
Abstract
Local patterns of genetic variation were analysed in the land snail Helix aspersa for 32 populations sampled within a patchy agricultural landscape: the polders of the Bay of Mont-Saint-Michel (France). This investigation examined the allele frequencies at four enzymatic markers and five microsatellite loci through the genotyping of 580 individuals. A strongly significant population genetic substructuring (mean FST=0.088, P<0.001) was found at the scale of the whole polders area (3050 ha) and both categories of markers displayed a similar magnitude of spatial genetic differentiation. We did not find any obvious effects of habitat fragmentation on the distribution of genetic variability. Despite the reality of habitat patchiness and environmental instability (related to farming practices), an isolation by distance process was clearly depicted, although selective pressures cannot be ruled out for one enzymatic locus. Overall, genetic drift, along with occasional long-distance episodes of gene flow, was presumably the most likely evolutionary force that shaped the observed pattern of genetic variation.
Similar content being viewed by others


Introduction
The distribution of neutral genetic variability within and among natural populations arises from the combined effects of processes like mutation, migration and random genetic drift, and the description of genetic structure in wild populations provides insights into the past and contemporary respective influences of such evolutionary forces (Slatkin, 1994; Hutchison and Templeton, 1999). Habitat fragmentation has been a strong feature of wild populations, especially where agricultural practices have dominated. One consequence of this mosaic of terrestrial landscapes is the subdivision of species into many arrays of localised populations, often of limited size, where mating is more likely to occur among related individuals (Frankham et al, 2002). Within the context of human-disturbed environments, an array of local populations, considered as a whole, can be expected to persist for a longer period of time than any one of its individual elements, assuming that the colonisation rate is sufficient to offset local extinctions (McCauley, 1993; Pannell and Charlesworth, 2000). This observation has led to the well-known concept of the metapopulation, consisting of a large set of transient populations with a regional persistence achieved by a balance between local extinctions and recolonisations (Hanski, 1999). Hence, metapopulation models can take many forms that may have different genetic and evolutionary consequences, largely depending on the spatial interconnection of local populations, the demographic processes associated with colonisation events (ie the source of the colonists and the rate of colonisation), and how extinction events are distributed in time and space (McCauley et al, 1995; Thrall et al, 2000; Whitlock, 2001; Charbonnel et al, 2002).
Terrestrial molluscs generally reside in patchy habitats likely to promote geographical structuring and have been considered as ideal organisms for studies of ecological genetics over a large range of spatial scales (eg Selander and Ochman, 1983; Arter, 1990; Bahl et al, 1996; Arnaud et al, 1999a, 1999b; Davison and Clarke, 2000; Armbruster, 2001; Goodacre, 2002, and references therein). They are easy to collect and mark, making both direct and indirect measurements of gene flow possible, and can exhibit extensive demographic and genetic population structure even on a microgeographical scale. This study describes the pattern of population genetic structure among local colonies of the land snail Helix aspersa (= Cornu aspersum), within the polders of the Bay of Mont-Saint-Michel (Western Brittany, France). The polders area consists in a mosaic of habitat patches of varying size and composition that includes a mixture of woodlots, pastures, farmlands and intensive agricultural zones delimited by a complex network of hedges and dikes. In this environment, H. aspersa occurs in patchily distributed colonies characterised by localised unpredictable extinction events, a situation presumably being characteristic of a metapopulation structure. For simplicity, we will use, throughout this study, the term ‘metapopulation’ in the broader view of a ‘population of populations’, a term allowing the possibility of extinction and recolonisation processes without requiring such events as a necessary characteristic (Hanski, 1999; see also Whitlock, 2001). By using a combination of enzymatic and microsatellite loci, the present study was designed to study the extent of spatial genetic differentiation among populations of H. aspersa in this human-disturbed landscape.
Materials and methods
Study field and sample collection
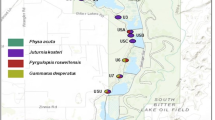
Specimens of H. aspersa were collected from 30 localities within the ‘polders’ of the Bay of Mont-Saint-Michel, located in northwestern France at the margin of Brittany and Normandy (48°36′N; 1°32′W) (see Figure 1). Two additional colonies were sampled at the base of the island of Mont-Saint-Michel. A total of 90% of the polder area (for an area of ∼3050 ha) is under intensive agriculture and the seminatural habitats, used as refuges by many animal species, are distributed over a landscape mosaic delimitated by a complex network of hedges and dikes (Paillat and Butet, 1996). The first dike was created in 1054, the building of the polders started in 1851, and the last dike was completed in 1933. Dikes are either planted with poplar or covered with naturally regenerated grass and shrubby vegetation. The polders area is bordered on its southern side by mixed woodlands, on its eastern side by the river ‘Couesnon’ and on its northern part by the last dike (‘Digue à Flot’) (Figure 1). Beyond this last dike, which protects the polders against the rising of water sea, the ‘Herbus’ area (salt meadow sheep) is periodically covered by sea during major high tides. Human disturbances (weeding, mowing, burning) often lead to some localised extinctions of H. aspersa colonies (Arnaud, 2000; Madec et al, 2000).

Map of sample locations numbered from 1 to 32. Sample sizes are indicated in italics under each population number. Full lines: local roads or overland routes. Dashes: dikes or hedges.
Individuals were sampled along borders of roads, under bridges, under leaf litter, but most often under solid supports that potentially constitute aestivating or hibernating refuges for snails (corrugated iron, boards, bricks and the like). To minimise genetic artefacts owing to substructuring within the sampling area (ie a Wahlund effect), samples were collected from areas as small as possible, usually less than 5 m × 5 m, which is smaller than the estimated neighbourhood size of H. aspersa (Guiller et al, 1994, 1998). A total of 580 individuals (mean sample size of 18.2) were genotyped for nine polymorphic markers.
Allozyme and microsatellite data
Four polymorphic enzymatic systems (Aat-1, Est-3, Lap-2 and Mdh-1) were used and allozyme genotypes were determined by means of polyacrylamide gel electrophoresis, as described in Guiller et al (1994). Five hypervariable microsatellite loci were examined and successfully multiplexed in a single PCR reaction (Ha2, Ha5, Ha10, Ha11 and Ha13). Each locus consisted of dinucleotide or tetranucleotide repeats either perfect, interrupted (tandem repeats interrupted by additional or substituted nucleotides) or compound (composed of stretches of different repeat motifs). DNA extraction, locus amplification and allele identification were performed as reported in Guiller et al (2000) with slight modifications. Briefly, forward primers of microsatellite loci were endlabelled with fluorescent dyes: FAM (Ha5 and Ha11), NED (Ha2 and Ha10) and HEX (Ha13). PCR amplifications were carried out in 25 μl volumes using a Biometra® thermocycler with one cycle of 5 min at 95°C; 30 cycles of 30 s at 94°C, 30 s at 54°C, 30 s at 72°C and one final cycle of 5 min at 72°C. The reaction contained 10 mM Tris-HCl (ph 9.0), 50 mM KCl, 1.5 mM MgCl2, 200 μm of each dNTP, stabilizers including BSA, 1.5 U of Taq DNA polymerase (Ready to Go®, Amersham Pharmacia Biotech.), 0.20 μm of each primer and ∼50 ng of template DNA. The PCR products were electrophoresed using a PE Applied Biosystems 310 Genetic Analyzer (Perkin-Elmer). DNA fragments were automatically visualised and sized using the ABI PRISM 310™ collection and GENESCAN 3.1™ software (Applied Biosystems) with the local southern sizing algorithm.
Statistical analyses
Allele frequencies, observed heterozygosities and unbiased estimates of expected heterozygosities under Hardy–Weinberg expectations were calculated for each population and locus using the software GENEPOP version 3.1d (Raymond and Rousset, 1995). Genotypic linkage disequilibrium was estimated to test for locus independence prior to other analyses. Within-sample deviation from Hardy–Weinberg equilibrium (HWE) was investigated using Fisher's exact tests at individual loci, over all loci and across loci and populations. The overall significance of multiple tests was evaluated by Fisher's combined probability test. Sequential Bonferroni corrections for simultaneous statistical tests were applied following Rice (1989). To quantify the levels of population subdivision, we used hierarchical partitioning of standardised genetic variance statistics according to Weir and Cockerham (1984). Numerical resampling was used to provide estimates of variance and confidence intervals for each locus (jackknifing) and across loci (bootstrapping) using FSTAT (Goudet, 1995). Tests for significant departure from zero were applied by permuting 5000 times alleles within (FIS) and among (FIT) samples, and multilocus genotypes among sample colonies (FST). To depict genetic affinities among populations of H. aspersa, a minimum spanning tree (MST) was calculated using the DCE distance (Cavalli-Sforza and Edwards, 1967). In addition to the above approaches, genetic structure was analysed using nonmetric multidimensional scaling (NMDS), which circumvents the linearity assumption of metric ordination methods. This nonparametric method takes the distances between objects and tries to approximate those distances in a reduced number of dimensions through an iterative fitting procedure (Guiller et al, 1998; Jordaens et al, 2000). The goodness of fit between the fitted and observed distances was measured by a stress test.
To test for isolation by distance (IBD), the relation between pairwise FST and geographical distance between sampled populations was analysed following the method suggested by Rousset (1997). The spatial distribution of genetic variability was also investigated using simple and directional spatial autocorrelation statistics (Sokal et al, 1987). All spatial autocorrelograms were constructed using AUTOCOR, a program written by A Bellido (University of Rennes 1).
Results
Genetic diversity, genotypic linkage and Hardy–Weinberg disequilibrium
Allelic diversity ranged from 2 (Aat-1) to 5 (Lap-2) alleles per enzymatic locus and from 6 (Ha2) to 32 (Ha13) alleles per microsatellite locus, with a mean number of observed alleles per population ranging from 1.97 to 9.62 across all loci (Table 1). Overall, mean levels of gene diversity were relatively high, especially for microsatellite loci, and ranged from 0.32 (Aat-1) to 0.83 (Ha11). Mean observed heterozygosities (Ho) per colony were close to those observed for mean gene diversity for both categories of markers (Table 1). Out of 1152 comparisons across the 32 colonies, only 50 (4.34%) significant genotypic linkage disequilibria were depicted, a proportion expected by chance alone. Multiple probability tests (Fisher's method) performed across all colony samples yielded only one significant value out of 66 pairs of loci, observed for Lap-2/Aat-1 (P<0.05, χ262=105.81) after Bonferroni correction. Genotype proportions at either allozyme or microsatellite loci were largely concordant with those expected under HWE. Out of 288 individual tests, only 16 (5.5%) showed a significant deviation from HWE assumptions, with departures involving either slight heterozygote deficiencies or excesses. Agreement with HWE expectations was detected by multilocus probability tests for all populations excepting samples 15 and 18 (P<0.001 and <0.05, respectively). Mean FIS estimates were not significantly different from 0 except for Aat-1 and Ha2 (FIS=0.085 and 0.058, respectively), and a slight excess of heterozygotes, seen as negative FIS values, is also observed for Lap-2, Mdh-1, Ha10 and Ha11 (Table 2). Altogether, these results seemed to corroborate a general lack of inbreeding as well as the absence of heterogeneous mixture of genetically distinct entities.
Spatial genetic differentiation
Estimates of population differentiation were positive and significantly different from 0, with an overall value of FST of 0.088 (P<0.001) (Table 2). Both categories of markers displayed similar degrees of substructuring and, although the number of loci per class of markers was low, FST estimates were not significantly different between allozymes and microsatellites (Mann–Whitney's U test, P=0.71). Estimates of fixation indices FIT were always positively significant (synonym of an overall Wahlund effect consecutive to the pooling of structured entities), except for the locus Lap-2.
Linear regression of FST/(1−FST) on the logarithm of geographical distance showed a positive correlation between population differentiation and geographical isolation when allozymes and microsatellites are combined (r=0.34, P<10−5) (Figure 2). However, scatterplots were suggestive of a great variance and a strong scattering of pairwise FST/(1−FST) estimates. When pooling different population samples following three groups (western, southeastern and northern groups, see below), a highly significant spatial differentiation remained at this level (FST=0.030, P<0.001). Such a result was unexpected under an island model without distance effect, but suggested an IBD effect.

Scatterplots of FST/(1−FST) estimates against the logarithm of geographical distances (m) separating each pairwise combination of populations, using both allozyme and microsatellite loci. Regression equation: y=0.0237x−0.0996; P<10−5 after 10 000 permutations (Mantel test).
Spatial autocorrelation analyses yielded in most cases quite stochastic correlogram profiles, with some positive significant autocorrelation coefficients at short distance classes (ie <2000 m), immediately followed by strong fluctuations of Moran's I involving either high positive or negative values with no relation with geographical distance (correlograms not shown). The salient and most striking feature of spatial genetic arrangement was displayed by the locus Est-3 that clearly departed from the others. For the two most common alleles, this locus showed a significant monotonic decrease in Moran's I values for all distance classes, consistent with an isolation by distance process or a cline (Figure 3a). However, a genetic gradient is rarely uniformly distributed in space. Indeed, the decomposition of the initial unidirec-tional correlogram in four distance/direction classes demonstrated that such a decline of similarity in allelic frequencies is only effective for a 90°/135° orientation, that is a southeast/northwest direction (Figure 3b and c).

Correlograms of Moran's I for the two most common alleles of the enzymatic locus Est-3 in the land snail H. aspersa: (a) unidirectional correlogram; (b, c) directional correlograms for alleles Est-3100 and Est-395, respectively. Directional correlograms were calculated after constructing distance/direction classes following four angle directions: 0, 45, 90 and 135°. I: Moran's I index; d (m): geographical distance in metres. For unidirectional correlograms, significant (P<0.05) individual autocorrelation coefficients are indicated by an asterisk.
Genetic relationships among colonies
NMDS ordination analysis, based on both allozyme and microsatellite markers, depicted a local spatial patterning since populations can be broadly divided into two distinct geographical areas, that is a western and a south-northeast group (Figure 4). The same population relationships hold when constructing a neighbour-joining tree (dendrogram not shown). The superimposition of MST on samples map clearly revealed a split between western and eastern colonies, with close genetic relationships between nearby colonies and somewhat counterintuitive associations between geographically distant colonies (Figure 5). The MST superimposition further indicated a ‘star-like’ grouping of local colonies, involving localities 18 and 30 as initial point of diffusion.

NMDS ordination of pairwise DCE genetic distance (Cavalli-Sforza and Edwards, 1967) between 32 populations of H. aspersa, using the information from five microsatellite and four enzymatic loci. Numbers refer to population labels presented in Figure 1. Stress value=0.052 after 100 iterations.

MST superimposed on geographical locations of 32 population samples of H. aspersa, based on the DCE distance (Cavalli-Sforza and Edwards, 1967) and using five microsatellites and four enzymatic loci. Branch thicknesses are proportional to the population divergence.
Discussion
Levels of gene diversity and extents of differentiation in H. aspersa
Land snail species are likely to be subdivided into interacting genetically subdivided neighbourhoods that generate and maintain a strong variability in continuously distributed populations (Bahl et al, 1996; Arnaud et al, 2001; Goodacre, 2002). Nonetheless, habitat fragmentation may reduce exchanges of individuals between adjacent neighbourhoods, leading to a reduced genetic diversity and to an increase of differentiation through random drift (Bahl et al, 1996; Frankham et al, 2002). In contrast to these predictions, the observed level of polymorphism in this population does not seem to be strongly depressed by the fragmentation into local semiisolated colonies, as is suggested by the relatively high polymorphism displayed by both categories of nuclear markers. As stressed by Peacock and Ray (2001), the loss of genetic variability is not a general scheme in metapopulations with high turnover, and high levels of polymorphism do not preclude a potential extinction–recolonisation dynamic (see also Thrall et al, 2000; Mossman and Waser, 2001). Moreover, inbreeding avoidance, jointly with multiple matings, could be an effective diversifying factor in Pulmonate land snails (Murray, 1964). Alternatively, some kinds of selective pressures could not be ruled out (see below). However, it is worth noting that (i) some populations were monomorphic for three of the four enzyme loci (see Table 1), a pattern suggestive of genetic erosion and (ii) there is no appropriate nonfragmented population with which the present results can be compared. More generally, only the most variable markers were used in our study. Hence, we could have an upward bias in the reported level of polymorphism, likely to distort our picture of the genetic variability in fragmented colonies of H. aspersa (see the review of Amos and Harwood, 1998).
A strongly significant local genetic differentiation was reported and the extent of genetic substructuring displayed by microsatellites was of the same order of magnitude as that observed with allozyme data, as was similarly documented by Estoup et al (1998) and Fréville et al (2001). The building of the polders was completed in 1933 (Paillat and Butet, 1996) and the generation time does not seem to exceed 2 years in this disturbed environment (Madec et al, 2000). Thus, owing to the presumed recent divergence of studied colonies as well as to the concordance of multilocus FST estimates, the observed genetic structure may be expected to be shaped, to a large extent, by gene flow and genetic drift rather than other pressures like mutation or selection whose local effects occur over a larger timescale (McCauley, 1993; Pannel and Charlesworth, 2000). The pattern of spatial arrangement observed for the locus Est-3 could be determined by the interaction of drift and gene flow, leading to the cumulative effects of a certain degree of IBD. However, drift or migration affect all loci similarly, and the striking clinal pattern demonstrated at the locus Est-3 is more likely to be related to selective pressures reflecting genetic adaptations to site-specific conditions. Indeed, Jordaens et al (1999) established, in other terrestrial gastropods (Carinarion spp.), that nonspecific esterase variation could be physiologically or environmentally influenced, sometimes leading to inflated genetic diversity estimates. Strong evidence for habitat-specific selection inferred from allozyme variation was also demonstrated in the land snail Cochlicopa lubrica (Armbruster, 2001). One possible ecological determining factor characterising the polder area could be the degree of salinity. However, given the absence of ecological field data, further hypotheses remain speculative.
Population genetic structure in H. aspersa
Low dispersal capabilities, associated with a sedentary behaviour intrinsic to land snails (homing behaviour), may contribute to a fine-scaled spatial structuring. Founder effects could also be involved in the strong differentiation displayed at some loci (eg Aat-1 or Ha2). Indeed, the turnover of local colonies is an important determinant of the genetic structure because it can enhance opportunities for both genetic drift and gene flow, depending either on the number of colonists relative to the number of recurrent migrants, and on whether the colonists arise from a single source or many sources (McCauley et al, 1995; Pannell and Charlesworth, 2000; Whitlock, 2001). Linear regression of pairwise estimates of genetic differentiation suggested an IBD for both types of markers. However, the observed genetic cleavage between western and eastern colonies may have possibly resulted from two independent colonisations of one or several sites, followed by a neighbourhood diffusion with ongoing gene flow within these two groups. By contrast, the strong differentiation of the two island colonies of the Mont-Saint-Michel, compared to those located in the polders, could have arisen from an independent and more ancient colonisation event, possibly mediated by monks who used this edible snail as an alimentary resource during scarcity times in the Middle Ages.
Owing to the restricted dispersal capability of land snails, the usual expectation would involve a continuous increase of genetic differentiation with geographical distance. However, geographically distant populations are sometimes genetically more similar than would be expected based on distance alone, suggesting a patchiness in the distribution of alleles (Davison and Clarke, 2000). Such observations cannot easily be explained by active dispersal. Owing to the presumed recent colonisation of the polders area by H. aspersa (the last dike and subsequent cultivated fields were created only 60 years ago), the time span since the introduction and separation of colonies is too short to allow an equilibrium state between the opposing forces of gene flow and drift (Slatkin, 1994; Hutchison and Templeton, 1999). Beyond stochastic effects, another possible interpretation could be that the foundation of new populations may have been facilitated by passive and accidental transport owing to human activities, which would not be restricted by the extent of geographical separation (spread by the circulation of farming scraps, agricultural engines or sheep flocks; JF Arnaud, pers. obs.).
As expected, the level of genetic differentiation among colonies was greater in the whole fragmented agricultural area than that found on a more restricted scale (Arnaud et al, 1999a, 2001). Nonetheless, the comparison between a fragmented and a nonfragmented landscape is not possible in this case study. It also brings into question whether landscape fragmentation increases the level of differentiation because of direct barrier effects, and/or more indirectly, through the distribution of individuals in space because of their limited dispersal capabilities (see Wiens, 1997). Applying more realistic geographical distances that take into account landscape components (roadside ditches, edges, cultivated fields) clearly gave a better fit to an IBD model (Arnaud, unpublished results). Similarly, the concept of a functional IBD process taking place along environmentally induced gene flow paths has been suggested in the land snail Arianta arbustorum (Arter, 1990).
In conclusion, the genetic substructuring among colonies of H. aspersa seems to be related to low short-range gene flow and, at least partially, to stochastic factors including occasional long-distance dispersal and landscape components. Habitat fragmentation may have significant effects on the genetic structure of subdivided populations because physical barriers are likely to impede gene flow (McCauley, 1993; Frankham et al, 2002). However, despite the reality of habitat patchiness and environmental instability, this study showed no clear evidence concerning the potential effects of fragmentation and human disturbances on the distribution of genetic variability (see also Mossman and Waser, 2001). Trends compatible with a metapopulation structure would require either direct investigations in the field, such as mark-release studies, or a temporal survey of genetic structuring. To address this question, the temporal evolution of the within-population genetic composition is currently being investigated in the polders area.
References
Amos W, Harwood J (1998). Factors affecting levels of genetic diversity in natural populations. Philos Trans R Soc London B 353: 177–186.
Armbruster GFJ (2001). Selection and habitat-specific allozyme variation in the self-fertilizing land snail Cochlicopa lubrica (O.F. Müller). J Nat Hist 35: 185–199.
Arnaud J-F (2000). Organisation spatiale de la Variabilité Génétique dans une Métapopulation de l'Escargot Terrestre Helix aspersa (Gastropoda: Pulmonata). PhD Thesis, Université de Rennes 1, France.
Arnaud J-F, Madec L, Bellido A, Guiller A (1999a). Microspatial genetic structure in the land snail Helix aspersa (Gastropoda: Helicidae). Heredity 83: 110–119.
Arnaud J-F, Madec L, Daguzan J (1999b). Spatial differentiation of allozyme frequencies in a subdivided population of the land snail Helix aspersa. J Moll Stud 65: 267–271.
Arnaud J-F, Madec L, Guiller A, Bellido A (2001). Spatial analysis of allozyme and microsatellite DNA polymorphisms in the land snail Helix aspersa (Gastropoda: Helicidae). Mol Ecol 10: 1563–1576.
Arter HE (1990). Spatial relationship and gene flow paths between populations of the alpine snail Arianta arbustorum (Pulmonata: Helicidae). Evolution 44: 966–980.
Bahl A, Pfenninger M, Bamberger H, Frye M, Streit B (1996). Survival of snails in fragmented landscapes. In: Settele J, Margules CR, Poschlod P, Henle K (eds) Species Survival in Fragmented Landscapes. Kluwer Academic Publishers: The Netherlands, pp 329–343.
Cavalli-Sforza LL, Edwards AWF (1967). Phylogenetic analysis: models and estimation procedures. Am J Hum Genet 19: 233–257.
Charbonnel N, Angers B, Rasatavonjizay R, Bremond P, Jarne P (2002). Evolutionary aspects of the metapopulation dynamics of Biomphalaria pfeifferi, the intermediate host of Schistosoma mansoni. J Evol Biol 15: 248–261.
Davison A, Clarke B (2000). History or current selection? A molecular analysis of ‘area effects’ in the land snail Cepaea nemoralis. Proc R Soc London B 267: 1399–1405.
Estoup A, Rousset F, Michalakis Y, Cornuet J-M, Adriamanga M, Guyomard R (1998). Comparative analysis of microsatellite and allozyme markers: a case study investigating microgeographic differentiation in brown trout (Salmo trutta). Mol Ecol 7: 339–353.
Frankham R, Ballou JD, Briscoe DA (2002). Introduction to Conservation Genetics. Cambridge University Press: Cambridge, UK.
Fréville H, Justy F, Olivieri I (2001). Comparative allozyme and microsatellite population structure in a narrow endemic plant species, Centaurea corymbosa Pourret (Asteraceae). Mol Ecol 10: 879–889.
Goodacre SL (2002). Population structure, history and gene flow in a group of closely related land snails: genetic variation in Partula from the Society Islands of the Pacific. Mol Ecol 11: 55–68.
Goudet J (1995). FSTAT (Version 1.2). A computer program to calculate F-Statistics. J Hered 86: 485–486.
Guiller A, Arnaud J-F, Vautrin D, Solignac M (2000). Highly polymorphic microsatellite markers in the land snail Helix aspersa (Mollusca: Gastropoda). Mol Ecol 9: 1191–1193.
Guiller A, Bellido A, Madec L (1998). Genetic distances and ordination: the land snail Helix aspersa in North Africa as a test case. Syst Biol 47: 208–227.
Guiller A, Madec L, Daguzan J (1994). Geographical patterns of genetic differentiation in the landsnail Helix aspersa Müller (Gastropoda: Pulmonata). J Moll Stud 60: 205–221.
Hanski I (1999). Metapopulation Ecology. Oxford University Press: Oxford, UK.
Hutchison DW, Templeton AR (1999). Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53: 1898–1914.
Jordaens K, Geenen S, Reise H, Van Riel P, Verhagen R, Backeljau T (2000). Is there a geographical pattern in the breeding system of a complex of hermaphroditic slugs (Mollusca: Gastropoda: Carinarion)? Heredity 85: 571–579.
Jordaens K, Van Riel P, Verhagen R, Backeljau T (1999). Food-induced esterase electromorphs in Carinarion spp. and their effects on taxonomic data analysis (Gastropoda, Pulmonata, Arionidae). Electrophoresis 20: 473–479.
Madec L, Desbuquois C, Coutellec-Vreto M-A (2000). Phenotypic plasticity in reproductive traits: importance in the life history of Helix aspersa (Mollusca: Helicidae) in a recently colonized habitat. Biol J Linn Soc 69: 25–39.
McCauley DE (1993). Genetic consequences of extinction and recolonization in fragmented habitats. In: Kareiva PM, Kingsolver JG, Huey RB (eds) Biotic Interactions and Global Change. Sinauer Associates Inc.: Sunderland, MA, pp 217–233.
McCauley DE, Raveill J, Antonovics J (1995). Local founding events as determinants of genetic structure in a plant metapopulation. Heredity 75: 630–636.
Mossman CA, Waser PM (2001). Effects of habitat fragmentation on population genetic structure in the white-footed mouse (Peromyscus leucopus). Can J Zool 79: 285–295.
Murray J (1964). Multiple mating and effective population size in Cepaea nemoralis. Evolution 18: 283–291.
Paillat G, Butet A (1996). Spatial dynamics of the bank vole (Clethrionomys glareolus) in a fragmented landscape. Acta Oecol 17: 553–559.
Pannell JR, Charlesworth B (2000). Effects of metapopulation processes on measures of genetic diversity. Phil Trans R Soc London B 355: 1851–1864.
Peacock MM, Ray C (2001). Dispersal in pikas (Ochotona princeps): combining genetic and demographic approaches to reveal spatial and temporal patterns. In: Clobert J, Danchin E, Dhondt AA, Nichols JD (eds) Dispersal. Oxford University Press: Oxford, UK, pp 43–56.
Raymond M, Rousset F (1995). GENEPOP (Version 1.2): a population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rousset F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Selander RK, Ochman H (1983). The genetic structure of populations as illustrated by Molluscs. In: Rattazzi MC, Scandalios JG, Whitt GS (eds) Isozymes: Current Topics in Biological and Medical Research. Alan R. Liss, Inc.: New York, pp 93–123.
Slatkin M (1994). Gene flow and population structure. In: Real LA (ed) Ecological Genetics. Princeton University Press: Princeton, NJ, pp 3–17.
Sokal RR, Oden NL, Barker JSF (1987). Spatial structure in Drosophila buzzatii populations: simple and directional spatial autocorrelation. Am Nat 129: 122–142.
Thrall PH, Burdon JJ, Murray BR (2000). The metapopulation paradigm: a fragmented view of conservation biology. In: Young AG, Clarke GM (eds) Genetics, Demography and Viability of Fragmented Populations. Cambridge University Press: Cambridge, UK, pp 75–95.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whitlock MC (2001). Dispersal and the genetic properties of metapopulations. In: Clobert J, Danchin E, Dhondt AA, Nichols JD (eds) Dispersal. Oxford University Press: Oxford, UK, pp 273–282.
Wiens JA (1997). Metapopulation dynamics and landscape ecology. In: Hanski I, Gilpin ME (eds) Metapopulation Biology: Ecology, Genetics, and Evolution. Academic Press: San Diego, CA, pp 43–62.
Acknowledgements
We thank JF Brookfield, MS Johnson, E Petit, F Viard and two anonymous referees for useful comments and critical reading on earlier versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arnaud, JF., Madec, L., Guiller, A. et al. Population genetic structure in a human-disturbed environment: a case study in the land snail Helix aspersa (Gastropoda: Pulmonata). Heredity 90, 451–458 (2003). https://doi.org/10.1038/sj.hdy.6800256
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800256
Keywords
This article is cited by
-
Unexpected and spatially structured genetic diversity of the relict population of the endangered corsican land snail Tyrrhenaria ceratina
Conservation Genetics (2023)
-
Population structure and genetic signs of population bottlenecks in the endangered Hawaiian tree snail Achatinella sowerbyana
Conservation Genetics (2014)
-
Phylogeography of an invasive land snail: natural range expansion versus anthropogenic dispersal in Theba pisana pisana
Biological Invasions (2012)
