can cover all known human viruses, we performed an in silico analysis and found that among 386 of all known human viruses (Supplementary Information S1), this set of 88 8-mer oligonucleotides can cover 327 (85%) of them. The remaining 59 unmatched human viruses are usually small human viruses (Supplementary Information S2), which includes human immunodeficiency virus 1 (HIV-1). To cover all know human pathogenic viruses, we also developed a new list of 81 9-mer oligos that do not match the top 2,000 most abundant human transcripts while cover all known human pathogenic viruses (see below In silico experiment).
Using the same computer program, we generated several sets of 7-, 8-, 9- and 10-mer oligonucleotides that selectively amplify non-human sequences during construction of the cDNA library (Supplemental Information S7-10), which is summarized in Table 3. As shown in Table 3, there are 197 9-mer oligonucleotides that do not match the sequence of 500 bp 3’end of all 86,248 human transcripts. To make this strategy work, we introduced ddNTP and used a mixture of ddNTP with normal dNTP (1% ddATP in dNTP solution) in the construction of cDNA library. ddNTP lacks the OH needed to continue the elongation of the DNA strand. When ddATP is added to the reaction, the elongation of the strand stops once the ddATP is added to the new strand. Using this set of 9-mer oligos, the likelihood to find a match in a random sequence is 49 × 9 /197 = 11,976 bp. Most human pathogens have larger genome sizes than this.
Preferential Amplification of Pathogenic Sequences (PATHseq)
As shown in Fig. 1, PATHseq procedure includes: (1) Total mRNAs are extracted and purified from clinical samples; (2) A primer (P1) is designed to specifically transcribe mRNA into first-strand cDNAs (anti-sense) while introducing a T7 promoter/primer sequence into the cDNA; (3) The ribonuclease activity of RNase H cleaves RNA in a DNA/RNA duplex, allowing the synthesis of secondary cDNA strands; (4) A set of 88 specific 8-mer oligonucleotides (Table 2) is used as primers for the synthesis of secondary cDNA strands. Because these primers do not amplify the 2,000 most abundant human mRNAs, about 72% of all human mRNAs is eliminated from amplification, preferentially amplifying non-human (pathogenic) sequences; (5) Using the T7 promoter introduced in step 2, the T7 RNA polymerase synthesizes RNAs with double-stranded DNA as template; (6) Because the T7 promoter is attached to the poly(A) end, newly generated RNAs are anti-sense; (7) Human reference cDNA library was created using the same set of 8-mer primers (Table 2), plus a poly d(T) primer (not P1 primer). Normal (non-pathogenic) human mRNAs are used as templates when the library is constructed. Sense strands of human reference cDNAs are separated using poly d(T) beads. The beads are further used as the solid phase for subtractive hybridization. Newly generated anti-sense human RNAs from step 6 are captured (hybridized) by these cDNAs and specifically degraded by RNase H in RNA-DNA hybrids. RNase H does not digest single or double-stranded DNA; (8) Pathogenic RNAs are greatly enriched because they do not hybridize to human reference cDNAs; (9) A T7 primer is used to synthesize the first cDNA strands; (10) Again, RNase H cleaves RNAs in a DNA/RNA duplex; (11) Synthesized RNAs are anti-sense; (12) Step 6 through12 form a cycle in which pathogenic RNAs are repeatedly enriched.

Schematic representation of the PATHseq (Preferential Amplification of Pathogenic Sequences) method.
(1) Total mRNAs from clinical sample, including human mRNAs and relatively scarce pathogenic mRNAs; (2) Total mRNAs are transcribed into first strand cDNAs with P1 primer; (3) RNase H cleaves RNAs in RNA-DNA duplex; (4) Reverse transcriptase (RT) synthesizes secondary cDNA strands with P2 primers; (5) T7 RNA polymerase synthesizes RNAs in the presence of T7 promoter; (6) Synthesized anti-sense RNAs; (7) Synthesized RNAs are hybridized to human reference (non-pathogenic) cDNA library coated on a solid phase. RNase H cleaves bound RNAs (human RNAs) in RNA-DNA duplex; (8) Pathogenic RNAs are enriched; (9) Reverse transcription; (10) RNase H cleaves RNAs in RNA-DNA duplex; (11) T7 RNA polymerase synthesizes RNAs; (12) New RNAs synthesized from enriched pathogenic RNAs are amplified 100-1000 fold.
Computational subtraction
To further analyze sequence data, we developed a computer program to subtract sequencing reads that match human sequences23,24 and assemble them into contiguous sequences for direct comparison with the GenBank databases of nucleic acids using BLASTN software25,26. Using this method, any non-matching sequences representing potential pathogenic sequences will be enriched and remain in the final dataset4,5,20. Figure. 2 shows the approach for computational subtraction. In Step 1, to ensure reasonably high quality of the reads, we trimmed the sequences at the 5’-end by 20 base pairs and also trimmed them by quality score from the 3’-end using 30 as the threshold score27. In Step 2, using a short read aligner called STAR28, we aligned the remaining reads on the human genome of primary assembly sequences unmasked that we fetched from ENSEMBL. Reads aligned to multiple loci in the reference human genome were also considered to be unmapped reads and were filtered out to reduce the false positive rate. In Step 3, to obtain longer non-human origin reads, unaligned reads were assembled using a de novo assembler named Trinity29. In Step 4, contigs were blasted against NCBI “nr” nucleotide sequence database for sequences that were similar to the de novo assembled unaligned sequences30,31. In Step 5, the assembled unaligned sequences were assigned to species by the taxonomic unit using the NCBI nucleotide sequence database search result and the taxonomy database32.

Workflow of identification of sequences with foreign origin.
Step 1 is of quality check of the reads, removing low quality reads and trimming low quality bases. Step 2 is to filter out unmapped reads on a host genome. Step 3 is of de novo assembling unmapped reads into contig sequences. Step 4 is to search the BLAST nucleotide sequence database for sequences similar to the contig sequences. Step 5 is to select contig sequences that are mapped on foreign origin sequences in the nucleotide sequence database.
Proof of concept 1 – enrichment of viral sequences from human cell line
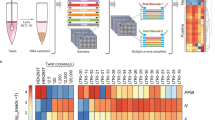
We tested the PATHseq method for the enrichment of pathogenic (viral) sequences from Kaposi’s sarcoma-associated herpesvirus (KSHV) (also known as human herpesvirus 8 or HHV-8)-infected BCBL-1 cells using two different approaches, quantitative real time PCR (qPCR) and next generation sequencing. For qPCR, we designed and optimized KSHV-specific primer sets to monitor the enrichment of viral transcripts compared with cellular house-keeping genes, beta-actin and GAPDH, as controls of background human sequences (Table 4). We also included two viral genomic DNA controls, KSHV-OriL and KSHV-OriR. These two viral genomic locations do not generate viral transcripts. As shown in Fig. 3A, we measured relative enrichment of various viral transcripts from at least three independent experiments compared with the cellular house-keeping gene control, beta-actin. Up to 164-fold enrichment (ORF50F1) could be enriched by PATHseq method. The different enrichment level among various viral genes can be explained as the result of viral transcript size and viral sequence matches with 8-mer oligonucleotides used in PATHseq method.

Enrichment of pathogenic sequences by PATHseq method.
(A) Quantitative real time PCR results indicating enrichment of viral transcripts by PATHseq method from KSHV/HHV-8 latently infected BCBL-1 cell line. ORFK8, ORF40, ORF50F1, ORF50F2, ORF50 and ORF57 are individual lytic viral genes. Actin and GAPDH are cellular house-keeping genes for control; KSHV-OriL and KSHV-OriR are two viral genomic DNA controls; Results are shown from at least three repeats with standard deviation. (B) Next generation sequencing results showing enrichments of viral sequencing reads by PATHseq method. Two independent runs using Illumina MiSeq system were performed.
We further tested the PATHseq method for the enrichment of KSHV viral sequences by next generation sequencing. As shown in Fig. 3B, we performed two independent NGS runs, each consisted of a control (non-enriched) and PATHseq-enriched sequencing libraries. For Run 1, 6 and 493 viral reads could be identified from a total of 11,552,534 and 12,356,296 sequencing reads for control and PATHseq libraries, respectively. For Run 2, 76 and 2,682 viral reads could be identified from a total 13,988,804 and 15,103,847 reads, respectively. The overall enrichments of KSHV viral reads among total sequenced reads increased from 0.0052% to 0.399% (76.8-fold) and 0.0543% to 1.7757% (32.7-fold), respectively.
Proof of concept 2 – enrichment of viral sequences from virus-spiked human samples
We tested the PATHseq method for the enrichment of viral sequences from human samples spiked with multiple viruses. Whole blood was obtained from a healthy blood donor and peripheral blood mononuclear cells (PBMC) were isolated from whole blood using standard procedure. Total RNAs from either PBMC or virus-infected cells were extracted and purified using Qiagen RNeasy Mini Kit. Total RNAs from infected cells were spiked into human PBMC RNAs with a ratio of 1 to 1,000 based on Nanodrop readings. After enrichment by the PATHseq method, quantitative real time PCR (qPCR) was performed with individual viruses, along with two cellular controls, beta-actin and GAPDH. As shown in Fig. 4, we measured relative enrichment of individual viral transcripts from at least three independent experiments compared with cellular house-keeping gene control, beta-actin, which was set to 1. Various human viruses were enriched differently from 31 fold (Influenza A virus) to 242 fold (for Human herpesvirus 6B). The different levels of enrichment for different viruses could be attributed to the fact that the list of 88 8-mer nucleotides used in the PATHseq method matches disproportionately to different viruses. As shown in Table 5, There are 66 8-mer oligonucleotides that match the sequence of human herpesvirus 6B and 57, 3, 2, 4 and 14 match sequences of human herpesvirus 8, hepatitis C virus, influenza A virus, human parainfluenza virus and human adenovirus C, respectively. In contrast, there are no oligonucleotides from the list of 88 8-mer oligos that match the genome sequence of human immunodeficiency virus. As a result, there was no enrichment (0.98 fold) of HIV viral sequences from spiked human sample as shown in Fig. 4.

Enrichment of spiked viral sequences.
Human mRNAs isolated from peripheral blood mononuclear cells (PBMC) were spiked with viral mRNAs from cell culture infected with human herpesvirus 6B (HHV-6B), human herpesvirus 8 (HHV-8), hepatitis C virus (HCV), Influenza A virus, human Parainfluenza Virus Type 1 (HPIV1), human immunodeficiency virus (HIV) and human Adenovirus Type C. Beta-actin and GAPDH are cellular controls. PATHseq method was performed to enrich the viral sequences. qPCRs were used to monitor the relative enrichments (in fold) of individual virus to cellular gene control, beta-actin. Results are shown from at least three repeats with standard deviation.
Clinical diagnosis of unknown infection
The PATHseq method was further tested by investigating an unknown clinical respiratory infection and successfully identified a new variant of Streptococcus pneumonia (ASVL_JC_001) from a clinical specimen with bronchitis & pulmonary inflammation33. Using PATHseq coupled with NGS, we generated a total of 16,031,250 sequencing reads and eventually identified 118,200 (0.73%) reads as S. pneumonia sequences (Fig. 5). These reads formed clusters enriched by the 88 8-mer oligonucleotides and further assembled into 2067 contig sequences. Sequencing analysis of this strain shows atypical features of S. pneumonia as it shows alpha-hemolytic colonies (Supplemental Information S3) and bile solubility but was resistant to optochin (Supplemental Information S4). Antimicrobial susceptibility testing shows that this strain was sensitive to cefotaxime, chloramphenicol, oxacillin, penicillin, tetracycline and vancomycin, but resistant to erythromycin and ethyl hydrocupreine (Taxo P) and partially resistant to sulfamethoxazole trimethoprim (Supplemental Information S4). Metabolic biochemical assay indicated that this variant behaved more like Streptococcus pneumonia than to closely related specie Streptococcus mitis (Supplemental Information S5). PCR assay of housekeeping genes for S. pneumonia indicated that this variant harbors genes encoding the virulence factors pneumolysin (ply) and the major autolysin (lytA), both of which are normally associated with pneumococci (Supplemental Information S6). Whole genome sequencing was performed on an Illumina MiSeq version 2 system33. The total genome of this variant is 2,092,532 base pairs long and has a GC content of 40.3%. We annotated the genome assembly by using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline34. Among a total of 83 contigs, 69 contigs harbor annotations of genes, CDS and RNAs. We found 2158 putative genes and 2051 CDS. We also found 5 rRNAs, 44 tRNAs and 1 ncRNA for this variant33.

Application of PATHseq method in identifying an unknown infection.
Initial raw sequencing reads were 16,031,250, after subtracting human sequences, 118,200 (0.7%) reads were identified as S. pneumonia sequences.
In silico experiment
We performed in silico experiment to test how likely the PATHseq method can enrich known human viral pathogens. We first generated a set of 199 human pathogenic viruses based on NCBI and ViralZone human viruses databases (viralzone.expasy.org/) (Supplementary Information S11). As summarized in Table 6, we generated the sets of 8-, 9- and 10-mer oligonucleotides that do not match the most abundant human transcripts at top 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000, 20000 and all 75987, respectively. For example, there are 329 8-mer oligonucleotides that do not match the top 1000 most abundant human transcripts. Among these 329 oligos, there are 62 that match at least one known human pathogenic virus and cover 86.9% of all human pathogenic viruses. Please note there are some minor differences in the total number of human transcripts between Table 5 (75,987) and Table 1 and 3 (86,248) because Table 5 used the UCSC Genome Browser assembly (ID: hg38), while Table 1 and 3 used ENCODE database16. Please also note that the total number of human viruses (386) (Supplementary Information S1) is different from the total number of human pathogenic viruses, which is 199 as listed in Supplementary Information S11. Overall, this in silico analysis indicates that by recruiting the 81 9-mer oligos (Table 6), we can exclude the top 2000 most abundant human transcripts while still covering 100% of known human pathogenic viruses including HIV, which was missed from coverage by 88 8-mer oligos (Table 5 and Fig. 4). By recruiting the 171 10-mer oligos, we can exclude the top 20,000 most abundant human transcripts while still covering more than 95% of known human pathogenic viruses. Because the rest 55,987 transcripts only count for less than 5% of all human transcripts, none of them would have more abundance than the viral genome.
Maximal number of sequencing samples
The maximal number of samples that can be loaded onto a sequencing run depends on two factors: 1) the minimal sequencing coverage required for successfully identifying a pathogen, usually the sequencing depth to achieve at least 1 RPKM (Reads per kilobase of transcript per million mapped reads) of pathogen sequences is required; 2) the capacity of the sequencing instrument. Using the same computer program we developed, we generated a total of 40 10-mer oligonucleotides that do not match the sequences to any of the total 161,999 human transcripts (Table 7). These oligos can be divided into two sets and used as adaptors for the construction of sequencing libraries. The maximum combination of these 20 + 20 adaptors can generate 400 (20 × 20 = 400) sample identifiers (barcodes) from A1 to T20. Therefore, a maximum total of 400 samples can be separately labeled by these two sets of adaptors, mixed into one sample run for sequencing and then separated by its own sample identifier (barcode) from A1 to T20 (Table 7).
Discussion
Next-generation sequencing technology provides broad detection of infectious agents in a sequence independent manner and is rapidly being adapted for the clinical diagnosis of human pathogens. However, NGS technology is still a research tool, rather than a diagnostic tool and cannot be used in current infectious disease diagnostic laboratories due to the scarcity of pathogen sequences in human clinical samples and necessary subsequent deep sequencing and intensive bioinformatics analysis in order to identify the pathogenic sequences. To solve this problem, many laboratories have developed various strategies. A ribosomal RNA depletion method is wildly used in RNA-sequencing (RNA-Seq) because large ribosomal RNA (rRNA) constitutes approximately 90% RNA species in total RNA35,36,37,38. We compared commercial rRNA depletion method (Epicentre’s Ribo-Zero rRNA Removal Core Kit, Cat. No. RZC110424) with our PATHseq method. The rRNA depletion method does not distinguish human RNA from pathogenic RNA, therefore, the amount of human RNA is still overwhelming in the final sequencing library. However, we believe the best way is to combine these two methods together, i.e. to recruit rRNA depletion prior to the application of PATHseq method, instead of polyA enrichment, because some viruses do not produce mRNA. Other methods for enrichment of pathogenic sequences include a capture-based approach using virus-specific DNA fragments as probes through hybridization39 and an approach using methyl-CpG binding domain (MBD) to separate methylated host DNA from microbial DNA based on differences in CpG methylation density40.
The PATHseq method provides an innovative way to preferentially amplify pathogenic sequences over host sequences through the use of non-human but pathogenic-specific short primers. Theoretically, these primers are short enough to broadly detect pathogens (viruses and bacteria) above a given threshold genome size. For example, the set of 88 8-mer primers has the probability to amplify any sequences larger than 5,958 bp (48 × 8 /88), which should include almost all human pathogens (both viruses and bacteria). However, we noticed that some small viruses including human immunodeficiency virus, could not be enriched by this set of 88 8-mer primers. In addition, some viruses, which do not produce mRNAs, would be missed by the PATHseq method. To address this problem, we further generated a list of 81 9-mer oligos (Table 6). These 9-mer oligos do not match the top 2,000 most abundant human transcripts while covering all known human pathogenic viruses. Using these short non-human primers instead of random primers in the construction of cDNA library, the PATHseq method enables efficient enrichment of pathogenic sequences. With significant enrichment of pathogenic sequences in the final sequencing library, it is possible that more samples can be put into a single run to reduce overall cost and turnaround time. The PATHseq method, in combination with NGS technology, has the potential to provide comprehensive and unbiased (sequence-independent) detection of human known as well as unknown (novel) pathogens.
Materials and Methods
BCBL-1 cell line
The body-cavity-based lymphoma cell line, BCBL-1, is latently infected with Kaposi’s sarcoma-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV-8) with average virus copy number ~50 per cell41. BCBL-1 is grown in RPMI1640 (HyClone Laboratories, Logan, Utah) supplemented with 10% fetal bovine serum (FBS) at 37 oC in a 5% CO2 condition.
Human specimen with spiked viruses
Single donor human whole blood was purchased from Fisher Scientific (Thermo Fisher Scientific, Waltham, MA, Cat. No. 50-177-224). Peripheral blood mononuclear cells (PBMC) were purified from whole blood using standard procedure42. Infecting cells with human herpesvirus 6B was described previously43. Hepatitis C patient serum was diagnosed and processed in this lab. Supernatants from infected cell cultures for Influenza A virus (Cat. No. VR-1736), Human Parainfluenza Virus Type 1 (HPIV1)(Cat. No. VR-94), Human Adenovirus Type C (Cat. No. VR-1) were purchased from ATCC. Total RNAs from either PBMC or supernatant of virus-infected cells were extracted and purified using Qiagen RNeasy Mini Kit (Qiagen Inc., Valencia, CA, Cat. No. 74104). mRNAs were purified using NEB Magnetic mRNA Isolation Kit (Cat. No. S1550S). Total RNAs from HIV transfected Jurket cells were described previously44. Ratio of spiked viral mRNAs to human PBMC mRNAs were 1 to 1,000 based on Nanodrop reading.
Clinical specimen with unknown respiratory infection
Sputum from the lower respiratory tract (bronchi and lungs) was obtained from a patient with an unknown clinical bronchitis & pulmonary inflammation at Alaska State Public Health Laboratories. All experiments were performed in accordance with relevant guidelines and regulations and were approved by the Institution Review Board of University of Alaska Fairbanks. Informed consent was obtained from the subject. Total RNAs were extracted and purified using Qiagen RNeasy Mini Kit (Qiagen Inc., Valencia, CA) with some modification. Briefly, sputum was suspended in 500 μl of lysis buffer with 20 μl of proteinase K (20 mg/ml) and incubated for 1 hour at 56 oC. The sample was further extracted with phenol/chloroform several times until there was no white protein layer between two phases. The clean aqueous phase was then processed according to Qiagen protocol. Purified total RNA was then used to prepare a sequencing library using the PATHseq method described below.
Procedure for coupling magnetic beads with oligonucleotides
Primer P1 was specifically designed to contain T7 promoter sequence, in addition to poly d(T) sequences (P1: 5’-ACGGCCTAATACGACTCACTATAGGGTTTTTTTTTTTTTTTTTTVN-3’). When synthesized, a primary amino group was attached to the 5’-end of P1 with a standard (C6) spacer arm (Integrated DNA Technologies, Coralville, IA). Modified primer P1 was manually coupled to Pierce NHS-activated magnetic beads (Thermo Fisher Scientific Inc., Rockford, IL), according to the manufacturer’s instruction. The final concentration of P1-coupled magnetic beads was 10 mg/ml.
Poly(A) mRNA isolation and first strand cDNA synthesis with P1-Magnetic Beads
Total RNA was extracted using Qiagen’s RNase Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instruction. 5 μg of total RNA was diluted with nuclease-free water to a final volume of 50 μl in a nuclease-free 0.2 ml PCR tube. 10 μl of P1-Magnetic Beads were washed twice with 100 μl of RNA Binding Buffer (1 M LiCl, 40 mM Tris HCl, pH 7.5, 2 mM EDTA and 0.1% Triton X-100). Beads were re-suspended in 50 μl of RNA Binding Buffer and added to the 50 μl of total RNA sample. Tubes were placed on the thermal cycler and heated at 65 °C for 5 minutes and held at 4 °C to denature the RNA and facilitate binding of the poly(A) mRNA to the beads. Tubes were removed from the thermal cycler and incubated at room temperature for 5 minutes to allow the RNA to bind to the beads. Tubes were then placed on the magnetic rack at room temperature for 2 minutes to separate the poly(A) mRNA bound to the beads from the solution, supernatant was removed and discarded. Beads were washed twice with 100 μl of Wash Buffer (150 mM LiCl, 20 mM Tris HCl, pH 7.5, 1.0 mM EDTA and 0.01% Triton X-100) to remove unbound RNA, each time with all the supernatant being removed and discarded. Beads were equilibrated with 100 μl 1x reverse transcriptase (RT) buffer and separated by magnetic field. Beads bound with poly(A) mRNA were re-suspended in 12 μl nuclease-free water, 4 μl dNTP mix (10 mM each), 2 μl of 10x RT buffer, 1 μl of RNAse inhibitor and 1 μl of M-MuLV reverse transcriptase were added to the solution. Mixture was incubated at 42°C for one hour and then inactivated at 90 °C for 3 min. Magnetic beads with first strand cDNA synthesis were separated and the supernatant being removed and discarded.
PATHseq
88 octamer (Table 2) and 197 nonamer (Extended Data Table 4) oligonucleotides were synthesized using Fisher Custom Oligos service (Thermo Fisher Scientific, Waltham, MA). Oligos were suspended in nuclease-free water at final concentration of 10 μM. The set of 88 octamer oligos were used in all experiments in this report. Normal peripheral blood mononuclear cells (PBMCs) were isolated from human whole blood of a single male donor (Thermo Fisher Scientific, Waltham, MA). Total reference RNAs were extracted using Qiagen RNeasy Kit. mRNAs were isolated from total RNAs using NEBNext Poly(A) mRNA Magnetic Isolation Module according to manufacturer’s protocols (NEB). Reference cDNA library was constructed using NEB First Strand Synthesis Protocol with M-MuLV Reverse Transcriptase42. cDNAs were purified using Qiagen MinElute Reaction Cleanup Kit. To perform PATHseq method, magnetic beads with first strand cDNA synthesis were equilibrated with 100 μl 1x PATHseq buffer (40 mM Tris-HCl, pH 7.9, 20 mM MgCl2, 10 mM DTT and 2 mM spermidine) and separated by magnetic field. Supernatant was removed and discarded. Beads were re-suspended in 8.4 μl nuclease-free water, then the following solutions were added: 2 μl of 10x PATH buffer, 2 μl Octamers (10 μM), 1 μl dNTP mix (25 mM), 3.2 μl rNTP mix (25 mM), 0.4 μl Pyrophosphase (Inorganic (E. coli), 100 units/ml, New England BioLabs, Ipswich, MA), 1 μl reference human cDNAs (0.1 μg/1 μl), 1 μl M-MuLV Reverse Transcriptase (NEB) and 1 μl T7 RNA polymerase (NEB). Final volume was 20 μl. Mixture were incubated at 40 °C for 3 hours with gentle agitation. PATHseq cDNA library was purified using Qiagen MinElute Reaction Cleanup Kit.
Next generation sequencing
Next generation sequencing was performed with Illumina MiSeq Desktop Sequencer version 2 (Illumina, San Diego, CA). Sequencing library was prepared using Nextera DNA Sample Preparation Kit (Illumina) according to the product’s guide. Sequencing run was carried out using Illumina MiSeq Reagent Kit v2 (500-cylces), which generates 24–30 million paired-end reads of 2 × 250 bp length, total output of 7.5-8.5 Gb.
Sequencing data analysis
Raw sequencing data was filtered by in-house scripts: 1) Remove reads with 3 N; 2) Remove reads contaminated by adapter (default: 15 bases overlapped by reads and adapter); 3) Remove reads with a certain proportion of low quality (20) bases (40% as default, parameter setting at 36 bp); 4) Remove duplication contamination.
Using a computer program called STAR28, quality sequencing reads were aligned against the human genome primarily assembled from ENSEMBL (http://uswest.ensembl.org/index.html). Reads aligned to multiple loci in the reference human genome were also considered as unmapped reads and filtered out to reduce the false positive rate. To obtain longer non-human origin reads, unaligned reads were further assembled using a de novo assembly computer program named Trinity29, resulting in larger contig sequences. The de novo assembled unaligned sequences were blasted against the nucleotide sequence database known as NCBI “nr” database. Finally, the assembled sequences were identified using the NCBI genomic BLAST database for “Microbes” including bacteria, fungi and viruses32.
Quantitative real time PCR (qPCR)
qPCRs were performed with ABI’s StepOnePlus Real-Time PCR Systems. Reactions were set up with ABI’s SYBR Select Master Mix (Life Technologies, Carlsbad, CA) according to product’s instruction.
Accession numbers
The genome sequence data of S. pneumonia strain ASVL_JC_0001 identified in this study has been deposited at DDBJ/EMBL/GenBank under the accession number JJMK01000000 and consists of sequences JJMK01000001 - JJMK01000083.
Additional Information
How to cite this article: Ge, F. et al. Preferential Amplification of Pathogenic Sequences. Sci. Rep. 5, 11047; doi: 10.1038/srep11047 (2015).
References
Metzker, M. L. Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46, 10.1038/nrg2626 (2010).
Shendure, J. & Ji, H. Next-generation DNA sequencing. Nat Biotechnol 26, 1135–1145, 10.1038/nbt1486 (2008).
Shendure, J. & Lieberman Aiden, E. The expanding scope of DNA sequencing. Nat Biotechnol 30, 1084–1094, 10.1038/nbt.2421 (2012).
Feng, H., Shuda, M., Chang, Y. & Moore, P. S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100, 10.1126/science.1152586 (2008).
Palacios, G. et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N Engl J Med 358, 991–998, 10.1056/NEJMoa073785 (2008).
Radford, A. D. et al. Application of next-generation sequencing technologies in virology. J Gen Virol 93, 1853–1868, 10.1099/vir.0.043182-0 (2012).
Barzon, L. et al. Next-generation sequencing technologies in diagnostic virology. J Clin Virol 58, 346–350, 10.1016/j.jcv.2013.03.003 (2013).
Chiu, C. Y. Viral pathogen discovery. Curr Opin Microbiol 16, 468–478, 10.1016/j.mib.2013.05.001 (2013).
Firth, C. & Lipkin, W. I. The genomics of emerging pathogens. Annu Rev Genomics Hum Genet 14, 281–300, 10.1146/annurev-genom-091212-153446 (2013).
Quinones-Mateu, M. E., Avila, S., Reyes-Teran, G. & Martinez, M. A. Deep sequencing: Becoming a critical tool in clinical virology. J Clin Virol 61, 9–19, 10.1016/j.jcv.2014.06.013 (2014).
Hoffmann, B. et al. Novel orthobunyavirus in Cattle, Europe, 2011. Emerg Infect Dis 18, 469–472, 10.3201/eid1803.111905 (2012).
Rosseel, T. et al. DNase SISPA-next generation sequencing confirms Schmallenberg virus in Belgian field samples and identifies genetic variation in Europe. PLoS One 7, e41967, 10.1371/journal.pone.0041967 (2012).
McMullan, L. K. et al. A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med 367, 834–841, 10.1056/NEJMoa1203378 (2012).
Yozwiak, N. L. et al. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl Trop Dis 6, e1485, 10.1371/journal.pntd.0001485 (2012).
Negredo, A. et al. Discovery of an ebolavirus-like filovirus in europe. PLoS Pathog 7, e1002304, 10.1371/journal.ppat.1002304 (2011).
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108, 10.1038/nature11233 (2012).
Consortium, E. P. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, 10.1038/nature11247 (2012).
Gonzalez-Porta, M., Frankish, A., Rung, J., Harrow, J. & Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol 14, R70, 10.1186/gb-2013-14-7-r70 (2013).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, 10.1038/35057062 (2001).
Feng, H. et al. Human transcriptome subtraction by using short sequence tags to search for tumor viruses in conjunctival carcinoma. J Virol 81, 11332–11340 (2007).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, 10.1093/bioinformatics/btr011 (2011).
Rizk, G., Lavenier, D. & Chikhi, R. DSK: k-mer counting with very low memory usage. Bioinformatics 29, 652–653, 10.1093/bioinformatics/btt020 (2013).
Weber, G., Shendure, J., Tanenbaum, D. M., Church, G. M. & Meyerson, M. Identification of foreign gene sequences by transcript filtering against the human genome. Nat Genet 30, 141–142 (2002).
Xu, Y. et al. Pathogen discovery from human tissue by sequence-based computational subtraction. Genomics 81, 329–335 (2003).
Huang, X. & Madan, A. CAP3: A DNA sequence assembly program. Genome Res 9, 868–877 (1999).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864, 10.1093/bioinformatics/btr026 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, 10.1093/bioinformatics/bts635 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, 10.1038/nbt.1883 (2011).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J Comput Biol 7, 203–214, 10.1089/10665270050081478 (2000).
Morgulis, A. et al. Database indexing for production MegaBLAST searches. Bioinformatics 24, 1757–1764, 10.1093/bioinformatics/btn322 (2008).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 38, D5–16, 10.1093/nar/gkp967 (2010).
Choi, S. C. et al. Draft Genome Sequence of an Atypical Strain of Streptococcus pneumoniae Isolated from a Respiratory Infection. Genome Announc 2, 10.1128/genomeA.00822-14 (2014).
Angiuoli, S. V. et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 12, 137–141, 10.1089/omi.2008.0017 (2008).
Chen, Z. & Duan, X. Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications. Methods Mol Biol 733, 93–103, 10.1007/978-1-61779-089-8_7 (2011).
Cloonan, N. et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5, 613–619, 10.1038/nmeth.1223 (2008).
Ruan, Y., Le Ber, P., Ng, H. H. & Liu, E. T. Interrogating the transcriptome. Trends Biotechnol 22, 23–30 (2004).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6, 377–382, 10.1038/nmeth.1315 (2009).
Bent, Z. W. et al. Enriching pathogen transcripts from infected samples: a capture-based approach to enhanced host-pathogen RNA sequencing. Anal Biochem 438, 90–96, 10.1016/j.ab.2013.03.008 (2013).
Feehery, G. R. et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One 8, e76096, 10.1371/journal.pone.0076096 (2013).
Chen, J., Ye, F., Xie, J., Kuhne, K. & Gao, S. J. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386, 290–302, 10.1016/j.virol.2009.01.031 (2009).
Chen, J. Serial analysis of binding elements for human transcription factors. Nat Protoc 1, 1481–1493 (2006).
Inagi, R. et al. Identification and characterization of human herpesvirus 8 open reading frame K9 viral interferon regulatory factor by a monoclonal antibody. J Hum Virol 2, 63–71 (1999).
Chen, J., Malcolm, T., Estable, M. C., Roeder, R. G. & Sadowski, I. TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. J Virol 79, 4396–4406 (2005).
Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Numbers P20GM103395 through Alaska INBRE and U54GM104944 through Mountain West Clinical and Translational Research Infrastructure Network (CTR-IN) Pilot Grant Program and by an equipment grant of Alaska State Public Health Laboratories. The clinical sample was collected and diagnosed at the Alaska State Public Health Laboratories. We thank staff at Alaska State Public Health Virology Laboratory for their technical assistance.
Author information
Authors and Affiliations
Contributions
J.C. designed the study, performed experiments, analyzed the data and wrote the paper. F.G., J.P. performed experiments and analyzed data. K.R. and B.J. provided clinical diagnosis. S.C.C. and M.L. performed bioinformatics analysis.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ge, F., Parker, J., Chul Choi, S. et al. Preferential Amplification of Pathogenic Sequences. Sci Rep 5, 11047 (2015). https://doi.org/10.1038/srep11047
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11047
This article is cited by
-
Investigation of a Canine Parvovirus Outbreak using Next Generation Sequencing
Scientific Reports (2017)
-
Enrichment of Viral Nucleic Acids by Solution Hybrid Selection with Genus Specific Oligonucleotides
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
