
Abstract
To thwart viral infection, the host has developed a formidable and integrated defense network that comprises our innate and adaptive immune response. In recent years, it has become clear that in an attempt to prevent viral replication, viral dissemination or persistent viral infection of the cell, many of these protective measures actually involve the induction of programmed cell death, or apoptosis. An initial response to viral infection primarily involves the innate arm of immunity and the killing of infected cells with cytotoxic lymphocytes such as natural killer (NK) cells through mechanisms that include the employment of perforin and granzymes. Once the virus has invaded the cell, however, a second host defense-mediated response is also triggered which involves the induction of a family of cytokines known as the interferons (IFNs). The IFNs, which are essential for initiating and coordinating a successful antiviral response, function by stimulating the adaptive arm of immunity involving cytotoxic T cells (CTLs), and by inducing a number of intracellular genes that directly prevent virus replication/cytolysis or that facilitate apoptosis. The IFN-induced gene family is now known to comprise the death ligand TRAIL, the dsRNA-dependent protein kinase (PKR), interferon regulatory factors (IRFs) and the promyelocytic leukemia gene (PML), all of which have been reported to be mediators of cell death. That DNA array analyses indicate that numerous cellular genes, many as yet uncharacterized, may similarly be induced by IFN, further emphasizes the likely importance that these cytokines have in the modulation of apoptosis. This likelihood is additionally underlined by the elaborate strategies developed by viruses to inhibit IFN-antiviral function and the mechanisms of cell death. Cell Death and Differentiation (2001) 8, 113–126
Similar content being viewed by others


Host defense against viral infection: innate immunity and apoptosis
The innate arm of immunity plays a critical role in the early control of viral infection as well as in the initiation and subsequent direction of the adaptive immune response. Typically, viral infection starts with local invasion, for example of an epithelial surface, and results in the infection of the target organ such as the skin, nervous system, pulmonary tract or immune system. Following entry, viruses then face a variety of hurdles, constituting cellular host defense that are designed to protect the organism and eradicate the infectious agent. One of the first lines of defense that confronts an invading virus before it has infected the cell involves neutralizing antibody, which binds to the envelope or capsid proteins of the pathogen and prevents viral attachment and entry into host.1 These antibodies can comprise ‘natural’ antibodies, which represent a spontaneous repertoire of circulating immunoglobulins that bind to virus and influence the infection of vital target organs as well as direct the virus to secondary lymphatic organs to accelerate and enhance immune responses.2 Similarly, complement activation may participate in antibody mediated viral immunity by coating the virus, by promoting phagocytosis, or by directly lysing those viruses with lipid membranes.3 Should the virus infect the cell, however, a formidable variety of cellular and humoral mechanisms designed to prevent viral dissemination are subsequently invoked. For example, antibody-dependent cell-mediated cytotoxicity (ADCC) involving large granular lymphocytes, or K cells, can be summoned which bind to antibody on virus infected target cells via their surface FC receptors and kill the cell. In addition, natural killer (NK) cells, which also do not appear to require prior contact with target antigens to develop cytolytic capacities, similarly appear to be potent effectors in ADCC. NK cells are thought to distinguish infected from uninfected cells via multiple histocompatibility complex class I (MHC) expression. Receptors (Ly49) on the surface of the NK cells recognize normal syngeneic cellular MHC class I molecules, which inhibit NK activity. Since MHC class I protein expression and host protein synthesis in general can be inhibited by many viruses, NK cells interacting with an infected cell would no longer be inhibited through their MHC-specific receptor. In this situation, another NK receptor (NKR-P1), which recognizes a wide variety of carbohydrate ligands found on many cells can trigger cytolysis of a target cell. In addition, NK cells express the Fc receptor (CD16) which recognize selected host Ig subclasses bound to viral antigens on the surface of infected cells and can kill such cells also.4 Both mechanisms of cell death likely involve the orderly elimination of infected cells through use of perforin and granzymes.5 Following interaction with the target, killer cells secrete the protein perforin onto cells infected with virus. Perforin, by calcium-dependent binding to phospholipid head groups and polymerization, forms transmembrane channels of sizes up to 16 nm. Processed granzymes are also secreted from the killer cell and can enter the target cell through repair endocytosis. This event ultimately causes downstream cellular pathways to be activated that results in the suicide of the cell by a mechanism referred to as apoptosis.6
Apoptosis
Apoptosis, or programmed cell death is a process in which cells activate intracellular death pathways to terminate themselves in a systematic way in response to a wide variety of stimuli.6 Morphologically, cells undergoing apoptosis appear to shrink and are swiftly engulfed by neighboring cells and phagocytes before there is any leakage of their contents. This process of cellular death is different to where cells die in response to injury. In the latter situation, damaged cells swell and burst, spilling out their contents and attracting lymphocytes, thereby eliciting an unwanted inflammatory response.7 Within the last few years it has become apparent that apoptosis plays an important role in development, the regulation of hematopoetic progenitor cells, in the elimination of cells that have sustained genetic damage or that undergo uncontrolled cellular proliferation, and in preventing viral replication.
Genetic analysis of the nematode Caenorhabditis elegans gave investigators a clue as to the molecules involved in the apoptotic process. For example, two loci ced-3 and ced-4 (ced for cell death abnormal) were found to be required for the apoptosis of 131 cells during worm development.8 Since the mechanisms of apoptosis are somewhat conserved, it was not very long before it was noticed that ced-3 was remarkably similar to a human protein referred to as interleukin-1-converting enzyme (ICE), and to a number of similarly structured proteins now known as caspases. These 30 to 60 kDa proenzymes are continuously made in the cell and are activated by proteolytically processing either autocatylytically or in a cascade by enzymes with similar characteristics.9 Once activated, caspases will cleave proteins containing specific tetrapeptide motifs harboring an aspartic acid residue. About 50 or so cellular substrates for the caspases have so far been identified, although many more likely exist. Most caspase targets included proteins that prevent the dismantling of the cell and degradation of the nucleus including DNA fragmentation. The eventual outcome of caspase activation generates the morphological hallmarks of apoptosis.
Apoptosis: death receptors
The perforin/granzyme pathway is one way in which CTLs and NK cells can eradicate virus infected cells through the use of apoptosis.10 Yet another important apoptosis-related mechanism of antiviral host defense involves CTLs that recognize and kill target cells by signaling though cell surface death receptors.11 Death receptors belong to the tumor necrosis factor (TNF) receptor family and contain a homologous sequence referred to as the ‘death domain (DD domain)’ in their cytoplasmic tail. The most well characterized death-receptors are CD95/Fas and TNFR1, although additional death receptors have been isolated and are referred to as DR3, 4, and 5.11,12 Numerous studies have indicated that FasL binds to Fas, TNF and lymphotoxin alpha bind to TNFR1, Apo3L/TWEAK binds to DR3 and Apo2L/TRAIL binds to DR4 and 5. Aside from killing virus-infected or malignant cells lymphocytes use death receptor induced killing to delete activated mature T cells at the end of an immune response. In the case of Fas-mediated killing, CTLs expressing Fas ligand (FasL) bind to Fas receptors on the target cell, clustering their intracellular death domains. An adapter molecule FADD (for Fas-Associated Death Domain) then binds to the receptors through its own death domain.13 FADD also contains a ‘death effector domain’ (DED domain) that functions as a caspase recruitment domain (CARD), found in several caspases. The preferred target of FADD is caspase-8 or FLICE, which following oligomerization, activates itself through self cleavage to activate downstream effector or executioner caspases such as caspase-3, thus committing the cell to apoptosis.14
Activation of all the death receptors can recruit FADD to initiate the death signaling process. However, in the case of TNFR1 and DR3, recruitment of FADD and the triggering of apoptosis is accomplished by another adapter protein TRADD (for TNFR-Associated Death domain). TNFR1/DR3-mediated cell death rarely occurs though, unless protein synthesis is inhibited.15 Activation of the TNFα pathway is also able to suppress apoptosis by utilizing the NF-κB and c-Jun NH2-terminal kinase (JNK)/AP-1 signaling pathways, to stimulate the expression of anti-apoptotic genes such as the inhibitors of apoptosis (IAPs) and TNFR-associated factors (TRAFs) which prevent caspase activation and thus cell death.16,17,18 The role of NF-κB in controlling cell death is becoming increasingly apparent. Recent data indicate that to activate TNF receptor mediated cell death, an intracellular death domain kinase RIP, must be recruited to the TNF receptor complex. In the presence of both TRAF2 and RIP, however, IKβ kinase complexes (IKK) are also recruited to the receptor and become activated.19,20 Upon stimulation by IKKs, IκBs becomes phosphorylated which is the signal for their subsequent ubiquitination and degradation by the proteosome pathway. This event, in turn, releases bound NF-κB complex allowing it to translocate to the nucleus and activate the transcription of specific genes responsible for cell survival (cIAPs, TRAFs, Bcl-xL) or even cell death (Fas).21,22 It has recently been shown that p53-mediated apoptosis requires the activation of NF-κB. However, the induction of NF-κB by p53 is apparently different from that mediated by TNFα, reportedly involves MEK1 and pp90rsk and may be cell-specific.23 Certainly, NF-κB likely plays an important role in modulating virus-mediated apoptosis (see below). For example a recent report demonstrated that NF-κB activity may be regulated by a number of viruses, such as by Epstein-Barr virus (EBV) through LMP-1, resulting in enhanced cell survival following infection.24,25
Apoptosis: mitochondria
Death receptor ligation can also elicit the help of cellular organelles such as the mitochondria. The involvement of the mitochondria in apoptosis first came to light when it was discovered that one of the critical factors required for the activation of caspase-3 was cytochrome c.26 Cytochrome c, released from the mitochondria, binds to a protein called Apaf-1, which turned out to be the homologue of C. elegans ced-4, inducing it to associate with pro-caspase-9.26,27 This event triggers caspase-9 activation and initiates the apoptotic cascade by processing executioner caspase-3. Other mitochondrial proteins such as SMAC/DIABLO or apoptosis inducing factor (AIF) can also be released by mitochondria to promote apoptosis in a caspase-dependent or independent manner, respectively.28,29,30 Direct damage to the mitochondria by viruses such as VSV, perhaps ceramides, ultraviolet (UV) radiation and drugs such as etoposide and staurosporin can induce mitochondrial damage perhaps through ruptured membranes or changes in intramitochondrial and cytosolic pH which modulate caspase activation. In many cases, the mitochondrial inner transmembrane potential (Δψm) collapses, which facilitates the release of cytochrome c.27 A number of proteins that reside in the inner and outer membranes of the mitochondria may form ion channels to regulate the efflux of cytochrome c. One of these regulatory proteins, Bcl-2 turned out to be the homologue of the anti-apoptotic C. elegans gene ced-9. At least 15 members of the Bcl-2 family with either pro-apoptotic properties such as Bax and Bid (activated by caspases) or anti-apoptotic properties such as Bcl-2 and Bcl-xL have been identified.31,32 Significantly, several Bcl-2-like proteins are encoded by many viruses, such as by EBV(BHRF1) and human herpes virus-8 (KSBcl-2) to inhibit mitochondrion triggered cell death.33,34,35,36 It is interesting to note that a recent report indicated that pro-apoptotic members of the Bcl-2 family, such as Bax can also function as survival factors, for example in response to fatal Sindbis virus infection. Such data implies that as yet unknown cell-specific factors likely play an important role in regulating mitochondria-induced apoptosis.37
The interferons (IFNs) and the modulation of immune responses
Interestingly, the activity of NK cells can increase up to 100-fold following exposure to a group of cytokines referred to as the interferons (IFNs).3,4 The IFNs are now considered to play a critical role in innate immunity to viral infection and aside from effectively preventing intracellular viral replication, can also mediate the activation and recruitment of the adaptive immune response. For example, MHC class I and II molecules, required for efficient CTL activity, are potently induced by IFN α/β.4 More recently, the existence of natural IFN producing cells (IPCs or CD4+ CD11c- type 2 dendritic cell, DC, precursors), has been reported which produce high amounts of type I IFN in response to viral infection.38,39,40,41 In addition to cytokines, such as TNFα and IL-3, viral infection was also found to promote the differentiation of IPCs into mature DCs. Virally stimulated mature DCs are then able to initiate adaptive immune responses, including antibody production, by strongly activating antigen-specific naïve T cells and by inducing them to produce type II IFN and IL-10.41
Since there is a delay of approximately 5 days before the initial adaptive immune response takes effect, the IFNs also have a critical role in controlling intracellular viral infection during this period. In the last few years, the importance of IFN antiviral action, including ability to modulate cell death has become increasingly apparent. This has been best emphasized by demonstrating that mice lacking a functional IFN system are extremely sensitive to lethal infection with numerous viruses such as vesicular stomatitis virus (VSV), semliki forest virus, and vaccinia virus infection.42,43,44
The IFNs and apoptosis
The IFNs were discovered in the late 1950s by their capacity to inhibit influenza virus replication.45 After over 40 years of research, however, the exact mechanism of IFN's antiviral activity still remains to be elucidated. What is apparent is that the IFNs embrace two main families referred to as type I (α/β) and type II (γ), although a third member referred to as ω has also been identified.46 Type I IFNs, induced by most cell-types such as leukocytes and fibroblasts, are clustered on the short arm of chromosome 9 and consist of several α genes and pseudogenes, and one β gene. In contrast, type II IFN consists of a single gene on chromosome 12 that is mainly secreted by Th-1 lymphocytes and NK cells. The IFNs can be induced by a number of stimuli including viruses and dsRNA, through mechanisms involving activation of interferon-regulatory factor (IRF-3), NF-κB and perhaps the dsRNA-dependent protein kinase (PKR) and the JNK2 pathway.47,48,49,50 Following secretion from the cells, these cytokines bind to species specific cell-surface receptors and initiate the induction of a number of responsive genes (>30) via signaling through the Janus protein kinase (JAK)/signal transducers and activators of transcription (STAT) pathway.51
Biochemically purified natural or recombinant IFN has little antagonistic effect on the viability of most cell-lines in culture and in many instances IFN treated cells are protected against the replication of a number of viruses such as vesicular stomatitis virus (VSV), vaccinia virus and encephalomyocarditis virus (EMCV).46 Indeed, hepatitis C virus infection and a number of virus-associated malignant diseases have been found to respond well to IFN treatment. However, in vivo administration of IFN as a general antiviral therapy, has been found to be more problematic and less efficient at eliminating virus infection in part due to toxic side effects that presently prevent optimum dose regimes from being implemented.52
Although it is clear that IFN can prevent the replication of a variety of viruses such as VSV without damaging the host, evidence also indicates that these cytokines can also greatly sensitize certain tissue cultured cells to apoptosis, for instance in response to dsRNA and influenza virus.53,54,55,56,57 The mechanism by which IFN appears to sensitize cells to apoptosis appears to be predominantly via the FADD/caspase-8 signaling, since IFN-induced cell death could be prevented by inhibitors of caspase-8, by dominant-negative variants of FADD or in cells lacking FADD, but not those lacking Apaf-1.55,56 Although the pathways governing IFN-induced apoptosis remain to be unraveled it is possible that as yet uncharacterized IFN-induced proteins may govern the regulation of death induced signaling complexes (DISCs) that comprise FADD/caspase-8.55
Currently, one model of IFN-induced antiviral action suggests that only a virus infected cell producing IFN, is likely to undergo apoptosis.57 In this model, IFN is able to function in an autocrine manner to induce the death of the infected cell. However, by also functioning in a paracrine manner, IFN is able to induce an antiviral state in uninfected cells, thus preventing virus replication and virus mediated apoptosis (Figure 1). A slightly different model favored by ourselves, proposes that IFN can establish an antiviral state that can result in either cell death or cell survival, depending on the stimulus, such as type of virus.55 This may explain why IFN is able to actually sensitize certain cells to influenza virus or dsRNA-mediated apoptosis but protect against VSV replication.55 Evidence indicates that IFN is able to inhibit virus replication and virus-induced apoptosis by mechanisms that likely involves the early blocking of viral transcription/translation as well as genome replication which could activate stress-related cell death (see below). Unfortunately, many of these models are based on in vitro studies and such results may be governed by cell-or tissue specific factors. Thus, the importance of IFN-mediated apoptosis in vivo has yet to be confirmed. In part, the mechanism of IFN-regulated cell survival has been complicated by the fact that numerous IFN-induced proteins remain to be characterized or even discovered. DNA array technology indicates that a considerable number of proteins are inducible by IFN.58 Some of these proteins have been implicated as playing a role in regulating apoptosis and are discussed below, though many more await characterization and may lend further light into the complex world of IFN action, including role in apoptosis.

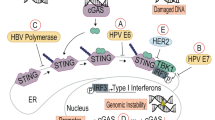
(A) Virus invades the cell and triggers the activation of proteins involved in the induction of interferon (IFN). In response to virus infection, IRF-3 becomes phosphorylated and translocates to the nucleus to cooperate with transcription factors to induce IFN-β. Viral dsRNA also activates JNK-2 and PKR, the latter assisting with IFN production perhaps through activation of the NF-κB pathway. Aside from inducing IFN, IRF-3, PKR and JNK-2 may also contribute towards regulating cell death or survival by inducing the activation of other genes. (B) Type I or II IFN binds to species specific cell surface receptors and through the Jak/STAT pathway induce the transcription of genes harboring IFN-stimulated response elements (ISRE) or gamma-activation sequence (GAS) in their promoters regions, respectively. Genes known to be induced by IFN and to play a role in apoptosis include TRAIL, PKR, IRF-1 and PML
The IFNs and apoptosis: the dsRNA connection
Knowing that influenza virus was a potent inducer of IFN, a number of reports emerged indicating that dsRNA complexes formed in the course of viral multiplication, as an intermediate or side product, were the actual inducers of IFN.59,60 Indeed, very small amounts of dsRNA were reported capable of influencing IFN production. For single stranded viruses, the formation of dsRNA in the infected cell may arise from replicative intermediates i.e. both positive and negative sense RNA. Alternatively, single-stranded RNAs may be capable of forming dsRNA duplexes.46,54 The presence of dsRNA has also been reported for DNA viruses such as vaccinia virus, adenovirus (ADV) and herpes simplex virus (HSV). Evidence indicates that complementary mRNAs from DNA viruses can be produced from genes transcribed in the opposite direction. Alternatively, in the case of viruses with dsRNA genomes, such as reovirus, the inducer of IFN may be the genome itself. Many viruses are known to be potent inducers of IFN, including VSV, Sendai virus and Newcastle disease virus.46 In the early 1970 s when the term apoptosis was first coined, Stewart et al. noted that dsRNA exhibited potent cytotoxic activity.46,47 This effect, which we now know is due to the cell undergoing apoptosis, was noted to be greatly exacerbated by IFN. Thus dsRNA is not only a potent inducer of IFN, but is also a potent inducer of cell death. Evidence now indicates that the two mechanisms may involve the same molecules, such as members of the interferon-regulatory factor (IRF) family and the dsRNA-dependent protein kinase, PKR (see below).

T lymphocytes can directly attack a virus infected cell and induce apoptosis through the perforin/granzyme pathway. Alternatively, T lymphocytes expressing death ligands such as FasL can trigger death receptor mediated apoptosis. In the case of a ligated Fas receptor, the death adapter FADD is directly recruited to activate caspase-8 (shown as triangles) and trigger the apoptotic cascade. This can involve caspase-8 directly activating executioner caspase-3. Alternatively, caspase-8 can recruit Bid, which can influence the efflux of cytochrome c to activate the apoptosome comprising Apaf-1 and pro-caspase-9, leading to cell death. In contrast, ligation of TNFR1, DR-3, 4 or 5 can lead to FADD-mediated cell death through the adapter TRADD. In addition, activation of receptors such as TNFR1 can also stimulate the NF-κB pathway through the TRAFs and RIP, which may contribute towards cell survival through transcriptional upregulation of factors such as the TRAFs themselves and possibly the IAPs. Since viruses are potent activators of apoptosis, these organisms have evolved a variety of strategies to prevent these events. For example, herpesviruses (vFLIPs), poxviruses (CrmA) and insect viruses (p35) encode products to inhibit efficient caspase activation, while gammaherpesviruses (BHRF1, KSBcl-2) inhibit mitochondrial input into the apoptotic process. DNA viruses such as herpes simplex virus (LANA), adenovirus (E1B-55K), papillomavirus (E6) and polyomavirus (SV-40; LT) can also inhibit p53-mediated apoptosis. Viruses such as EBV (LMP1) and human T-lymphotropic virus I (HTLV-1; tax) may also activate the NF-κB pathway to stimulate the expression of cell survival factors
The IFN regulatory factors (IRFs)
The IRFs are a family of transcription regulators originally identified during studies of the transcriptional regulation of the human IFN-β that are induced by dsRNA, viruses or in many cases by IFN itself.61 There are now considered to be more than 10 or so members of this family, including IRFs1-7, 8 (ICSBP), IRF-9 (ISGF3γ/p48) and even virus-encoded forms of the IRF proteins.62,63,64,65 Nearly all IRFs share homology in the N-terminal region and bind similar DNA motifs found in most IFN-inducible genes (IFN stimulated response element (ISRE)), MHC class I promoters (IFN consensus sequence (ICS)) or in the IFN-β promoter (positive regulatory domains (PRD)). Activation of the IFN-β gene has been extensively studied and cellular protein synthesis is not required for this cytokine's induction. A number of distinct cis-acting DNA sequences exist within the promoter of IFN-β gene and in response to dsRNA or certain viruses, several transcription factors are recruited to this region to form a synergistic multicomponent transcriptional enhancer complex.62,63 This enhanceosome consists of ATF2/cJun heterodimers, IRF-1 and 3, HMGI/(Y) the NF-κB heterodimer p50/p65 and CBP/p300.66 Candidate molecules deemed important in initiating IFN-β transcription, in response to virus/dsRNA are IRF-3, JNK2 and NF-κB, although how these are activated by viruses is not yet fully clear. Following expression and secretion, IFN-β then stimulates the induction of ISGF3 and IRF-7 through the Jak/STAT pathway, which in turn contributes to the transcriptional induction of IFN-α genes.67
In response to viral infection or dsRNA, IRF-3 is phosphorylated on specific C-terminal serine and threonine residues causing it to translocate to the nucleus.62,63,65,68 Aside from being involved in the transcription of IFN-β, IRF-3 also induces the activation of a number of target genes including the chemokine RANTES. A recent study demonstrated that expression of constitutively active IRF-3 was toxic to both Jurkat and human embryonic kidney 293 cells. By developing a system in which the expression of constitutively active IRF-3 was inducibly controlled it was established that IRF-3 is a potent mediator of apoptosis.69 Wild-type IRF-3 expression alone was also found to augment paramyxovirus-induced apoptosis whereas expression of a truncated dominant-negative form of IRF-3 blocked virus-induced apoptosis. The mechanism of IRF-3 mediated apoptosis also remains to be determined but may predominantly involve the DISC that comprises caspase-8 since inhibitors of caspase-8 rather than caspase-9 more effectively inhibited cell death.69 Possibly, IRF-3 may induce an unknown gene(s) that contributes to apoptosis via modulation of the DISC and may prove to be an important modulator of virus mediated apoptosis.
Other members of the IRF family have also been shown to play a key role in regulating viral replication, apoptosis and even tumorigenesis.61 IRF-1 and 2 were originally discovered as transcription factors that following activation by dsRNA, viruses and other cytokines, could play a role in the regulation of IFN-β. In addition, IRF-1 (an activator) and IRF-2 (a repressor) are also involved in the regulation of other genes such as IFN-α and MHC class I.70,71 Mice lacking IRF-1 have been shown to be extremely sensitive to viral infection and are susceptible to developing tumors. Support that IRF-1 utilized apoptosis to mediated tumor suppression came from studies using embryonic fibroblasts lacking IRF-1. For example, expression of oncogenic c-ras caused normal fibroblasts but not IRF−/− fibroblasts to undergo death when combined with inhibitors of cellular proliferation or when treated by anticancer drugs or UV irradiation.72,73 Similarly, IRF-1 has been reported to mediate DNA-damage-induced apoptosis in T lymphocytes and to regulate cell cycle arrest and apoptosis in primary hepatocytes in response to IFN-γ signaling. Myeloid cells but not thymocytes derived from ICSBP (IRF-8) deficient mice also show resistance to apoptosis induced by DNA damage.74 Collectively, these data indicate that the IRFs contribute a significant role in the regulation of virus-mediated, host defense related apoptosis.
Interferon regulation: JAKs and STATs
Signal transduction involving the Jaks and STATs are not unique to the IFN-signaling pathway and other cytokines utilize the same or related proteins. However, following IFN-α/β binding to the type I IFN-receptors, JAK1 and TYK2 are recruited and undergo tyrosine phosphorylation. This event leads to the phosphorylation and activation of STAT1 and STAT2, which form heterodimers and translocate to the nucleus where in conjunction with p48 they form the transcription complex ISGF3 which regulates the transcription of genes such as PKR and IRF-1 that contain IFN response elements (ISREs) in their promoters.51 In contrast, stimulation of the IFN-γ receptors recruits JAK1 and JAK2 and the STAT1 homodimers to activate genes containing a gamma-activated sequence (GAS) element.51 Deficiency in JAK1 results in perinatal death and JAK1−/− cells are unresponsive to many types of cytokines while mice lacking STAT1 are unable to resolve infections by many viruses such as VSV.42,44,75 Recently, STAT1 has been shown to act as a TNFR1-signaling molecule to suppress NF-κB activation.76 STAT-1 may also be required for the expression of ICE (caspase-1) which was reported to be necessary for IFN-γ induced apoptosis. Possibly, STAT1 can play a role in efficient caspase expression since low levels of these apoptotic proteins were observed in STAT1 deficient cells. Interestingly, transfected STAT1 variants that could not dimerize retained the ability to restore caspase expression, suggesting that the apoptotic functions of STAT1 differ from those functions of the molecule required to induce gene expression.77,78 Although little is known about the role of the other members of the JAK/STAT family in the regulation of apoptosis, the finding that these signaling proteins may be required for the optimal expression of several pro-apoptotic genes indicates that they may have multiple roles in the cell. In this regard, it is interesting to note that in many instances, viruses appear to cause the constitutive activation of the STATs, though whether this is an inadvertent effect or whether viruses utilize their transcriptional properties to enhance their own replication is not clear. What is more clear is that many viruses such as Sendai virus and adenovirus block this pathway to avoid the deleterious consequences that IFN-induced gene production can have on their replication.79,80,81,82
The dsRNA-dependent protein kinase, PKR
Biochemical characterization of proteins from IFN-treated cells led to the identification of a protein kinase that became phosphorylated when introduced to dsRNA.59 Once activated, the kinase was found to potently inhibit protein synthesis in reticulocyte lysates. This kinase, of approximately 68 kDa in human cells, is now referred to as PKR, for protein kinase RNA-dependent, and is a serine/threonine kinase found mainly, although not exclusively, in the cytoplasm of most cell types, in a latent form.83 Two dsRNA binding domains reside in the amino terminus region of the kinase and interaction with dsRNA modifies the conformation of PKR permitting it to undergo autophosphorylation on a number of sites. This event enables PKR to catalyze the phosphorylation of substrate targets, the most well characterized being the translation initiation factor eIF2α, resulting in the inhibition of protein synthesis in the cell.59 Evidence indicates, however, that PKR may have a plethora of targets other than eIF2α. For example, PKR has been shown to be involved in the regulation of NF-κB perhaps through interacting with one of the IkB kinases, IKKβ.47,49,84 NF-κB is potently activated by dsRNA and plays a key role in the induction of IFN-β production. In addition to these roles, PKR has been reported to function in the tumor suppressor effects of IRF-1, in the modulation of STAT-1 activity, and perhaps in growth factor and cytokine regulated signal transduction pathways.85,86,87,88,89
Attempts to further study the importance of PKR in interferon-mediated anti-viral function initiated a number of studies aimed at overexpressing the kinase in mammalian cells in the absence of other interferon-induced or viral gene products. However, it rapidly became clear that PKR exerted a strong growth suppressive and toxic effect on the host, in the absence of virus infection.90,91 In contrast, murine fibroblasts expressing a catalytically inactive dominant-negative PKR variant were readily isolated and became malignantly transformed.92,93 However, an indication that PKR could mediate apoptosis came from studies with recombinant vaccinia viruses expressing wild-type PKR or a defective PKR variant.94 Viruses expressing the functional gene but not the PKR variant were found to cause the rapid apoptosis of HeLa cells. Other data showed that cells expressing catalytically inactive PKR variants were less resistant to influenza induced apoptosis, and that inactivation of PKR with antisense RNA rendered U937 cells resistant to EMCV induced apoptosis.95,96,97 Studies on murine fibroblasts lacking PKR also indicated resistance to TNF-induced apoptosis.98 Collectively, these data provided powerful evidence that PKR was a pro-apoptotic gene that could mediate dsRNA and virus induced programmed cell death.99
One potential mechanism of PKR-mediated apoptosis could involve the eIF2α pathway and the inhibition of proteins synthesis. For example, recombinant vaccinia viruses expressing an eIF2α variant [Ser 51->Ala] unable to be phosphorylated by PKR was protected from virus-induced apoptosis.84 Conversely, transient expression of a variant of eIF2α, that mimics phosphorylated eIF2α [Ser->51Asp] was reported to cause apoptosis in COS-1 cells.100 It is plausible that PKR mediated inhibition of protein synthesis may deplete the cells of short lived proteins involved in suppressing cell death. However, treatment of cells with levels of cycloheximide capable of inhibiting protein synthesis does not necessarily induce rapid apoptosis unless another apoptotic signal such as Fas or TNF receptor ligation is performed.56 In contrast, PKR-mediated apoptosis may work in a manner similar to the GCN2 paradigm in yeast, where the amount of unphosphorylated eIF2α governs whether translation of certain mRNAs will occur at an authentic initiation codon or at alternative upstream non-coding open reading frames.101
Further clues as to the mechanisms of PKR induced apoptosis has come from cells that inducibly express PKR. In these situations, fibroblasts either undergo apoptosis or are rendered extremely susceptible to treatment with dsRNA or certain viruses such as influenza virus.55,56 Activation of PKR was also found to correlate with the expression of the death receptor Fas which appeared to be able to escape the translational block imposed by PKR. Further investigations by our own laboratory revealed that cells lacking FADD but not the mitochondria related Apaf-1 were resistant to dsRNA and PKR-induced apoptosis, clearly implicating the importance of the FADD/caspase-8 death signaling process in this pathway.55 In addition, HeLa cells expressing dominant-negative FADD variants appear resistant to dsRNA-induced cell death and specific inhibitors of caspase-8 block dsRNA-mediated cell death.102 Presently, the mechanism of PKR-induced Fas expression and FADD-mediated death is unclear. Since PKR has been proposed to play a role in mediating dsRNA-signaling and in activating the NF-κB pathway, it is plausible that PKR may induce the transcription of several death promoting molecules that harbor NF-κB recognition motifs in their promoters.47 In this regard, Fas and FasL are known to be induced by NF-κB.103,104 However, NF-κB is also known to elicit the transcriptional induction of a number of anti-apoptotic genes, such as the cIAPs. One model of PKR-mediated cell death could involve the inhibition of proteins synthesis through eIF2α, and concomitant activation of NF-κB, a combination known to induce cell death. A further interesting observation noted in cells expressing a dominant negative variant of PKR, was that a number of pro-apoptotic genes such as Fas, FADD, TRADD, caspase-8 and Bax and Bad were transcriptionally suppressed.56 The mechanism is again unclear but could involve a gain-of-function effect in which the PKR variant interacts with signaling molecules of the transcriptional regulators of such genes.
Taken together, it is clear that although the exact mechanisms remain to be elucidated, PKR is able to effectively regulate apoptosis in response to viral infection. Moreover, the importance of this kinase in innate immunity has recently been demonstrated using mice deficient in PKR. Such PKR−/− animals were extremely sensitive to intra nasal infection with VSV and influenza virus and exhibited defective IFN-signaling in response to both virus and dsRNA.50 Thus, along with members of the IRF family, PKR is likely an important mediator of dsRNA and virus-induced cell death.
The 2′,5-oligoadenylate/RNaseL pathway
Important dsRNA activated, IFN-inducible anti-viral proteins also include thermostable isozymes that use ATP to produce 2′5′ oligoadenylates with 5′-terminal triphosphate residues (2-5A).60 The 2-5A compounds were found to induce the dimerization and resultant activation of a latent endonuclease, RNAse L that is responsible for degrading RNA.60 Both 2-5A and RNase L are IFN inducible and have been suggested to function in the antiviral effects of IFN against encephalomyocarditis virus (EMCV), reovirus and vaccinia virus.105 The importance of this system in mediating virus-induced apoptosis, however, is unclear. The 2′,5A synthetases, prevalent in most cells, exist in three main forms of molecular weight, 40, 69 and 100 kDa respectively. In contrast, there appears to be only one RNaseL gene product of molecular weight 95 kDa.60,105,106 Inducible overexpression of human RNaseL in murine fibroblasts has been found to induce apoptosis, by an as yet uncharacterized mechanism.107 In addition, dominant-negative variants of RNaseL rendered cells resistant to dsRNA and poliovirus induced apoptosis. Recombinant vaccinia viruses expressing RNaseL have also been found to increase the apoptosis of infected cells, an effect that could be blocked by Bcl-2. Thymocytes and fibroblasts from mice with a targeted disruption of the RNaseL gene are reportedly resistant to a variety of apoptotic stimuli, including FasL, staurosporine and anti-CD3.108 Collectively, it is plausible that following virus infection, the 2′,5A/RNaseL system could contribute to apoptosis by degrading mRNAs for anti-apoptotic proteins or cell survival factors.
IFN and the induction of death receptor and ligands:TRAIL
Recent data has reported the induction of the death ligand TRAIL in both CD4 and CD8 peripheral blood thymocytes following treatment with type I IFN. Type I IFNs were also able to significantly increase the cytotoxic activity of anti-CD3-stimulated thymocytes against carcinomas in a TRAIL-dependent manner.109 Type II IFNs have also been reported to induce TRAIL in fibroblasts but not in thymocytes and to selectively kill virus infected cells.110 At least two receptors can interact with TRAIL, DR-4 and 5, although it is now apparent that TRAIL can also bind to two decoy receptors prevalent in certain cells (DcR1 and DcR2). The regulation of TRAIL may constitute an important component of IFN-mediated host defense against both malignant and viral disease by sensitizing cells to apoptosis.111 Significant excitement originally surrounded the use of TRAIL as an antitumor agent since many types of cancer have been shown to be sensitive to the cytotoxic effects of TRAIL in vitro and in vivo. However, though original reports indicated that most normal cells appear resistant to this death ligand, recent data indicates that TRAIL can potently induce the apoptosis of primary human hepatocytes.112 Although little data exists to indicate that type I IFNs induce other members of the death receptor family, IFN-γ has been reported to induce Fas in glioma, adenocarcinoma and naïve T-cells.113,114
PML, nuclear bodies and viruses
Almost 100% of APL cases are associated with chromosomal translocations [t(15;17)] involving what is referred to as the PML gene and the retinoic acid receptor alpha (RARα), leading to the production of a PML-RAR alpha chimeric protein.115 PML-RAR alpha chimeras are thought to interfere with both the PML and retinoid-X-receptor (RXR) pathways, thus acting as a dominant-negative mutant, the net result of which involves stabilizing co-repressor-histone acetyl transferase complexes. Retinoic acid receptors are nuclear hormone receptors that act as RA-inducible transcriptional activators while PML belongs to a family of RING finger proteins and is IFN-inducible.115 PML is typically concentrated into discrete speckled nuclear structures called nuclear bodies (NBs) or PML oncogenic domains (PODs). Although the function of the NBs remain to be clarified, these structures are known to contain a number of other IFN-inducible genes such as Sp100 and ISG20 as well as other proteins including PIC1/SUMO, CBP, Rb1 and even pro-apoptotic Bax.116 Significantly, the IFNs are able to increase the size of the NBs in cells. Upon RA treatment PML-RARα chimeras re-acquired their natural localization and kill APL cells. It is also noteworthy that PML has also been proposed to control MHC expression and confer resistance to viral infection.117 NB's are frequently targeted by DNA viruses such as herpesviruses and adenovirus, perhaps to disrupt their function.118 Certainly, overexpression of PML in rat or mouse fibroblasts induces rapid cell death and correlates with NB formation.119 Ablation of the PML gene by gene targeting also revealed that PML is essential for the tumor suppressive activity of retinoic acid and is required for the efficient induction of apoptosis induced not only by IFN but also by Fas and TNFα.120 Such data indicate that NBs and their proteins may constitute important components of IFN anti-proliferative and anti-viral action in the cell.
Cellular host defense, p53 and viral counteractions
The IFNs are not the only cellular host defense proteins that can influence apoptosis to prevent viral propagation. For example, the tumor suppressor protein p53 is a transcriptional regulator that can also induce apoptosis in response to virus infection, as well as in response to many other stimuli such as DNA damage or unscheduled DNA synthesis.121 The mechanisms of p53-mediated apoptosis remain to be determined but stabilization and post-translational modifications of p53, involving phosphorylation and acetylation, can trigger p53 mediated cell death.121 p53 target genes involved in apoptosis include Bax, Fas, perhaps TRAIL receptors and even NF-κB.23,122,123,124 Since the induction of apoptosis early after infection would severely limit virus production and reduce or even eliminate the spread of progeny virus in the host, most animal viruses have evolved strategies to evade or delay early apoptosis in an attempt to allow production of high yields of progeny virus. This includes the suppression of IFN action, as is discussed briefly below. Examples include DNA viruses such as human adenovirus and human papillomaviruses (HPV) which need to induce DNA synthesis in quiescent host cells, in order to facilitate their replication.82 In the case of adenovirus, the early region 1A (E1A) product and the E7 product of HPV are largely responsible for this action and consequently inducing p53-mediated apoptosis. As a result, these viruses have also devised strategies for dealing with p53. For example, the early region 1B (E1B) 55 kDa protein (E1B-55K) of ADV inhibits p53, while HPV E6 and SV40 t-antigen performs a similar task.36,125 Recently, HHV8 has also been found to encode a p53 inhibitor referred to as LANA.126
In addition, viruses are also known to encode products that directly block the apoptotic signaling cascade. For example adenovirus also encodes E1B-19K, a product functionally analogous to Bcl-2 that can influence mitochondrial induced cell death as well as apoptosis mediated through Fas and TNFR-1.82 Other viruses that encode products to prevent caspase activation include cowpox virus crmA and insect baculovirus encoded p35. EBV encodes a Bcl-2 homologue, BHRF1 as well as LMP1, which may induce expression of endogenous Bcl-2 and NF-κB.36,125 Finally, viral homologues of inhibitors of apoptosis referred to as Flips have similarly been reported.127,128
Viral inhibition of IFN function
Presumably, since the IFNs are also able to influence cell death, many of the genes encoded by viruses that may block apoptosis could most probably affect IFN-mediated antiviral action. For example, recent data from our laboratory indicates that crmA potently inhibits IFN-induced apoptosis as well as apoptosis induced by dsRNA.102 In addition, the IFN system is also known to be suppressed by many viruses. The induction of IFN itself may be inhibited by preventing the function of the IRF family or PKR. In one example, IRF-3 is known to be inhibited by papilloma virus E7 and HHV-8 is known to encode several versions of the IRFs, which interfere with normal IRF function.62,129
Numerous reports have also emerged over the years to indicate that PKR is a favorite target of viruses.130,131 Indeed, viruses have independently developed many strategies to inhibit PKR. These include synthesizing proteins that sequester dsRNA activators, as in the case of vaccinia virus E3L and reovirus sigma 3 (σ3) as well as producing proteins that impede PKRs ability to bind to target substrates, such as in the case of vaccinia virus encoded K3L.130,131 Adenovirus has devised a different way of inhibiting PKR, namely by producing RNA, VAI, which is a highly structured inhibitor which competes with dsRNA activators to bind to PKR, but fails to cause PKR autophosphorylation. In addition to these viruses, inhibitors of PKR have been reported from studies using influenza virus, HIV-1, hepatitis C virus and poliovirus, amongst others. Should IFN be produced, then viruses such as vaccinia virus are known to encode decoy IFN receptors to interfere with IFN-signaling while adenovirus E1, and Sendai virus viruses target the Jak/STAT pathway.79,82,132 Finally, many IFN-inducible genes themselves, such as the MHC molecules are controlled by viruses such as cytomegalovirus (CMV) following infection of the cell.133
It is not yet clear whether certain viruses, especially non-enveloped, non-lytic viruses, such as ADV require apoptosis to facilitate dissemination of progeny virions. In the case of the Sindbis virus, cellular overexpression of Bcl-2 allowed the virus to establish a persistent infection rather than enter a lytic phase.134,135 A number of in vitro studies, however, indicate that in many cases, lytic viral replication is not affected by regulating apoptosis. Nevertheless, virus induced apoptosis may also have a role in the development of diseases, for example AIDS which correlates with the depletion of CD4+T cells due to the replication of HIV-I.136 In part, HIV-1 induced apoptosis may again involve the FADD/caspase-8 pathway.137,138 For example, the HIV-1 transcription factor Tat product has been reported to play a role in stimulating FasL expression.137 Speculatively, these data infer that some viruses may carefully control both the suppression and induction of apoptosis to manipulate the lytic to latency switch and thus optimize their replication and dissemination.
IFN-mediated apoptosis in the treatment of viral and malignant disease
While the mechanisms of IFN-mediated apoptosis remain to be fully elucidated, it is clearly apparent that these cytokines do have beneficial effect as a therapy against many types of malignant and viral disease. Both type I and II IFN, is able to effectively induce apoptosis in a number of documented malignancies including herpesvirus-associated lymphomas, APL, non-small-cell lung cancer, non-melanoma skin cancer and glioma.139,140,141,142 In addition, IFN has potent effects against hepatitis C virus, currently the major etiological cause of hepatocellular carcinoma, and a viral infection, which effects 140 million people worldwide. The mechanisms of IFNs actions against HCV (as well as HCV-mediated suppression of IFN action) also remain to be elucidated.52 However, IFN's effects against lymphoproliferative diseases are better characterized. These malignancies are associated with gammaherpesvirus infection. Such viruses, which include EBV and HHV-8 are predominantly detected in non-Hodgkin's lymphomas (NHL), malignancies which frequency occur in immunosuppressed organ transplant patients, children with hereditary immunodeficiencies and acquired immunodeficiency (AIDS) patients.143 EBV-positive lymphomas and HHV-8 positive primary effusion lymphomas (PEL) which occur in the setting of severe immunodeficiency of advanced HIV infection, as well as HTLV-1 mediated ATL, have been found to be susceptible to apoptosis mediated by combinations of IFN and the nucleoside analogue AZT.142,144,145 Recent data has further indicated that HHV8 positive PEL cells express high levels of TRAIL in response to IFN treatment. Despite the induction of TRAIL, however, apoptosis was only potentiated when IFN was combined with AZT. The mechanism of apoptosis likely involved activation of the FADD/caspase-8 pathway since the apoptotic effect was inhibited by dominant-negative FADD and specific inhibitors of TRAIL signaling.146 Tumor cells derived from patients resistant to IFN and AZT treatment were found to exhibit high level expression of Bcl-2 or to be defective in TRAIL expression.142 It is possible that the effects of IFN are impeded through EBV encoding inhibitors of apoptosis such as LMP-1 and BHRF1.24,147 In contrast, the increased sensitivities of HHV-8 positive PELs to AZT may be due to the presence of a viral thymidine kinase which may phosphorylate AZT and enhance the apoptotic effect mediated through DISCs. Presumably, the combined effects of IFN and AZT override mechanisms devised by HHV-8 to subvert host defense which may include vIRFs and KSBcl-2.36 Whether these combination therapies work against other forms of viral and malignant disease remains to be seen.
Viruses as oncolytic, apoptosis-inducing agents
Another strategy of killing tumors or infected cells actually uses viruses either to kill cells by introducing suicide genes into the malignant population or by stimulating host defense mechanisms governing the regulation of apoptosis/cytolysis as a result of high level virus replication. Examples of ‘suicide gene’ strategies include the herpes simplex virus thymidine kinase (HSVtk)/gancyclovir system.148 However, viruses, usually retroviruses or adenovirus, have also been developed that can introduce cytokines that stimulate the adaptive immune response, and that may also stimulate apoptosis as described earlier. It is thought that HSVtk phosphorylates the nontoxic drug, GCV, causing chain termination and single-stranded breaks upon incorporation into DNA, inducing apoptosis. Interestingly, HSVtk/GCV treatment was found to induce p53 accumulation and the increased expression of Fas, TNFR1, leading to FasL-independent, Fas receptor oligermirization-mediated death.149 Such cells were also strongly sensitized to FasL and TRAIL mediated cell death and was found to be mediated through FADD. This effect is similar to the mechanisms of cell death induced by IFN and AZT and one plausible mechanism to explain the death of PEL cells by IFN and AZT could involve HHV-8tk activating AZT to induce DNA damage and modulate DISC associated cell-death.149
Finally, although tumors are likely defective in a number of key host defense pathways, a number of reports have indicated that viruses can be used to directly induce the cytolysis of malignant cells in vivo and may be useful as a treatment against cancer.148 Recently, we and others showed that VSV could not replicate efficiently in cells unless PKR or the IFN-pathway was defective.150,151 If these key host defense pathways were impaired then VSV was found to rapidly replicate to high levels and induce the apoptosis of the host cell. Of interest is that it is well documented that VSV replicates extremely efficiently in numerous types of tissue cultured cell-lines, possibly inferring that host defense mechanisms involving the IFNs in these cells are down regulated.50 Our data further indicated that VSV could induce apoptosis very effectively in many tumorigenic cells, in vitro, including those cells defective in p53, myc and ras or overexpressing Bcl-2.151,156 Importantly, VSV could also induce apoptosis of xenografted tumors in vivo, greatly reducing the growth of the tumor mass.156 Other virus related therapies being used to kill tumor cells include adenovirus, which replicates in cells defective in p53 and reovirus, which similarly replicates in cells harboring suppressed PKR.152,153
Summary
Although unraveling the mechanisms of cellular defense and apoptosis has proven to be a complex task, it has become clear that the IFNs are critical for coordinating the adaptive immune response and ultimately for effectively protecting the host. In addition, aside from playing a role in shaping T-cell responses, numerous studies have now implicated that a variety of IFN regulated genes, such as members of the IRF family, PKR, TRAIL, PML utilize apoptosis to exert some of their antiviral and tumor suppressor function. Although many studies have indicated that the IFNs are positive mediators of cell death, however, there are instances where both type I and type II IFNs may actually prevent apoptosis. For example, IFN-α/β has been shown to act as survival factors for activated T cells which may prevent such cells from undergoing cell death during infections.154 In addition, type II IFN has been shown to prevent macrophages from undergoing apoptosis by blocking the cell cycle via the induction of p21waf1.155 Therefore, through the induction of their many genes, it is likely that the IFNs can exert multiple effects in the cell and can concomitantly elicit a protective antiviral, antiproliferative or even antiapoptotic state as well as prime cells for apoptosis. The balance between life and death presumably depends on possible co-stimuli, such as type of virus or drug, cytokine, as well as type of cell or tissue and even perhaps the nature of the malignant disease. Comprehending the mechanisms of IFN-mediated action, including role in regulating apoptosis, however, should greatly facilitate our knowledge of viral pathogenesis as well as potentially generate new strategies for the treatment of viral and malignant disease.
Abbreviations
- NK:
-
natural killer
- IRFs:
-
interferon regulatory factors
- PML:
-
promyelocytic leukemia gene
- CTLs:
-
cytotoxic T cells
- PKR:
-
dsRNA-dependent protein kinase
- TNF:
-
tumor necrosis factor
References
Biron CA . 1999 Initial and innate responses to viral infections–pattern setting in immunity or disease. Curr. Opin. Microbiol. 2: 374–381
Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H and Zinkernagel RM . 1999 Control of early viral and bacterial distribution and disease by natural antibodies. Science 286: 2156–2159
Ahmed R and Biron CA (eds.). . Immunity to Viruses Lippincott-Raven, Philadelphia 1998
Biron CA, Nguyen KB, Pien GC, Cousens LP and Salazar-Mather TP . 1999 Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17: 189–220
Podack ER . 1999 How to induce involuntary suicide: the need for dipeptidyl peptidase I [comment]. Proc. Natl. Acad. Sci. USA 96: 8312–8314
Kerr JF, Wyllie AH and Currie AR . 1972 Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26: 239–257
Raff M . 1998 Cell suicide for beginners [news]. Nature 396: 119–122
Horvitz HR . 1999 Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 59: 1701s–1706s
Salvesen GS . 1999 Programmed cell death and the caspases. Apmis 107: 73–79
Froelich CJ, Dixit VM and Yang X . 1998 Lymphocyte granule-mediated apoptosis: matters of viral mimicry and deadly proteases. Immunol. Today 19: 30–36
Ashkenazi A and Dixit VM . 1998 Death receptors: signaling and modulation. Science 281: 1305–1308
Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y and Nagata S . 1991 The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66: 233–243
Chinnaiyan AM, O'Rourke K, Tewari M and Dixit VM . 1995 FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505–512
Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME and Dixit VM . 1996 FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85: 817–827
Baker SJ and Reddy EP . 1998 Modulation of life and death by the TNF receptor superfamily. Oncogene 17: 3261–3270
Wang CY, Mayo MW, Korneluk RG, Goeddel DV and Baldwin Jr AS . 1998 NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281: 1680–1683
Arch RH, Gedrich RW and Thompson CB . 1998 Tumor necrosis factor receptor-associated factors (TRAFs)–a family of adapter proteins that regulates life and death. Genes Dev. 12: 2821–2830
Deveraux QL and Reed JC . 1999 IAP family proteins–suppressors of apoptosis. Genes Dev. 13: 239–252
Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M and Liu Z . 2000 The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity 12: 419–429
Pimentel-Muinos FX and Seed B . 1999 Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity 11: 783–793
Foo SY and Nolan GP . 1999 NF-kappaB to the rescue: RELs, apoptosis and cellular transformation. Trends Genet. 15: 229–235
Chen C, Edelstein LC and Gelinas C . 2000 The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell. Biol. 20: 2687–2695
Ryan KM, Ernst MK, Rice NR and Vousden KH . 2000 Role of NF-kappaB in p53-mediated programmed cell death. Nature 404: 892–897
Feuillard J, Schuhmacher M, Kohanna S, Asso-Bonnet M, Ledeur F, Joubert-Caron R, Bissieres P, Polack A, Bornkamm GW and Raphael M . 2000 Inducible loss of NF-kappaB activity is associated with apoptosis and Bcl-2 down-regulation in Epstein-Barr virus-transformed B lymphocytes. Blood 95: 2068–2075
Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J and Kieff E . 2000 NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 97: 6055–6060
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X . 1997 Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489
Green DR and Reed JC . 1998 Mitochondria and apoptosis. Science 281: 1309–1312
Verhagen AM, PG E, Pakusch LM, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ and Vaux DL . 2000 Identifcation of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53
Du C, Fang M, Li Y, Li L and Wang X . 2000 Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM and Kroemer G . 1999 Molecular characterization of mitochondrial apoptosis-inducing factor [see comments]. Nature 397: 441–446
Luo X, Budihardjo I, Zou H, Slaughter C and Wang X . 1998 Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Adams JM and Cory S . 1998 The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326
Hardwick JM . 1997 Virus-induced apoptosis. Adv. Pharmacol. 41: 295–336
Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS and Hardwick JM . 1997 A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 94: 690–694
Henderson S, Huen D, Rowe M, Dawson C, Johnson G and Rickinson A . 1993 Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 90: 8479–8483
Roulston A, Marcellus RC and Branton PE . 1999 Viruses and apoptosis. Annu. Rev. Microbiol. 53: 577–628
Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S and Hardwick JM . 1999 Inhibition of virus-induced neuronal apoptosis by Bax. Nat. Med. 5: 832–835
Cella M, Salio M, Sakakibara Y, Langen H, Julkunen I and Lanzavecchia A . 1999 Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 189: 821–829
Verdijk RM, Mutis T, Esendam B, Kamp J, Melief CJ, Brand A and Goulmy E . 1999 Polyriboinosinic polyribocytidylic acid (poly(I:C)) induces stable maturation of functionally active human dendritic cells. J. Immunol. 163: 57–61
Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S and Liu YJ . 1999 The nature of the principal type 1 interferon-producing cells in human blood [see comments]. Science 284: 1835–1837
Kadowaki N, Antonenko S, Lau JY and Liu YJ . 2000 Natural interferon alpha/beta-producing cells link innate and adaptive immunity [In Process Citation]. J. Exp. Med. 192: 219–226
Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M and Schreiber RD . 1996 Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84: 431–442
Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM and Aguet M . 1994 Functional role of type I and type II interferons in antiviral defense. Science 264: 1918–1921
Durbin JE, Hackenmiller R, Simon MC and Levy DE . 1996 Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84: 443–450
Isaacs A and Lindemann J . 1957 Virus Interference: The interferons. Prog. Roy. Soc. Biol. (Lond.) 147: 258–273.
Stark GR, Kerr IM, Williams BR, Silverman RH and Schreiber RD . 1998 How cells respond to interferons. Annu. Rev. Biochem. 67: 227–264
Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J and Karin M . 1999 JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity 11: 721–731
Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C and Williams BR . 1997 Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 16: 406–416
Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M and Weissmann C . 1995 Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 14: 6095–6106
Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR and Barber GN . 2000 Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13: 129–141
Darnell Jr JE . 1998 Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. J. Interferon Cytokine Res. 18: 549–554
Polyak SJ and Gerotto M . 2000 The molecular basis for responsiveness to anti-viral therapy in hepatitis C. Forum (Genova) 10: 46–58
Stewart WED, De Clercq E, Billiau A, Desmyter J and De Somer P . 1972 Increased susceptibility of cells treated with interferon to the toxicity of polyriboinosinic-polyribocytidylic acid. Proc. Natl. Acad. Sci. USA 69: 1851–1854
Stewart WED, De Clercq E and De Somer P . 1973 Specificity of interferon-induced enhancement of toxicity for double- stranded ribonucleic acids. J. Gen. Virol. 18: 237–246
Balachandran S, Roberts PC, Kipperman T, Bhalla KN, Compans RW, Archer DR and Barber GN . 2000 Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 74: 1513–1523
Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K and Barber GN . 1998 Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 17: 6888–6902
Tanaka N, Sato M, Lamphier MS, Nozawa H, Oda E, Noguchi S, Schreiber RD, Tsujimoto Y and Taniguchi T . 1998 Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 3: 29–37
Der SD, Zhou A, Williams BR and Silverman RH . 1998 Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95: 15623–15628
Lengyel P . 1987 Double-stranded RNA and interferon action. J. Interferon Res. 7: 511–519
Kerr IM . 1987 The 2-5A system: a personal view. J. Interferon Res. 7: 505–510
Taniguchi T, Tanaka N and Taki S . 1998 Regulation of the interferon system, immune response and oncogenesis by the transcription factor interferon regulatory factor-1. Eur. Cytokine Netw. 9: 43–48
Pitha PM, Au WC, Lowther W, Juang YT, Schafer SL, Burysek L, Hiscott J and Moore PA . 1998 Role of the interferon regulatory factors (IRFs) in virus-mediated signaling and regulation of cell growth. Biochimie 80: 651–658
Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M and Lin R . 1999 Triggering the interferon response: the role of IRF-3 transcription factor. J. Interferon Cytokine Res. 19: 1–13
Nguyen H, Hiscott J and Pitha PM . 1997 The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 8: 293–312
Lin R, Heylbroeck C, Pitha PM and Hiscott J . 1998 Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18: 2986–2996
Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM and Maniatis T . 1998 Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo [published erratum appears in Mol Cell 1999 Jun;3(6):813]. Mol. Cell 1: 507–518.}
Marie I, Durbin JE and Levy DE . 1998 Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17: 6660–6669
Schafer SL, Lin R, Moore PA, Hiscott J and Pitha PM . 1998 Regulation of type I interferon gene expression by interferon regulatory factor-3. J. Biol. Chem. 273: 2714–2720
Heylbroeck C, Balachandran S, Servant MJ, DeLuca C, Barber GN, Lin R and Hiscott J . 2000 The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 74: 3781–3792
Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW and Taniguchi T . 1994 Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 77: 829–839
Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S and Taniguchi T . 1995 An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen- activated T lymphocytes. Nature 376: 596–599
Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S and Taniguchi T . 1997 DNA damage-induced apoptosis and Ice gene induction in mitogenically activated T lymphocytes require IRF-1}. Leukemia 11. Suppl 3: 439–440
Kano A, Haruyama T, Akaike T and Watanabe Y . 1999 IRF-1 is an essential mediator in IFN-gamma-induced cell cycle arrest and apoptosis of primary cultured hepatocytes. Biochem. Biophys. Res. Commun. 257: 672–677
Gabriele L, Phung J, Fukumoto J, Segal D, Wang IM, Giannakakou P, Giese NA, Ozato K and Morse III HC . 1999 Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein [published erratum appears in J Exp Med 1999 Dec 6;190(11):following 1722]. J. Exp. Med. 190: 411–421
Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson Jr EM and Schreiber RD . 1998 Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 93: 373–383
Wang Y, Wu TR, Cai S, Welte T and Chin YE . 2000 Stat1 as a component of tumor necrosis factor alpha receptor 1-TRADD signaling complex to inhibit NF-kappaB activation. Mol. Cell. Biol. 20: 4505–4512
Chin YE, Kitagawa M, Kuida K, Flavell RA and Fu XY . 1997 Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol. 17: 5328–5337
Kumar A, Commane M, Flickinger TW, Horvath CM and Stark GR . 1997 Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases [see comments]. Science 278: 1630–1632
Komatsu T, Takeuchi K, Yokoo J, Tanaka Y and Gotoh B . 2000 Sendai virus blocks alpha interferon signaling to signal transducers and activators of transcription [In Process Citation]. J. Virol. 74: 2477–2480
Chen H, Lee JM, Wang Y, Huang DP, Ambinder RF and Hayward SD . 1999 The Epstein-Barr virus latency BamHI-Q promoter is positively regulated by STATs and Zta interference with JAK/STAT activation leads to loss of BamHI-Q promoter activity. Proc. Natl. Acad. Sci. USA 96: 9339–9344
Bovolenta C, Camorali L, Lorini AL, Ghezzi S, Vicenzi E, Lazzarin A and Poli G . 1999 Constitutive activation of STATs upon in vivo human immunodeficiency virus infection. Blood 94: 4202–4209
Wold WS, Doronin K, Toth K, Kuppuswamy M, Lichtenstein DL and Tollefson AE . 1999 Immune responses to adenoviruses: viral evasion mechanisms and their implications for the clinic. Curr. Opin. Immunol. 11: 380–386
Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR and Hovanessian AG . 1990 Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62: 379–390
Gil J, Alcami J and Esteban M . 1999 Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Mol. Cell. Biol. 19: 4653–4663
Mundschau LJ and Faller DV . 1995 Platelet-derived growth factor signal transduction through the interferon-inducible kinase PKR. Immediate early gene induction. J. Biol. Chem. 270: 3100–3106
Cuddihy AR, Li S, Tam NW, Wong AH, Taya Y, Abraham N, Bell JC and Koromilas AE . 1999 Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol. Cell. Biol. 19: 2475–2484
Wong AH, Tam NW, Yang YL, Cuddihy AR, Li S, Kirchhoff S, Hauser H, Decker T and Koromilas AE . 1997 Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 16: 1291–1304
Kirchhoff S, Koromilas AE, Schaper F, Grashoff M, Sonenberg N and Hauser H . 1995 IRF-1 induced cell growth inhibition and interferon induction requires the activity of the protein kinase PKR. Oncogene 11: 439–445
Chatterjee-Kishore M, Wright KL, Ting JP and Stark GR . 2000 How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 19: 4111–4122
Chong KL, Feng L, Schappert K, Meurs E, Donahue TF, Friesen JD, Hovanessian AG and Williams BR . 1992 Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 11: 1553–1562
Barber GN, Wambach M, Wong ML, Dever TE, Hinnebusch AG and Katze MG . 1993 Translational regulation by the interferon-induced double-stranded-RNA-activated 68-kDa protein kinase. Proc. Natl. Acad. Sci. USA 90: 4621–4625
Koromilas AE, Roy S, Barber GN, Katze MG and Sonenberg N . 1992 Malignant transformation by a mutant of the IFN-inducible dsRNA- dependent protein kinase. Science 257: 1685–1689
Meurs EF, Galabru J, Barber GN, Katze MG and Hovanessian AG . 1993 Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 90: 232–236
Lee SB and Esteban M . 1994 The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology 199: 491–496
Takizawa T, Ohashi K and Nakanishi Y . 1996 Possible involvement of double-stranded RNA-activated protein kinase in cell death by influenza virus infection. J. Virol. 70: 8128–8132
Yeung MC, Liu J and Lau AS . 1996 An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc. Natl. Acad. Sci. USA 93: 12451–12455
Williams BR . 1999 PKR; a sentinel kinase for cellular stress. Oncogene 18: 6112–6120
Der SD, Yang YL, Weissmann C and Williams BR . 1997 A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl. Acad. Sci. USA 94: 3279–3283
Jagus R, Joshi B and Barber GN . 1999 PKR, apoptosis and cancer. Int. J. Biochem. Cell Biol. 31: 123–138
Srivastava SP, Kumar KU and Kaufman RJ . 1998 Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 273: 2416–2423
Hinnebusch AG . 1994 The eIF-2 alpha kinases: regulators of protein synthesis in starvation and stress. Semin. Cell Biol. 5: 417–426
Barber GN . unpublished.
Chan H, Bartos DP and Owen-Schaub LB . 1999 Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol. Cell. Biol. 19: 2098–2108
Kuhnel F, Zender L, Paul Y, Tietze MK, Trautwein C, Manns M and Kubicka S . 2000 NFkappaB mediates apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis. J. Biol. Chem. 275: 6421–6427
Zhou A, Hassel BA and Silverman RH . 1993 Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72: 753–765
Rebouillat D and Hovanessian AG . 1999 The human 2′,5′-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 19: 295–308
Diaz-Guerra M, Rivas C and Esteban M . 1997 Activation of the IFN-inducible enzyme RNase L causes apoptosis of animal cells. Virology 236: 354–363
Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C and Silverman RH . 1997 Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16: 6355–6363
Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K and Yagita H . 1999 Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 189: 1451–1460
Sedger LM, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, Cosman D and Wiley SR . 1999 IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 163: 920–926
French LE and Tschopp J . 1999 The TRAIL to selective tumor death [news; comment]. Nat. Med. 5: 146–147
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR and Strom SC . 2000 Apoptosis induced in normal human hepatocytes by tumor necrosis factor- related apoptosis-inducing ligand [see comments]. Nat. Med. 6: 564–567
Roth W, Wagenknecht B, Dichgans J and Weller M . 1998 Interferon-alpha enhances CD95L-induced apoptosis of human malignant glioma cells. J. Neuroimmunol. 87: 121–129
Tsushima H, Imaizumi Y, Imanishi D, Fuchigami K and Tomonaga M . 1999 Fas antigen (CD95) in pure erythroid cell line AS-E2 is induced by interferon-gamma and tumor necrosis factor-alpha and potentiates apoptotic death. Exp. Hematol. 27: 433–440
Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, Grosveld F, Pandolfi PP, Pelicci PG and Dejean A . 1995 The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 11: 871–876
Grotzinger T, Sternsdorf T, Jensen K and Will H . 1996 Interferon-modulated expression of genes encoding the nuclear-dot- associated proteins Sp100 and promyelocytic leukemia protein (PML). Eur. J. Biochem. 238: 554–560
Zheng P, Guo Y, Niu Q, Levy DE, Dyck JA, Lu S, Sheiman LA and Liu Y . 1998 Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature 396: 373–376
Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M and Parkinson J . 1998 The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72: 6581–6591
Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC and de The H . 1998 PML induces a novel caspase-independent death process [see comments]. Nat. Genet. 20: 259–265
Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R and Pandolfi PP . 1998 PML is essential for multiple apoptotic pathways [see comments]. Nat. Genet. 20: 266–272
Gottlieb TM and Oren M . 1998 p53 and apoptosis. Semin. Cancer Biol. 8: 359–368
Yin C, Knudson CM, Korsmeyer SJ and Van Dyke T . 1997 Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385: 637–640
Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G and el-Deiry WS . 1997 KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene [letter]. Nat. Genet. 17: 141–143
Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R and Weissberg P . 1998 Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis [see comments]. Science 282: 290–293
Teodoro JG and Branton PE . 1997 Regulation of apoptosis by viral gene products. J. Virol. 71: 1739–1746
Friborg Jr J, Kong W, Hottiger MO and Nabel GJ . 1999 p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402: 889–894
Chaudhary PM, Jasmin A, Eby MT and Hood L . 1999 Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18: 5738–5746
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME and Tschopp J . 1997 Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386: 517–521
Ronco LV, Karpova AY, Vidal M and Howley PM . 1998 Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 12: 2061–2072
Katze MG . 1995 Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol. 3: 75–78
Samuel CE . 1993 The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 268: 7603–7606
Mathews MB . 1995 Structure, function, and evolution of adenovirus virus-associated RNAs. Curr. Top. Microbiol. Immunol. 199: 173–187
Huard B and Fruh K . 2000 A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur. J. Immunol. 30: 509–515
Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE and Hardwick JM . 1993 Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 361: 739–742
Hardwick JM . 1998 Viral interference with apoptosis. Semin. Cell Dev. Biol. 9: 339–349
Pantaleo G and Fauci AS . 1995 Apoptosis in HIV infection [comment]. Nat. Med. 1: 118–120
Peter ME, Ehret A, Berndt C and Krammer PH . 1997 AIDS and the death receptors. Br. Med. Bull. 53: 604–616
Bartz SR and Emerman M . 1999 Human immunodeficiency virus type 1 Tat induces apoptosis and increases sensitivity to apoptotic signals by up-regulating FLICE/caspase-8. J. Virol. 73: 1956–1963
Bazarbachi A, El-Sabban ME, Nasr R, Quignon F, Awaraji C, Kersual J, Dianoux L, Zermati Y, Haidar JH, Hermine O and de The H . 1999 Arsenic trioxide and interferon-alpha synergize to induce cell cycle arrest and apoptosis in human T-cell lymphotropic virus type I-transformed cells. Blood 93: 278–283
Lokshin A, Mayotte JE and Levitt ML . 1995 Mechanism of interferon beta-induced squamous differentiation and programmed cell death in human non-small-cell lung cancer cell lines. J. Natl. Cancer Inst. 87: 206–212
Rodriguez-Villanueva J and McDonnell TJ . 1995 Induction of apoptotic cell death in non-melanoma skin cancer by interferon-alpha. Int. J. Cancer 61: 110–114
Lee RK, Cai JP, Deyev V, Gill PS, Cabral L, Wood C, Agarwal RP, Xia W, Boise LH, Podack E and Harrington Jr WJ . 1999 Azidothymidine and interferon-alpha induce apoptosis in herpesvirus-associated lymphomas. Cancer Res. 59: 5514–5520
Feigal EG . 1998 AIDS-associated malignancies: research perspectives. Biochimica et Biophysica Acta 1423: C1–C9
Gill PS, Harrington Jr W, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA, Bernstein-Singer M, Espina BM, Cabral L, Allen S et al.1995 Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N. Engl. J. Med. 332: 1744–1748
Harrington Jr WJ, Cabral L, Cai JP, Chan ASS and Wood C . 1996 Azothymidine and interferon-alpha are active in AIDS-associated small non-cleaved cell lymphoma but not large-cell lymphoma [letter]. Lancet 348: 833
Toomey NL, Wood C, Deyer VV, Boise LH, Scott, Lui L, Podack E, Barber GN and Harrington Jr WJ . 2001 The mechanism of IFN targeted therapy against HHV-8 associated lymphoma involves the induction of TRAIL-mediated apoptosis. (submitted).
Knecht H, Berger C, McQuain C, Rothenberger S, Bachmann E, Martin J, Esslinger C, Drexler HG, Cai YC, Quesenberry PJ and Odermatt BF . 1999 Latent membrane protein 1 associated signaling pathways are important in tumor cells of Epstein-Barr virus negative Hodgkin's disease. Oncogene 18: 7161–7167
Steele TA . 2000 Recent developments in the virus therapy of cancer. Proc. Soc. Exp. Biol. Med. 223: 118–127
Beltinger C, Fulda S, Kammertoens T, Meyer E, Uckert W and Debatin KM . 1999 Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 96: 8699–8704
Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N and Bell JC . 2000 Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6: 821–825
Balachandran S and Barber GN . 2000 Vesicular stomatitis virus therapy of tumors. IUBMB Life 50: 135–138
Coffey MC, Strong JE, Forsyth PA and Lee PW . 1998 Reovirus therapy of tumors with activated Ras pathway [see comments]. Science 282: 1332–1334
Alemany R, Balague C and Curiel DT . 2000 Replicative adenoviruses for cancer therapy. Nat. Biotechnol. 18: 723–727
Marrack P, Kappler J and Mitchell T . 1999 Type I interferons keep activated T cells alive. J. Exp. Med. 189: 521–530
Xaus J, Cardo M, Valledor AF, Soler C, Lloberas J and Celada A . 1999 Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity 11: 103–113.
Balachandran S, Porosnicu M and Barber GN . 2001 The oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras or Myc function and involves the induction of apoptosis. J. Virol. (in press)
Acknowledgements
Appreciation is expressed to Laura Saunders for assistance with this review.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J M Hardwick
Rights and permissions
This article is cited by
-
MiR-3976 regulates HCT-8 cell apoptosis and parasite burden by targeting BCL2A1 in response to Cryptosporidium parvum infection
Parasites & Vectors (2023)
-
The expanded inhibitor of apoptosis gene family in oysters possesses novel domain architectures and may play diverse roles in apoptosis following immune challenge
BMC Genomics (2022)
-
Urolithin A inhibits enterovirus 71 replication and promotes autophagy and apoptosis of infected cells in vitro
Archives of Virology (2022)
-
mTORC2 confers neuroprotection and potentiates immunity during virus infection
Nature Communications (2021)
-
Molecular basis of IRGB10 oligomerization and membrane association for pathogen membrane disruption
Communications Biology (2021)
