
Abstract
Study design:
Case report.
Objectives:
We report on a 52-year-old male patient with tumefactive demyelination of the spinal cord.
Setting:
University Hospital and Jessenius Faculty of Medicine, Comenius University, Martin, Slovakia.
Background:
In contrast to relatively frequent tumefactive fulminant lesions in the brain, cases affecting the spinal cord in isolation have been reported less frequently.
Methods:
Description of the case report.
Results:
Clinical, neuroradiological and necropsy findings are described in a 52-year-old man with tumefactive fulminant demyelination of the spinal cord. Progression of the demyelination process produced paraplegia, mild paresis of the right upper limb, neurogenic bladder and sensitive loss over 2 weeks. MRI scans revealed several ovoid lesions in cervical segments and tumefactive T2-hyperintense signals with oedema and post-contrast enhancement located in thoracic segments Th3 to Th6. Cerebrospinal fluid (CSF) examination displayed lymphomonocytic pleocytosis with normal proteinorhachia, positive CSF oligoclonal IgG bands (OCB) and elevated IgG index (1.55). Serum anti-AQP4-Ab was not tested. Stored frozen CSF samples were later repeatedly examined with negative findings of anti-AQP4-Ab. Treatment with high-dose methylprednisolon and plasma exchange had limited effect. Immunosuppressive medication was interrupted because of an acute urinary infection. The patient died suddenly because of pulmonary embolism as a secondary complication. Histopathology of the spinal cord confirmed active demyelination. We considered that tumefactive demyelination could be a variant of neuromyelitis optica.
Conclusion:
Our case could be anti-AQP4-Ab-negative longitudinally extensive transverse myelitis, a variant of neuromyelitis optica.
Similar content being viewed by others

Introduction
Case report
We report on a 52-year-old male patient with rapid worsening of motor functions. On admission he had a 5-day history of sensory deficit, including paresthesias of distal limbs with irradiation to the upper thoracic segments. Motor disability was also gradually worsening. Over the first 10 days his neurological state had deteriorated further and he became paraplegic. He was not able to sit up or rotate his body over when lying because of truncal muscle paresis. In addition, his right upper limb became mildly weaker. Sensory examination showed a complete deficit up to the Th4 level and paresthesias of the right upper limb. He was suffering from a complete neurogenic bladder.
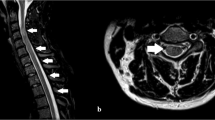
Magnetic resonance imaging (MRI) scans of his brain with gadolinium were negative; repeated exploration showed discrete, rare, nonspecific lesions (Figures 1a–c). MRI of his spinal cord revealed tumefactive longitudinally extensive T2-hyperintense signals with oedema located in thoracic segments Th3 to Th6, and several lesions in the cervical cord from C1 to C6 (Figures 1d–f).

T2-weighted MRI of the brain showing a few small nonspecific lesions in subcortical frontal and occipital regions (a). T1-weighted sagittal and contrast-enhanced T1-weighted MRI of the brain were negative (b, c). T1-weighted (d) and contrast-enhanced T1 (e) MRI of the spinal cord at baseline of the disease showed negative results. T2-weighted spinal cord MRI revealed small hyperintense lesions with irregular pattern in C3 and C6 segments, and T2-hyperintense signal with oedema located in thoracic segments Th3 to Th6, indicating a tumefactive lesion (f).
Evoked potentials showed demyelination of all somatosensory tracts. Visual tracts were tested only by Flash-VEP and results were borderline; we evaluated them as negative.
Review of the system was negative and routine laboratory tests were within normal limits, with no signs of whole-body inflammation. Cerebrospinal fluid (CSF) showed pleocytosis (290 lymphocytes and monocytes) with normal proteinorhachia. Positive CSF oligoclonal IgG bands (OCB), class2,4 were detected and the IgG index was elevated (1.55). Serial examination of anti-AQP4-Ab from different CSF samples showed negative results (semiquantitative indirect immunofluorescence).
Treatment started immediately after admission to hospital, 15 days after symptom onset (4.75 g of methylprednisolon over 10 days and after a short pause 1 g methylprednisolon daily for 5 days). Peroral methylprednisolon at 8 mg per day was continued. Ceftriaxone and acyclovir were administered for the first 7 days as empirical treatment until laboratory results revealed no active neuroinfection. Because of failure to recognize the efficacy of the provided treatment, plasma exchange was indicated (2000 ml volume in 5 repeats in 10 days). At the end of those procedures, the patient was able to sit up on the bed with assistance, but severe paresis and sensory loss of distal limbs remained almost unchanged. At that time his expanded disability status scale value was 8.5, confirming severe disability. The patient continued physiotherapy.
Serial MRI revealed rapid enlargement and post-contrast enhancement (Figures 1d–f and Figures 2a–c). The last MRI, performed after the plasmapheresis treatment, documented progression of the spinal demyelination process. However, the patient became febrile. Immunosuppressive treatment was interrupted for a while. Unfortunately, the patient died 5 weeks after the onset because of cardiorespiratory failure that followed massive pulmonary embolism, despite continual use of anti-aggregants.

(a) T1-weighted and contrast-enhanced MRI of the spinal cord after 4 weeks revealed progression, and enlargement of the cervical lesions from C1 to C6. In thoracic segments Th3 to Th6 T1-hyperintense signal indicates a tumefactive lesion. Pathology of the spinal cord showed extensive demyelination of the white matter in dorsal and lateral roots (positive luxol fast blue) and a sharp demarcation between the perilesional white matter and the lesion edge. (b) T2-weighted MRI of the spinal cord after 4 weeks showed progression of demyelination in the cervical segments; the lesions were ovoid, longitudinal and dorsally oriented. There was T1-hyperintense signal in the upper thoracic segments. In histopathology protein-laden macrophages signed by CD68 antigen were frequent in demyelinated lesions. Inflammatory infiltrates (granulocytes and eosinophils) were located within the lesion, being concentrated around vessels. Thickening of the vessel wall was occasionally found in inflamed areas. (c) T2-weighted MRI of the thoracic spinal cord after 4 weeks showed enlargement of the tumefactive demyelination (Th3 to Th5). Histopathology found axons to be preserved. Necrosis and cavitation in the spinal cord, involving both grey and white matter, were not present.
Pathology of the spinal cord showed extensive demyelination of white matter in dorsal and lateral roots (positive luxol fast blue) (Figure 2a). We found a sharp demarcation between the perilesional white matter and the lesion edge (Figure 2a). Protein-laden macrophages signed by CD68 antigen were frequent in the demyelinated lesions (Figure 2b). Inflammatory infiltrates (granulocytes and eosinophils) were located within the lesion, being concentrated around vessels (Figure 2b). Thickening of the vessel wall was occasionally found in inflamed areas (Figure 2b). Reactive astrogliosis was found to be present (Figure 2c). Axons seemed preserved (Figure 2c). Necrosis and cavitation in the spinal cord, involving both grey and white matter, typical for neuromyelitis optica (NMO), were not present. Histology of tissue samples from periventricular and subependymal zones of the brain was normal. Hypothalamic, brain stem and spinal canal lesions were not seen.
Discussion
Development of the patient’s disability was indisputably rapid and we found him to have been negligent towards initial treatment. We are aware of a lack of knowledge about the final outcome of the patient because of his unexpected death. The features of patient disease evoke several hypotheses about the phenotype of his disease.
Progressive worsening of neurological function in our patient has allowed us to name the course ‘fulminant’ tumefactive subtype of demyelination of the spinal cord. Our results interfere with a recently published review article that describes a fulminant demyelinating disease as a range of inflammatory demyelinating disorders associated with rapid progression to disability within several days to weeks, culminating in the need for hospital admission and aggressive therapy for an acute attack.5
Among fulminant demyelinating diseases, acute disseminated myelitis can be one of the possible diagnoses because of pleocytosis in CSF and the tumefactive pattern of lesions. However, acute disseminated myelitis is only rarely located in the spinal cord. We excluded it because of lack of typical symptoms (fever and whole-body inflammation, headache, seizures or aphasia and confusion).5 CSF results and radiographic features did not support that diagnosis. The variant, acute haemorrhagic leukoencephalitis (Hurst disease), could also be excluded because of the absence of haemorrhage. Serology tests of viral and bacterial agents were nonsignificant. Clinical symptoms, MRI and pathological findings made diagnosis of Marburg variant, Balo concentric sclerosis, myelinoclastic diffuse sclerosis impossible.
Tumefactive spinal cord lesions are very often diagnosed as NMO or NMO-spectrum disorders. Since 2006, the diagnostic criteria for NMO have included the following: optic neuritis; acute myelitis; and 2 or 3 of the following supporting criteria— (1) contiguous cord lesions that are long, spanning three or more vertebral segments; (2) MRI failing to meet multiple sclerosis (MS) diagnostic criteria; and /or (3) anti-AQP4-Ab positivity.6
Our patient was diagnosed with isolated myelitis, which was longitudinally extensive, spanning more than three segments in the thoracic spinal cord. The findings were suggestive of NMO. We found anti-AQP4-Ab negativity in 2 CSF samples; however, they were taken after the first dose of corticosteroids. Negativity of anti-AQP4-Ab might have been caused by detection sensitivity of indirect immunofluorescence, which was reported to be rather low.7 Especially for anti-AQP4-ab, testing should be preferably performed on blood samples.7 The detection rate in CSF varies largely; negativity of anti-AQP4-Ab in CSF was reported to be rather the rule than the exception.7 The diagnostic value of anti-AQP4-Ab in the CSF may become reduced after longer storage of frozen CSF samples. Another possible reason could be the fact that prevalence of anti-AQP4-Ab seems to be higher in female patients and in patients with relapsing disease.7 Therefore, our patient met one direct and two out of three supporting diagnostic criteria for NMO.
Despite the positivity of CSF oligoclonal bands (type2), positivity of IgG index, and negativity of anti-AQP4-Ab, tumefactive demyelination as a variant of MS is still questionable. The significant pleocytosis found in our case supports a NMO-spectrum disorder.
On the other hand, we should consider a lack of knowledge about the final outcome of the patient, which leaves diagnosis of MS open. A recently reported case of tumefactive demyelination located in the spinal cord has shown MRI pictures revealing an abnormally increased signal extending more than three segments within the lower cervical and upper thoracic spinal cord. The case was assessed as MS on the basis of negative anti-AQP4-Ab, positive OCB and the presence of new clinical relapse.1 Maezawa et al.3 have also described isolated spinal cord demyelination mimicking glial tumour, which was finally diagnosed as relapse-remitting multiple sclerosis, based on another relapse with development of a new lesion in the brain stem and histopathological proof of acute demyelination.
Histopathological samples of our patient were classified as active demyelinated lesions with myelin loss and phagocytic parenchymal macrophages containing myelin degradation products. Macrophages signed by CD68 antigens were the most common in the demyelinated lesions. In our histopathological samples, glial fibrillary acidic protein-positive astrocytes were distributed throughout the demyelinated lesions. The axons were relatively preserved. Demyelination in NMO extends across multiple sections of the spinal cord6, 8 as was found in our case. We did not observe necrosis and cavitation across the spinal cord, but the structure of penetrating vessels in inflamed areas might evoke NMO,6, 8 as they were hyalinized in the affected areas.
Conclusion
In our patient we found tumefactive demyelination in the spinal cord, which could be a variant of NMO. Tumefactive forms of spinal cord demyelination have been shown to overlap in clinical, radiological and laboratory features.
References
Makary MS, Kirch CF . Tumefactive demyelinating disease with isolated spinal cord involvement. Acta Radiologica 2014; 3: 1–3.
Yaghi S, Gokdin M, Sethi H . Tumefactive demyelination of the spinal cord. Acta Neurol Belg 2010; 110: 206–208.
Maezawa H, Takano M, Nagai S, Iida H, Tachibana S . Spinal multiple sclerosis mimicking a spinal cord tumor: a case report. No Shinkei Geka 1995; 23: 1007–1010.
Andersson M, Alvarez-Cermeño J, Bernardi G, Cogato I, Fredman P, Frederiksen J et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: a consensus report. J Neurol Neurosurg Psychiatry 1994; 57: 897–902.
Rahmlow MR, Kantarci O . Fulminant demyelinating diseases. Neurohospitalist 2013; 3: 81–91.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG . Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66: 1485–1489.
Trebst C, Jarius S, Berthele A, Friedemann P, Schippling S, Wildemann B et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014; 61: 1–16.
Lucchinetti CF, Gavrilova RH, Metz I, Parisi JE, Scheithauer BW, Weigand S et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain 2008; 131: 1759–1775.
Acknowledgements
MUDr Ema Kantorová PhD's work has been supported by Project ESF ITMS kód: 26110230067 and Project ITMS 26110230071 “Virtuálna a simulačná výučba ako nová forma vzdelávania na JLF UK v Martine”. MUDr Štefan Sivák PhD's work has been supported by Project ESF ITMS kód: 26110230067 and Project ITMS 26110230071 “Virtuálna a simulačná výučba ako nová forma vzdelávania na JLF UK v Martine”. MUDr Juraj Marcinek PhD, MUDr Kamil Zeleňák PhD, MUDr Jozef Michalik, Prof MUDr Egon Kurča PhD, Prof MUDr Lukáš Plank CSc and MC Kryštof Kantor declare no potential conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kantorová, E., Marcinek, J., Zeleňák, K. et al. Tumefactive demyelination of the spinal cord: a case report. Spinal Cord 53, 877–880 (2015). https://doi.org/10.1038/sc.2015.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2015.52
