
Abstract
CD133, a 120 KDa glycoprotein is a transmembrane glycoprotein which has been recently used as a cancer stem cell (CSCs) marker in a variety of carcinomas. CD133+ cells possess strong tumorigenicity, responsible for tumor initiation and maintenance. Therefore, the goal of our study was to develop a novel CD133 humanized antibody as a promising target for cancer therapy. CD133 purified proteins were used for panning the naive human-semi-synthetic Tomlinson I + J phagemid library. The second extracellular domain (loop1) and the third extracellular domain (loop2) of CD133 were expressed in E. coli. In this study, we adopted a novel five-round selection strategy based on moderate stringent selection during the first rounds. This unique strategy was aimed at avoiding the loss of rare phages with high affinity to target proteins. After the five rounds of specific panning, six phage-antibody clones which specifically recognized recombinant human CD133 protein were obtained. The desirable phage clone named CD133-scFv-1 was cloned into the expression vector, then induced and purified. We show that CD133-scFv-1 and commercial murine antibody 293C3 could compete with each other in the indirect competitive immunoassay. Our work may lay the groundwork for future studies involving biological functions and applications of the CD133 humanized antibody.
Similar content being viewed by others


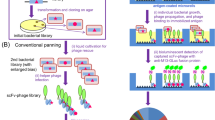
Introduction
Emerging evidence has demonstrated that malignant tumors are a frequently-occurring disease that represents a threat to human health. Surgery in combination with chemotherapy and radiotherapy constitute the major treatments used against tumorigenesis by medical researchers. Despite the current available tools for handling of the tumors, patients frequently manifest side effects, tumor recurrence and metastasis. According to the cancer stem cells (CSCs) model of tumorigenesis, a small subpopulation of tumor cells, the CSCs, possess the exclusive capabilities of self-renewal and self-maintenance1. These CSCs have been reported to be responsible for tumor initiation, metastasis and resistance to current therapy and other mechanisms related to tumor expansion or maintenance.
As the first identified member of the prominin family, CD133 is a penta-transmembrane glycoprotein2. To date, functions and specific ligands of the prominin family proteins are still not well-understood. A compelling number of studies has shown that CD133 is a marker for CSCs in a wide variety of tumors3, including pancreatic adenocarcinoma4,5,6, colon carcinoma5,7,8,9, hepatocellular carcinoma10,11,12 and neural tumors13. Thus, this molecule may serve as a promising novel target in cancer therapy. Recent studies using murine monoclonal CD133 antibodies have shown that these antibodies possess tumor quantitative and non-invasive in vivo imaging properties7,14, making them strong candidates for future cancer therapy. Therefore, we were determined to develop humanized antibodies against CD133.
Amongst the most commonly used methods for generating monoclonal antibodies of human origin phage display is widely accepted15,16. Such an in vitro selection technology provides several advantages over conventional hybridoma methods, including high efficiency and a broader spectrum of antigens. Phage single–chain antibody fragment (scFv) libraries are proven powerful tools for researchers who aim at obtaining tumor specific antibodies17,18,19.
Here, we describe the development of recombinant humanized antibodies against CD133 - a cell marker for CSCs in a wide variety of tumors - using the scFv Tomlinson I + J libraries, surpassing the need for coupling the antigen to a protein for animal immunization. Furthermore, we developed a competitive ELISA (cELISA), which allows a rapid, simple and economical quantification of recombinant CD133. Our present data contribute to the further study of biological functions and applications of the CD133 humanized antibody.
Results
Expression of purified recombinant CD133-Hexahistidine fusion protein
After induction by 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), the E. coli BL21 containing plasmid pGEX-4T-1-CD133-loop 1-Hexahistidine or pGEX-4T-CD133-loop 2-Hexahistidine was able to express the desirable amount of recombinant protein. These 55 KDa CD133-loop 1-Hexahistidine plus pGEX-4T-1-CD133-loop2-Hexahistidine were then purified with Ni-NTA sepharose. As shown in Figure 1, these two purified recombinant protein exhibited little amounts of unwanted bands in the sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Thus, our results clearly show the successful expression of purified recombinant CD133-Hexahistidine fusion protein.

Purified recombinant protein CD133-loop1-Hexahistidine/CD133-loop2-Hexahistidine).
The 12% SDS-PAGE gel shows on lane 1, the purified CD133-loop1-Hexahistidine fusion protein, whereas lane 2 is the purified CD133-loop2-Hexahistidine fusion protein. Molecular weight markers are indicated at the left margin.
Isolation of scFv phages against recombinant CD133
Biopanning against the recombinant CD133 was performed using the Tomlinson I and J libraries. 100 μl of antigens with a concentration ranging from 50 μg/mL to 5 μg/mL were added to the 96-well flat-bottom microtitre plates for each round of selection. After five rounds of panning, the recovery of phages through successive rounds of selection is depicted in Table I.
As expected, the yield of CD133 specific phages in the first round of selection was very low, with 0.575 × 104 cfu/mL recovered by panning. Amplification of these viruses and their subsequent reselection resulted in significantly increases in recoveries (by more than 1000 times). For the first three rounds, eluted scFv phages increased more than 105 times after amplification in XL1Blue E. Coli. These results demonstrated that viral particles eluted with trypsin maintained their infectivity properties. This was taken as a preliminary evidence of the effective enrichment of phages with the capacity to bind to recombinant CD133.
Specificity of scFv phages against recombinant CD133
The specificity of polyclonal phages after each round of panning was verified by enzyme linked immunosorbent assay (ELISA) using recombinant CD133 in microtitre plates. Approximately 1.0 × 1012 phage particles were added to each well, where coated with 10 μg/mL plus 100 μl per well overnight under 4 C and then washed 3 times following the ELISA protocol. The polyclonal ELISA was carried out 3 times (Figure 2). ScFv phages from the unselected library and helper phage VCSM13 elicited weak signals in the experiments. However, after 4 rounds of panning, phages from the fourth or fifth round showed significantly positive signals against CD133 - even diluted 45 times (Figure 2). Altogether these data suggest that the affinities of phages were increased round by round, demonstrating that the panning strategy is really effective.

Binding of selected phage-scFv antibodies from 5 rounds panning to recombinant CD133 by polyclonal phage ELISA.
Polyclonal phages were rescued at each round of panning against CD133-loop1 and CD133-loop2. Culture supernatants containing approximately 1012 phage particles were analyzed using colony forming unit per milliliter (cfu/mL). Absorbance values are represented as the mean ± standard deviation (SD) for three independent determinations. Error bars show the standard deviation for each set of data. The background binding (negative control) values of the individual phage-bound scFv to the support solution (PBS) were subtracted from the total binding values, thus these data represent specific binding. Due to the fact that negative control values differed for each phage clone, they are not indicated on the figure.
Specificity of individual scFv-phage clones measured by ELISA
101 individual phage colonies were picked at random from plates used for the titration of fifth round viral recoveries. The results of monoclonal phage-ELISA revealed that 87 clones showing positive binding to CD133 (absorbance against CD133/absorbance against negative control >2.1), as indicated in Figure 3A + B. Only 6 of these clones, designated as 1, 4, 5, 10, 14 and 18, showed a higher binding ability to the antigen (absorbance against CD133/absorbance against negative control >8.0). Therefore, these 6 clones were selected for further analysis. These data demonstrated that various phages from the fifth round exhibited different antigen binding abilities and most of phages (86.1%) possessed CD133-specificity.

Binding of 101 selected phage-scFv antibodies to recombinant CD133 by monoclonal phage ELISA.
(A + B) 101 individual clones' specificities to recombinant CD133 are shown. Representation of the absorbance (OD450 nm) using the horseradish peroxidase-conjugated detection system (HRP-mouse-anti-M13, purchased from Pharmacia) for the detection of the binding of phage-expressed scFv to recombinant CD133. Absorbance values are represented as the mean ± SD for three independent determinations. The background binding (negative control) values of the individual phage-bound scFv to the solution (PBS) were subtracted from the total binding values, thus these data represent specific binding. Due to the fact that negative control values differed for each phage clone, they are not indicated on the figure.
Identification of antigen-specific scFv clone by DNA sequencing
We performed PCR amplification (using pIT2-vector specific primers) of the scFv genes which yielded products of the desired ≈900 bp size for all the clones. However, amongst the selected S16-binding clones (4, 5, 10, 14 and 18), all contained amber (TAG) stop codons, except clone number 1, the CD133-scFv-1 clone (Figure 3).
We blasted our data with the NCBI database which confirmed that the nucleotide sequences were human VH and VL sequences. The complete nucleotide sequences of the heavy and light chain variable region were determined for CD133-scFv-1 (Figure 4). These results indicated that the coding sequences shared an identical nucleotide sequence giving a VH fragment of 118 amino acids and a VL fragment of 100 amino acids residues. Hence, CD133-scFv-1 phages were further designated into express vector pET22b.

Binding of 6 selected phage-scFv antibodies to recombinant CD133 by monoclonal phage ELISA.
Reanalysis of 6 selected clones (6 strongly positive clones from previous 101 clones). Phage-infected E. coli XL1Blue culture supernatants containing approximately 1012 phage particles per milliliter were analyzed in this assay. Absorbance values are the means of three independent determinations.
Expression of soluble CD133-scFv-1
In our study, we obtained the CD133-scFv-1 specific for CD133. However, the amounts of scFv-pIII fusion protein expressed by vector PIT2 plus HB2151 E. coli were not sufficient for further analysis. Hence, CD133-scFv-1 phages were further designated into express vector pET22b, which possessed a hexahistidine tag and infected E. coli BL21 (DE3) in order to induce the expression of suitable scFv protein. As shown in Figure 5, the purification of bacterial lysis showed CD133-scFv-1, with the size of approximately 31 KDa (Equation 1). Moreover, the results using SDS-PAGE demonstrated that the new recombined express vector allowed us to efficiently differentiate CD133-scFv-1 from other unwanted proteins.

Induced protein expression of recombinant plasmids and the purification of scFv-1.
Lane 1: inclusion body of BL21 (DE3)/pET22b; Lane 2: periplasm of IPTG induced BL21 (DE3)/pET22b-scFv1; Lane 3: purified CD133-scFv-1. Molecular weight markers are indicated at the left margin.
Development of a competitive ELISA for the quantification of recombinant CD133
After comparison with the results obtained with the indirect competitive immunoassay (data not shown), we calculated that the optimal dilution of 293C3 for use in the competitive ELISA (cELISA) is 1:60 (0.833 μg/L) and CD133-scFv-1 is 1:10 (6.1 μg/mL). The results obtained yielded a useful dynamic range and well analytical sensitivity of the assay.
The inhibition of the binding of 293C3 to CD133 antigen was measured in order to evaluate the usefulness of CD133-scFv-1. The indirect competitive immunoassay of 293C3 blocking and CD133-scFv-1 binding data is summarized in Figure 6. In these cELISA, we detected the murine binding by HRP-goat-anti-mouse antibody, at an absorbance of 450 (OD450) nm. According to Equation 2, the inhibition was among 0.001 to 69.65%. As figure 6 shows, more inhibition was observed due to the greater weight ratio of CD133-scFv-1 to 293C3. From these data, we could reason that CD133-scFv-1 effectively competes with commercial murine monoclonal antibody 293C3.

Competitive ELISA of murine monoclonal antibody and purified human scFv (Binding at OD450).
(A) Control 1: 50 μl 293C3 and 50 μl 3% BSA-PBS (uninhibited control); Control 2: only antigen and secondary antibody (HRP-goat-anti-mouse antibody). (B) Percentage of inhibition (%I) against the different weight ratio.
Moreover, Figure 7 shows inhibition of the binding of CD133-scFv-1 to CD133 - measured in order to compare the binding mode of CD133-scFv-1 with murine antibody 293C3. Since scFvs isolated from Tomlinson I + J libraries show significant protein A affinity, we applied HRP-protein A as the secondary antibody in these assays to detect the CD133-scFv-1 binding. Thus, we could evaluate the inhibition of the murine antibody by the absorbance of OD450 nm. Inhibition ranged from 30.34% to 99.9% due to the different weight ratio. When one compares the results of the samples with the mixed first antibody (293C3 and CD133-scFv-1) with the samples without 293C3, it is remarkable the bigger weight ratio of the murine antibody to CD133-scFv-1 and thus the weakening in the binding CD133-scFv-1 to the antigen. Altogether we can conclude that CD133-scFv-1 and 293C3 could compete with each other in cELISA.

Competitive ELISA of purified human scFv and murine monoclonal antibody: Binding of CD133-scFv-1 binding at OD450.
(A) Control 1: 50 μl murine monoclonal antibody (0.833 μg/mL) and 50 μl 3% BSA-PBS (uninhibited control); Control 2: only antigen and secondary antibody (HRP-protein A). (B) Percentage of inhibition (%I) against the different weight ratio.
Specificity of CD133-scFv-1
To assess the specificity of our clone CD133-scFv-1, we investigated the specificity of CD133-scFv-1 in the hepatoma cell line PLC/PRF/5 with two different experimental approaches. In summary, cells were infected with a lentivirus expressing an shRNA targeting CD133 or control shRNA, after which we performed immunoblotting analyses (Figure 8A) or flow cytometry analysis (Figure 8B). Both techniques confirmed that, CD133-scFv-1 could be employed as the detector to reflect the variation of the levels of cell CD133 surface protein (Figure 8A + B). Altogether, these data suggest that CD133-scFv-1 is able to bind natural CD133 protein on cell surface.

Immunoblotting and flow cytometry analysis confirms the specificity of CD133-scFv-1.
(A) The proteins of PLC/PRF/5 cells (transfected with CD133 shRNA or control shRNA) were subjected to immunoblot analysis using anti-CD133-scFv-1 (1:50) or anti-β-actin (1:5000). (B) To determine changes in CD133-scFv-1 cell binding affinity, transfected PLC/PRF/5 cells were stained with anti-CD133-scFv-1/PE and cells were sorted using a flow cytometer. Ten thousand cells were sorted per experiment (n = 3).
Discussion
The present data is the first description of soluble scFv recombinant antibody of human origin specific against CD133, a surface protein involved in tumor initiation, maintenance and metastasis2,20.
Since the Tomlinson I + J library is constructed on the basis of human antibody genes, the recombinant antibodies obtained are potentially involved in the human-therapeutic application without non-human antibody reactions, such as HAMA reactions in the treatment of patients with tumors21. Therefore, the key advantages of using this library over other technologies during screening for recombinant human antibodies seem obvious.
In order to isolate human scFv targeting extracellular loops, we coated gradually fewer quantities of recombinant CD133-loop1/CD133-loop2 on the 96-well flat-bottom microtitre plates. According to the screening principle, the less antigen results in the more potential to get antibodies with high affinities. Thus we adopted a novel and unique 5-round screening strategy. To avoid the loss of specific phages, during the first round of panning, antigens were relatively abundant and unspecific phages were removed non-stringently. After amplification with XL1Blue E. Coli, there was fewer amount of antigen together with more stringent washing in the following rounds of screening. In our study, we found that VCSM13 helper phages in the negative control well were not completely depleted until the washing procedure was repeated 5 times. The results of polyclonal ELISA demonstrated that, eluted phages in the first 3 rounds, containing many unspecific and helper phages cannot represent effective enrichments of scFv-phages. Besides, the results using the ELISA and phage titers of each round suggest that polyclonal phages possess increasing specificity to the recombinant CD133, confirming the enrichments of the phages of interest. At the end of the fifth round of panning, 101 monoclonal phage were obtained, of which 87 clones (86.1%) show specificity towards CD133-loop1 or CD133-loop2 (P/N > 2.1). Six strongly positive phages were picked for further study.
However, 2 of the selected scFv antibodies selected elicited amber stop codons in conjunction with 3 other phages which had shift mutations or deletions of VL. The presence of the amber stop codons in the clones required the use of the E. coli TG1 or XL1Blue suppressor strain to express these scFvs in soluble form as a fusion to gIIIp. Only phage number 1, named CD133-scFv-1 could be induced by soluble scFv. Theoretically, the combination of suppressor strain HB2151 and plasmid pIT2 could produce abundant soluble scFv22. Unfortunately, in the present study, the quantities ant the extremely elaborate purification of desirable scFv was rather discouraging. Therefore, the sequence of CD133-scFv-1 was cloned into another express vector pET22b and transformed in BL21 (DE3).
The combination of BL21 (DE3) and pET22b not only produced a significant increase of amounts of CD133-scFv-1 but also avoided the scFv-gIIIp fusion protein from desired scFv. Since the original induced CD133-scFv-1 and scFv-gIIIp fusion proteins shared the same hexahistidine tag and only excluding phage components can help to better understand physico-chemical or biological properties of scFv.
Analysis of reducing SDS-PAGE suggests that the majority of soluble CD133-scFv-1 was present in the bacterial inclusion bodies. Different concentration of IPTG (ranging from 0.5 mM to 2.0 mM) resulted in obsolete various amount of desirable scFv. Even in the absence of IPTG, transformed E. coli BL21(DE3) could express some scFv. In the scFv ELISA, CD133-scFv-1 exhibited specificity to CD133. Besides, in the indirect competitive immunoassay, CD133-scFv-1 and commercial murine monoclonal antibody 293C3 could compete with each other in binding with recombinant CD133 antigen. Furthermore, we have demonstrated by immunoblotting and flow cytometry analysis that CD133-scFv-1 is highly specific to CD133 protein.
Thus, we could reasonable come to the conclusion that epitopes recognized by those two antibodies maybe partly overlapped or located within the scope of steric hindrance. Although high affinities to targets may exhibited by scFv fragments23, several reports found that scFv affinities lower than their corresponding full-length IgG formats24,25,26. Some directed-evolutionary methods, such as error-prone PCR and affinity maturation could be applied to further improve the affinity of CD133-scFv-1.
CD133-scFv-1 VH and VL amino acid sequence were blasted to the respective sequences by NCBI (National Center for Biotechnology Information) online BLAST (http://www.ncbi.nlm.nih.gov/) and identified (Table II). The result indicated that the coding sequences of CD133-scFv-1 belongs to human nucleotide sequence giving a VH fragment of 118 amino acid residues and a VL fragment of 100 amino acid residues.
In the present study, we isolated and obtained a soluble scFv targeting CD133. This human CD133-scFv-1 could compete with commercial antibody 293C3 in recognition and binding CD133 antigen. This study may represent an important step in the detection and characterization of CD133+ CSCs in clinical samples and further utilized in novel cancer therapies. For example, CD133-scFv-1 can be conjugated to drugs, cytotoxins, suitable radionucleotides or functional nanoparticles, to achieve the desirable specific cell subpopulation killing functions27. In the future, these coupled agents could perform the elimination of tumor initiating cells contributing to the improvement of therapeutic effects such as suppression of recurrence.
Methods
General materials
pGEX-4T-1 with a hexahistidine tag at the C-terminus was used from our library. Ni-NTA sepharose was purchased from Qiagen (Shangai, China). Mouse anti-human CD133 monoclonal antibody (293C3) was purchased from Miltenyi Biotec (Bergisch Glabdach, Germany).
Libraries, helper phage and bacterial strains
VCSM13 helper phage and Escherichia coli (E. coli) XL1Blue were all kindly provided by Dr. Zhu (Key Laboratory of Antibody Technique of Ministry of Health, NJMU, Nanjing, Jiangsu, China). Express vector pET22b and Escherichia coli (E. coli) BL21 (DE3) were all kindly provided by Dr. Zhang (School of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu, China). The human single-fold scFv Tomlinson I + J libraries (I.M.Tomlinson, G. Winter, http://www.geneservice.co.uk/products/proteomic/datasheets/tomlinsonIJ.pdf, KM13 helper phage and Escherichia coli (E. coli) TG1-Tr and HB2151 were purchased from Centre for Protein Engineering, MRC Laboratories, Cambridge, United Kingdom). Both libraries are based on a single human framework for VH (V3-23/DP-47 and JH4b) and Vκ (O12/O2/DPK9 and Jκ1) which encodes the most common human canonical structure. The size of the Tomlinson I library is 1.47 × 108 phagemid clones in E. coli TG1 cells, while that of the Tomlinson J library is 1.37 × 108. The libraries have a high proportion of functional antibodies, with 96% (I) and 88% (J) of clones containing inserts. The antibodies are displayed as scFv fragments on the minor coat protein pIII of filamentous phage, cloned in an ampicillin resistant phagemid vector (pIT2). The phagemid clones are maintained and propagated in T-phage resistant E. coli TG1-Tr. Antibody fragments were cloned into a vector pET22b, which downstream to the scFv genes, contains sequences for a hexahistidine tail, which are useful for the detection of scFv fragments with anti-his tag antibodies and for their purification by nickel chelate affinity chromatography. Furthermore, this vector can be strongly inducible with isopropyl L-D-thiogalactosidase (IPTG).
Purification of recombinant CD133-Hexahistidine fusion protein
The nucleotide sequences of two extracellular loops (178aa–434aa and 507aa–793aa) within human CD133 were cloned into pGEX-4T-1 which contains a hexahistidine tag at the C-terminus. After the analysis of the sequence, E. coli BL21 transformed with the recombinant plasmid were induced with 0.1 mM IPTG. Then bacterial lysis was purified with Ni-NTA sepharose. Furthermore, purified recombinant CD133-Hexahistidine fusion protein was then analyzed using 10% SDS-PAGE.
Preparation of phage antibodies for panning
A total of 500 mL of the glycerol stock of the I and J libraries were inoculated into 200 mL of 2 × TY broth containing28 1% glucose and 100 μg/mL ampicillin and incubated with shaking at 37 C to an optical density at 600 nm (OD600) of 0.4–0.6 (1–2 h). VCSM13 helper phage (2 × 1011 particles) was added to 50 mL culture and the mixture was incubated in a 37 C water bath for 30 minutes. Infected cells were pelleted, resuspended in 100 mL 2× TY broth containing 0.1% glucose, 100 mg/mL ampicillin and 50 mg/mL kanamycin and incubated overnight at 30 C. Phage particles were concentrated from each culture supernatant by precipitation with 20 mL polyethylene glycol (PEG) 6000 in 2.5 M NaCl (20% w/v) and stored at −70 C with 15% glycerol, as described by the MRC protocol.
Polyclonal and monoclonal scFv-phage ELISA
Both polyclonal and monoclonal scFv-phage ELISAs were also performed according to the producer's protocol. Briefly, after the fifth round of panning, individual clones were grown in 3 mL SB containing 100 mg/mL ampicillin, 50 mg/mL kanamycin and 30 mg/mL tetracycline. Then phages were produced by addition of VCSM13 helper phages. Recombinant CD133-loop1 and CD133-loop2 were used to coat 96-well ELISA plates for competitive immunoassays, respectively. Recombinant CD133 was diluted to a final concentration of 10 μg/mL in 0.01 M phosphate-buffered saline pH 7.2 (PBS). One hundred microliters of the solution was used for coating each well of the ELISA plates (Maxisorp; Nunc, Roskilde, Denmark) at 4 C overnight. The plates were thereafter washed three times with 350 μl of washing solution (PBS). The plates were blocked at 37 C for 2 h with 200 μl of 3% (w/v) albumin from bovine serum (BSA) in PBS and then washed 3 times. Next, purified CD133-scFv-1 50 μl/well diluted 1:10 in 3% (w/v) BSA-PBS was added. Subsequently, triplicates of 100 μl phage dilutions were added to the sample and the plate was incubated at 37 C for 2 h. After removal of unbound material (3 washes with 0.05% Tween 20-PBS), 100 μl peroxidase-conjugated murine anti-M13 (Pharmacia, USA) diluted 1:3000 in PBS-T was added to each well and incubated for 1 h at 37 C. After 3 repeated PBS-T washes, the amount of bound peroxidase was determined using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate and the plates were incubated in absence of light at room temperature. The enzyme-substrate reaction was stopped by adding 50 μl 1 M sulfuric acid to each well. Background binding of the soluble scFv to the ELISA plate was determined by using antigen-free wells and applying the phage ELISA. The optical density (OD450) was measured at 450 nm using a microtitre plate reader (BIO-Rad, Hercules, California, USA).
Expression of soluble CD133-scFv-1
Isolated pIT2 phagemids were sequenced using the pHEN seq (5′-CTA TGC GGC CCC ATT CA-3′) and LMB3 (5′-CAG GAA ACA GCT ATG AC-3′) sequence primers and clones with desirable sequences were picked out then cloned into another vector pET22b. E. coli BL21(DE3) were transformed the new recombinant plasmid and induced with 1 mM IPTG. The bacterial lysis was purified with Ni-NTA sepharose and analyzed by 12% SDS-PAGE.
Indirect competitive immunoassay (ELISA)
Indirect competitive immunoassay was performed exactly as the protocol of polyclonal or monoclonal ELISA previously described. After antigens were coated, plates underwent blocking and washing. Same quantities (dilution 1:60 in 3% (w/v) BSA-PBS) of murine antibody 293C3 50 μl/well were added. Then 50 μl CD133-scFv-1 was added to the sample (ranging from 6.1 μg/mL to 0.381 μg/mL). We used as non-inhibited control only 3% BSA-PBS. The weight ratio of scFv to murine antibody varied from 0.457 to 7.323, all of which was performed in multiple two-fold dilutions. After removal of unbound material (5 washes with 0.05% PBS-T), 100 μl peroxidase-conjugated anti-mouse immunoglobulin (Sigma, USA) diluted 1:3000 in PBS-T was added to each well and incubated for 1 h at 37 C. After 5 repeated PBS-T washes, the amount of bound peroxidase was determined with TMB substrate and the plates were incubated in absence of light at room temperature. The enzyme-substrate reaction was stopped by addition of 50 μl sulfuric acid 1 M to each well. The optical density (OD450) was measured at 450 nm using a microtitre plate reader (BIO-Rad).
Next, the purified CD133-scFv-1 50 μl/well diluted 1:10 in 3% (w/v) BSA-PBS was added. Subsequently, triplicates of 50 μl 293C3 added sample (ranging from 0.833 μg/mL to 0.052 μg/mL, with non-inhibited control added only 3% BSA-PBS) all of which were performed in multiple two-fold dilutions. These were added and incubation followed at 37 C for 2 h. The results obtained for each assay were expressed as percentage of inhibition (%I), which was calculated according to the Equation 1:

When evaluating the blocking of murine 293C3 by CD133-scFv-1, we set almost all parameters identical to the competitive immunoassay described above, with the first antibody mixture consisted of 293C3 50 μl/well diluted 1:60 in 3% (w/v) BSA-PBS plus triplicates of 50 μl CD133-scFv-1 (ranging from 6.1 μg/mL to 0.381 μg/mL and non-inhibited control added only 3% BSA-PBS, while weight ratio of murine antibody to scFv varied from 0.008 to 0.137) all of which we performed in multiple two-fold dilutions. Then we adjusted peroxidase-conjugated protein A (1:3000 diluted, Sigma-Aldrich, St. Louis, Missouri, USA) as the secondary antibody. The results obtained for each assay were expressed as percentage of inhibition (%I), calculated according to the Equation 2:

Hepatoma cell line and cell culture
PLC/PRF/5 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich, St Louis, MO) containing 10% heat-inactivated fetal bovine serum (FBS) (Hyclone, Logan, UT) and supplemented with 100 IU/mL penicillin G and 100 μg/mL streptomycin (Sigma-Aldrich). The cell line was incubated at 37 C in a humidified atmosphere containing 5% CO2.
Construction of Lentiviral Vector for Silencing of PLC/PRF/5 CD133
Small interfering RNAs (siRNAs) targeting the human CD133 shRNA oligonucleotide sequences (CD133#1 5′-GGAUACACCCUACUUACUAAA-3′, CD133#2 5′-GCACUCUAUACCAAAGCGUCA-3′) were generated using stable lentivirus expression vectors by the Shanghai GeneChem. The RNAi sequences against human CD133 were cloned into the pGCL–GFP (GenScript). The sequence of lentiviral vector containing human CD133 shRNA was confirmed by PCR and sequencing analyses. A negative control lentiviral vector containing NS shRNA was also constructed by a similar process (NS lentivirus, 5′-CGTACGCGGAATACTTCGA-3′). PLC/PRF/5 cells were plated in 6-well plates (3 × 104 cells/well) and cultured overnight. On the day of the experiment, cells were infected with CD133 shRNA lentivirus or NS lentivirus. The medium was changed with fresh medium the next day. Infection efficiencies were determined by GFP fluorescence. 72 hours later, cells were harvested for protein extraction or flow cytometry.
Immunoblotting
The PLC/PRF/5 cells were lysed in RIPA buffer, then proteins were fractionated by SDS-PAGE and transferred to a PVDF membrane (Immobilon-P, Millipore). The membrane was subjected to immunoblot analysis using anti-CD133, anti-β-actin or CD133-scFv-1 at 4°C overnight. Then we employed HRP-conjugated protein A or HRP-goat-anti mouse as secondary antibodies. Visualization was performed using the TANON-5200 system (Bio-tanon, Shanghai).
Flow cytometry
Cells were stained with CD133-scFv-1 at 4 C or 30 min. PE-protein A employed as a secondary antibody. Stained cells were analyzed on a FACsCalibur flow cytometer using Cell Quest software (BD Biosciences, Mountain View, CA).
References
Clarke, M. F. et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66, 9339–9344 (2006).
Miraglia, S. et al. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization and molecular cloning. Blood 90, 5013–5021 (1997).
Ma, S. & Guan, X. Y. MiRegulators in cancer stem cells of solid tumors. Cell Cycle 10, 571–572 (2011).
Kim, M. P. et al. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One 6, e20636 (2011).
Hermann, P. C. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323 (2007).
Marhaba, R., Klingbeil, P., Nuebel, T., Nazarenko, I. & Buechler, M. W. et al. CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med 8, 784–804 (2008).
Tsurumi, C. et al. Non-invasive in vivo imaging of tumor-associated CD133/prominin. PLoS One 5, e15605 (2010).
Vermeulen, L. et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A 105, 13427–13432 (2008).
Schneider, M., Huber, J., Hadaschik, B., Siegers, G. M., Fiebig, H. H. et al. Characterization of colon cancer cells: a functional approach characterizing CD133 as a potential stem cell marker. BMC Cancer 12, 96 (2012).
Hagiwara, S. et al. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer 106, 1997–2003 (2012).
Ma, S. Biology and clinical implications of CD133(+) liver cancer stem cells. Exp Cell Res 319, 126–132 (2013).
Colombo, F. et al. Evidence of distinct tumour-propagating cell populations with different properties in primary human hepatocellular carcinoma. PLoS One 6, e21369 (2011).
Hemmati, H. D. et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A 100, 15178–15183 (2003).
Jin, Z. H. et al. Basic studies on radioimmunotargeting of CD133-positive HCT116 cancer stem cells. Mol Imaging 11, 445–450 (2012).
Dubel, S., Stoevesandt, O., Taussig, M. J. & Hust, M. Generating recombinant antibodies to the complete human proteome. Trends Biotechnol 28, 333–339 (2010).
Thie, H., Meyer, T., Schirrmann, T., Hust, M. & Dubel, S. Phage display derived therapeutic antibodies. Curr Pharm Biotechnol 9, 439–446 (2008).
Thie, H. et al. Rise and fall of an anti-MUC1 specific antibody. PLoS One 6, e15921 (2011).
Perez-Martinez, D., Tanaka, T. & Rabbitts, T. H. Intracellular antibodies and cancer: new technologies offer therapeutic opportunities. Bioessays 32, 589–598 (2010).
Dobson, C. L. et al. Human monomeric antibody fragments to TRAIL-R1 and TRAIL-R2 that display potent in vitro agonism. MAbs 1, 552–562 (2009).
Yin, A. H. et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90, 5002–5012 (1997).
Gelderman, K. A., Tomlinson, S., Ross, G. D. & Gorter, A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol 25, 158–164 (2004).
Tomlinson, I. & Holliger, P. Methods for generating multivalent and bispecific antibody fragments. Methods Enzymol 326, 461–479 (2000).
Griffiths, A. D. & Duncan, A. R. Strategies for selection of antibodies by phage display. Curr Opin Biotechnol 9, 102–108 (1998).
Schaefer, J. V. & Pluckthun, A. Transfer of engineered biophysical properties between different antibody formats and expression systems. Protein Eng Des Sel 25, 485–506 (2012).
Zhu, X. et al. Identification of internalizing human single-chain antibodies targeting brain tumor sphere cells. Mol Cancer Ther 9, 2131–2141 (2010).
Renaut, L. et al. Affinity maturation of antibodies: optimized methods to generate high-quality ScFv libraries and isolate IgG candidates by high-throughput screening. Methods Mol Biol 907, 451–461 (2012).
Weissleder, R., Kelly, K., Sun, E. Y., Shtatland, T. & Josephson, L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol 23, 1418–1423 (2005).
Sambrook, J. & Russell, D. W. Preparation of Lysates Containing Fusion Proteins Encoded by Bacteriophage lambda Lysogens: Lysis of Bacterial Colonies. CSH Protoc 2006, (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China No. 30871108. We would like to thank Dr. Francisco Javier Cubero (UKA Aachen, Aachen, Germany) for his help with the editing and the interpretation of the data.
Author information
Authors and Affiliations
Contributions
J.X. performed the experiments, the revision and wrote the manuscript. Y.Z., J.Q. and Y.Z. collaborated in the libraries and wrote parts of the manuscript. X.Z. analyzed data obtained from the libraries and prepared figures. J.Z. and G.Z. supervised the experiments, wrote the paper, reviewed the manuscript and provided the funding.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Xia, J., Zhang, Y., Qian, J. et al. Isolation, identification and expression of specific human CD133 antibodies. Sci Rep 3, 3320 (2013). https://doi.org/10.1038/srep03320
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03320
This article is cited by
-
Production of a Ribosome-Displayed Mouse scFv Antibody Against CD133, Analysis of Its Molecular Docking, and Molecular Dynamic Simulations of Their Interactions
Applied Biochemistry and Biotechnology (2024)
-
Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and anti-CD133 CAR-T combination therapy
Cancer Immunology, Immunotherapy (2021)
-
Periscope: quantitative prediction of soluble protein expression in the periplasm of Escherichia coli
Scientific Reports (2016)
-
Soluble Expression and Characterization of a New scFv Directed to Human CD123
Applied Biochemistry and Biotechnology (2016)
-
Characterization and applications of Nanobodies against human procalcitonin selected from a novel naïve Nanobody phage display library
Journal of Nanobiotechnology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
