
Abstract
Enhancing lithium-ion battery technology in terms of specific capacity and charging time is key for the advancement of the electrification of transportation. Particularly for fast charging, inhomogeneous deposition of metallic lithium, for example on commercial graphite or metallic lithium anodes, leads to cell degradation and safety issues. To understand the underlying mechanisms and develop counter-measures, non-invasive online detection techniques providing satisfactory time resolution are crucial. Here, we demonstrate in operando pulse electron paramagnetic resonance to observe transient processes during pulsed fast charging in cells with metallic lithium anodes. Sampling timescales of 100 ms enable real-time monitoring of the formation and evolution of porous lithium during and after charging pulses. It was observed that the generated morphology continued to evolve after the end of a charging pulse, whereby surface features were fusing with a time constant that was slower than their formation.
Similar content being viewed by others


Introduction
Rechargeable lithium-ion batteries are a key enabling technology for electric vehicles due to their high gravimetric and volumetric capacity1,2,3. A strategy to further increase the stored energy is to use lithium metal anodes, which promise a tenfold increase of specific energy density compared to state-of-the-art graphite anodes4. So far, the formation of lithium microstructures upon cycling has prevented its widespread use because of a limited lifetime due to Coulombic efficiency loss and considerable likelihood of short circuits5,6.
Lithium microstructures are formed intrinsically during electrochemical deposition and removal from a metallic surface4, denoted as plating and stripping, respectively. Depending on the cell chemistry, cycle age, and charging protocol, microstructures of different morphologies are found; whiskers, moss, and dendrites are discriminated when using liquid carbonate-based electrolytes7. Their appearances can be strongly affected by the applied current density (CD). For example, high CDs result in a large number of deposition nucleation sites, facilitating smooth lithium plating8,9 but may also cause needle-like dendrites, which are responsible for rapid cell degradation and ultimately short-circuiting. The exposure of freshly formed metallic lithium to electrolyte results in decomposition products referred to as solid electrolyte interphase (SEI). Decomposition products are found to be inorganic species covering the metallic surface10. The morphology and defective nature of the SEI contribute to irregular lithium growth11.
Towards safe application of lithium metal anodes, the interplay of cell characteristics and CD has to be investigated to optimise homogeneous plating and suppression of dendrites. A promising perspective is the recent qualitative finding of self-heating-induced anode healing12,13. Post-testing revealed smoothed structures upon using large CDs of 10–15 mA cm−2, which induce extensive surface diffusion through high local temperatures.
To actively monitor the emergence and evolution of microstructures, non-invasive diagnostics during operation are crucial. Electron paramagnetic resonance (EPR) is one suitable technique. Using microwave (MW) excitation, EPR enables low-energy probing of realistic cell designs14. In comparison to the closely related nuclear magnetic resonance15,16 technique, EPR is more selective regarding surface detection due to a lower skin depth at higher frequency and shows higher sensitivity due to the larger gyromagnetic ratio of electron spins compared to nuclear spins14. EPR observes spin transitions of lithium conduction electrons in an externally applied magnetic field17,18,19. The resonance signal linewidth depends on the apparent relaxation time of the electrons, which is largely affected by their mobility in different environments17,18. If conduction electrons leave the surface region with a thickness of about 1.1 µm in our experiments, they are not excited or detected anymore. Within porous lithium structures, electrons are located comparatively long in the surface region sensitive to EPR excitation because the diameter of these structures is on the order of or below the skin depth and the interconnected lithium metal structure confines diffusion away from the surface. Thus, the apparent spin relaxation time is increased in comparison to a bulk sample. The relaxation time constants become sufficiently long to allow room temperature pulse EPR (pEPR) experiments, which surpass the acquisition rate of previously demonstrated continuous-wave EPR14,20,21. pEPR is based on MW pulses lasting only a few nanoseconds followed by detection of the spin free induction decay (FID)22.
Electron spin relaxation times are largely affected by the particle size and, thus, can be used to estimate structure dimensions21,23. In pEPR experiments, morphology-dependent relaxation times facilitate a simple signal separation by selecting a detection window optimised for a particular morphology of interest. Long-lived signals from microstructured lithium can be selectively detected by placing an acquisition window at the tail end of the FID (Fig. 1a). Such boxcar detection enables fast repetition of the experiment, with an associated temporal resolution that is sufficient to observe microstructure dynamics in a regime relevant for fast battery charging. For a comparison of published sampling rates of established techniques for online battery applications see Supplementary Table 1.

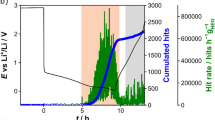
a Time line of each pEPR acquisition. A microwave pulse (grey bar) generates measurable electron spin coherence, which is decaying with a characteristic relaxation time. The signal is detected by integrating a part of the FID (blue bar). b In operando cell containing a symmetric Li/Li setup. Diameter d and thickness θ of compartments are given. The cylindrical shape enables insertion into an X-band pulse EPR resonator. c Exemplary electrochemical dataset applied to a freshly assembled cell. An identical pulse charging protocol (inset) was used throughout. pEPR relaxation and nutation behaviour was studied in regions where no current was passing.
Here, in operando pEPR is demonstrated as a selective technique for real-time detection of the surface morphology of lithium that is plated or stripped during cycling of a symmetric lithium metal cell. Transient morphology changes of electrodeposited lithium that depend on the applied CD are proposed based on the pEPR data.
Results and discussion
In operando EPR
Figure 1b shows the utilised reusable quartz glass housing accommodating a symmetric lithium cell. The dynamics of deposited lithium was studied using a pulse charging protocol with a repetition unit consisting of a 3 s current pulse and a subsequent 18 s resting period. Overall, nine identically prepared cells with varying pulse CDs from 1.75 to 7.00 mA cm−2 are analysed. In operando pEPR measurements are performed during electrochemical treatment at three periods indicated by grey, orange, and green bars in Fig. 1c. To compare all nine cells, these positions are identical regarding the areal charge, corresponding to identical amounts of deposited lithium. These three regions vary in duration for each cell since by using a higher pulse CD yet identical charging protocol, the respective areal charge is reached at different times. The regions where no current is flowing for more than 10 min (Fig. 1c) were used for more time-consuming in situ acquisition of the full FID and nutation spectra. The signal amplitude was constant over this waiting period, indicating the absence of side reactions or slower transient changes than the ones reported.
Upon pulse charging, lithium is removed from one electrode and plated onto the other. We verified that no pEPR signal is contributed by the stripping electrode (Supplementary Fig. 1), leaving only one electrode contributing to the in operando pEPR signal. During a single current pulse, 1015–1016 lithium atoms are deposited on the electrode. As the sensitivity limit of EPR is typically estimated to be around 1012 electrons, EPR sensitivity is sufficient for online detection. However, the onset of observable signal occurred only about 5–10 min after pulse charging initiation. This is consistent with the formation of initial lithium deposits that are still recognised as bulk species by EPR14. The delayed appearance of the signal is exploited when investigating the stripping process upon reversing the current direction, as signal interference from microstructured lithium on the second electrode is delayed as well. To provide comparable results, the electrochemical cell was positioned identically within the pEPR spectrometer for all cells investigated (Supplementary Fig. 2).
Figure 2 shows in operando pEPR data within one electrochemical repetition unit, detected via boxcar integration as described above. During the plating (stripping) pulse, the pEPR integral is increasing (decreasing) corresponding to an increase (decrease) in amount of detected microstructured lithium. During the resting period after a stripping pulse, the pEPR integral remains invariable (Fig. 2b). After a plating pulse, however, the integral declines, showing a monoexponential decay with a time constant of several seconds (Fig. 2a). A time-resolved measurement of the FID shows that this decay can be mainly attributed to a change in electron relaxation time (Supplementary Note 1, Supplementary Fig. 3, Supplementary Table 2), which effectively reduces the measured integral (Fig. 1a). We preclude residual flowing current to be responsible as the flanks of the current pulses are distinctly shorter than the exponential decay of the signal (Fig. 2a). Heating due to electrical resistance during the current pulse and subsequent cooling is also precluded since the relaxation time of conduction electrons in lithium is known to be largely independent of temperature17,18. Consequently, a dynamic change in particle morphology is presumed to be responsible. The metal particle dimension is known to alter the relaxation time, which was already observed to change with charging time by cwEPR14. The dynamic process is assumed to initialise during the current pulse, explaining the slightly non-linear EPR signal increase albeit constant lithium deposition rate (Supplementary Figs. 4, 5, Supplementary Note 2, Supplementary Table 3).

a, b Exemplary datasets with applied current densities of 1.75 mA cm−2 (red), 3.50 mA cm−2 (green), and 5.25 mA cm−2 (purple). For clarity, pEPR data was normalised to the interval [0,1]. Plating region II (a) is compared with the stripping region (b), visualised by schematic drawings. The inset in a shows a magnification and contains monoexponential fits (coloured traces).
In analogy to Fig. 2a, plating datasets from all investigated cells at plating regions I (grey) and II (orange) were fitted monoexponentially, with the time constants displayed in Fig. 3. Boxcar integration of FIDs with changing electron relaxation times (Fig. 1a) makes it difficult to directly attribute the fitted exponential decay constant to a real physical process. Nonetheless, based on the hypothesis of a dynamic process, it gives a phenomenological measure of the rate of the process, whose time constant consistently decreases with increasing CD. For plating region II, representing more pre-existing microstructured lithium, the dynamic process slows down, particularly for large CDs.

Current densities ranging from 1.75 to 7.00 mA cm−2 were analysed at plating region I (black spheres) and plating region II (orange spheres), corresponding to a deposited specific charge of 0.18 and 0.25 mAh cm−2, respectively. Error bars originate from the standard deviation of the fit. Lines are guides to the eye. The grey hatched area indicates current densities without sufficient pEPR signal intensity, thus no data are available.
Important factors for the pEPR experiments at various CDs are the electron relaxation times and the effect of the MW pulse. The acquired FIDs can be fitted with a stretched exponential function (Supplementary Note 3, Supplementary Fig. 6), which is commonly used to model a distribution of relaxation times24. The observed relaxation times are similar for all CDs. The effect of the MW pulse is probed by varying its duration, referred to as a nutation experiment, whereby local MW field strengths can be estimated via the electron nutation frequency distribution (Supplementary Note 4, Supplementary Fig. 7). For instance, the average MW field is reduced due to the skin effect or changed by induced eddy currents evoked by bulk lithium25,26. For all cells, the nutation frequencies spread over roughly 20 MHz but show no apparent correlation with the CD. Upon using a MW pulse duration of 16 ns, the signal response was maximised.
Electron microscopy
SEM images of lithium microstructures on electrodes formed during electrochemical pulse plating at CDs of 2.10 and 5.95 mA cm−2 are shown in Fig. 4a, b, respectively. Although lithium deposited at both low and high CDs can be described as mossy, the microstructures substantially differ in dimension. At high CDs, smaller structures occur in agreement with previous findings8. However, electron relaxation times within all cells are similar (Supplementary Fig. 6), hence microstructures formed at high CDs need to be well interconnected to prevent a bottleneck for electron conduction and a corresponding relaxation time increase. These connections prevent a localisation of conduction electrons, giving rise to faster apparent electron relaxation than expected for small microstructures23, as was also observed for dendrites in a harvested separator21.

Current pulses of a 2.10 mA cm−2 and b 5.95 mA cm−2 lasting 3 s were alternated with resting periods of 18 s.
Dynamic evolution of electrodeposited lithium
It is known that lithium nucleation is affected by local surface irregularities, the SEI layer, and electrolyte kinetics8,11, ultimately leading to irregular structures as shown in Fig. 4. However, lithium mobility at the surface27 might result in restructuring and possibly smoothing, which has previously been proposed based on electron microscopy data12. The deposition of amorphous microstructures after initial nucleation, detected via cryo-TEM28,29, followed by a transition to a crystalline form might further provide a thermodynamic driving force. Accordingly, the observed pEPR data can be explained by fusing of freshly deposited lithium microstructures with their surroundings, resulting in more thermodynamically stable structures. Correspondingly, decreasing electron relaxation times following a plating pulse indicate that the structures become more compact and electrically better connected, thereby reducing the probability of forming electrically disconnected dead lithium and slowing down the associated cell degradation14. The hypothesis is further supported by post-test electron microscopy images showing particles that appear partially fused (Supplementary Fig. 8). A competitive process is the SEI layer formation that might ultimately terminate the fusing process.
This model is sketched in Fig. 5. The CD largely affects the size of deposits and the evolution of their morphology (Fig. 4). When using low CD pulses, the SEM image shows comparatively large lithium microstructures. Due to a lower overpotential, the activation barrier to form nucleation sites is overcome less often leading to fewer of these sites which grow larger in size. The resulting metallic domains are thicker and less branched. After the current pulse, restructuring of the fresh lithium takes longer for larger deposits as their surface to volume ratio is low and lithium diffusion is mostly driven at the surface27. Utilising high CD pulses, a large number of irregular lithium nucleation sites are formed electrochemically due to a high overpotential. Their comparably high surface area and close distance of the lithium metal structures grown out of them promotes accelerated fusing, leading to earlier pEPR signal equilibration. Additionally, the fusing rate differs depending on whether considering plating region I or II. As the charging protocol is identical, their only difference is the amount of pre-existing microstructure. In general, fresher SEI layers and local electric field maxima lead to the deposition of lithium on pre-existing microstructures as opposed to the bulk surface11. At earlier stages of plating, the total surface area of the existing lithium microstructure is comparably small. Hence, in region I the distance between fresh lithium deposits along the surface is likely to be smaller. The fusing process of adjacent deposits is accelerated due to more diffusion pathways at their surface to form a final stable structure. At later stages of plating, fresh lithium deposits are statistically more dilute. The proposed mechanism is less likely to occur.

The yellow shaded area depicts the development of Li microstructure on a large time scale. Blue shaded areas show exemplary current pulses leading to freshly deposited Li (pink). Grey shaded areas show the result of deposited Li fusing to a more stable configuration during the resting period. Particularly for small microstructures, this process is accelerated by ripening of adjacent Li deposits (orange circles).
Conclusion
In conclusion, we present the application of in operando pEPR for the detection of electrochemically plated lithium microstructure with a temporal resolution of 100 ms. This technique is especially suitable to investigate fast-charging of lithium-ion batteries and to test prevention strategies of battery failure4 such as electrolyte engineering, artificial SEI generation, electrode host material variations, or charging protocol optimisation. pEPR was shown to be capable of real-time monitoring dynamic processes, which inherently require in operando techniques. Specifically, pEPR was used to track fusing of freshly deposited lithium on a metallic lithium anode with its surroundings. The process was found to last several seconds after deposition of fresh lithium until equilibrium was reached. The fusing rate was correlated with the applied pulse CD and the pre-existing lithium microstructure. The awareness of fusing processes will complement the fundamental understanding of lithium deposition and help to design safe metallic lithium anodes.
Methods
Battery cell design
Li metal ribbon was purchased from Sigma Aldrich with 0.38 mm thickness and a purity of 99.9% trace metal basis. Electrode discs with a diameter of 3 mm were punched out and positioned centrally at either side of a Celgard 3401 separator with 4 mm diameter and 25 µm thickness between the quartz glass half cells. The quartz glass cell compartments were manufactured by LightFab GmbH via laser induced etching. Copper wires from Goodfellow with 0.075 mm diameter and 99.99+% purity were used as current collectors. The two half cells were pressed together by PFA rings at the top and bottom insets. The separator was wetted with 5 µL of the standard electrolyte LP30 (1 M LiPF6 in 1:1 v/v ethylene carbonate:dimethyl carbonate). The assembled cell was pushed into a Wilmad CFQ EPR tube with an outer diameter of 5 mm and sealed air-tight. All procedures involving metallic Li were performed in an Ar-filled glovebox with water and oxygen levels below 0.1 ppm.
Electrochemistry
Experiments were performed with a Biologic SP-200 potentiostat. For all experiments, pulse charging/discharging with CDs ranging from 1.75 to 7.00 mA cm−2 is applied. A fixed current pulse duration of ton = 3 s is alternated with a resting period of toff = 18 s, resulting in a ratio toff/ton = 6. The fresh cell was used immediately after assembly without any electrochemical pre-treatment.
For time-resolved measurements, plating is firstly carried out up to 0.22 mAh cm−2 followed by a 10–20 min resting period for additional EPR experiments which require static electrochemical conditions (addressed in Supplementary Figs. 6, 7). Secondly, plating is continued to 0.31 mAh cm−2 followed by another resting period. Ultimately, the current direction is reversed to detect stripping. A detailed exemplary electrochemical dataset can be found in Supplementary Fig. 9 with experiment specifications in Supplementary Table 4 and explanation in Supplementary Note 5.
Pulse EPR spectroscopy
EPR data was acquired on a Bruker ElexSys E580 X-band spectrometer in an ER 4118X-MD5 resonator. For in operando experiments, a homebuilt setup was designed, which is compatible with Bruker’s Flexline series: A holder for the probehead, a probehead tube which was wrapped with copper foil, and a sample holder were 3D-printed in-house with a Prusa i3 MK3S (details in Supplementary Fig. 10). To minimise exterior disturbances, the spectrometer as a whole was covered with a silver blanket. Cells inside quartz glass tubes of 5 mm outer diameter were inserted into the resonator from below and centred in the cavity. Li electrodes were positioned parallel to the static magnetic field (Supplementary Fig. 2). A constant flow of nitrogen gas tempered at 20 °C directed towards the resonator provided temperature stabilisation.
During electrochemical charging/discharging, time-resolved measurements were performed by adjusting the magnetic field to resonance and the phase in such a way that signal is merely existent as real part. Following a MW pulse with pulse length tp = 16 ns, the integrated intensity of the FID in a window of 400 ns was recorded. Acquisition started 380 ns after the pulse was applied. MW pulses were generated using a X-band 1 kW travelling-wave tube with 6 dB attenuation. Shot repetition time (srt) was set to 100 µs and 1000 scans were averaged.
Time-resolved pEPR experiments were conducted during electrochemical treatment. To increase the sensitivity, six pulse charging repetition units at the indicated states of charge were monitored (Supplementary Fig. 11).
EPR data analysis
The datasets are divided into packages of 210 data points, corresponding to a repetition unit of a current pulse and a subsequent resting period lasting 21 s. For each package, the minimal value is subtracted and 3–6 packages are summed together, omitting packages with high noise level. Noise occurs irregularly from ambient sources due to incomplete shielding of the MW resonator. To compare datasets at different CDs, the summed Intensity I is finally normalised to the interval [0, 1] by the formula
with \({\mathrm{min}}(I)\) and \(\max \left( I \right)\) denoting the minimum and maximum value of I. For plating datasets, monoexponential fits were performed in the resting period range from 3 to 21 s (Fig. 2a inset), using
with τ being analysed as the exponential decay constant. Datasets that are not displayed in the main text are given in Supplementary Fig. 12.
SEM
A Quanta FEG 650 scanning electron microscope (FEI) was used to record SEM images, detecting secondary electrons. Electron beam acceleration voltage was 5–10 keV (specified in figure caption). Lithium electrodes were obtained from cells identically prepared as for EPR experiments described above. Assembled cells were pulse charged (ton = 3, toff = 18 s) at CDs of 2.10 and 5.95 mAh cm−2 up to 0.31 mAh cm−2, in order to represent the lower and upper current regime of the EPR experiments. Directly after electrochemical Li deposition, the cells were transferred back to the glovebox and disassembled. Harvested electrodes were washed three times with 150 µL dimethyl carbonate (DMC) and positioned on a SEM sample holder with carbon adhesive. Residues of DMC were removed by drying under dynamic vacuum for 5 min. Subsequently, the sample holder was transferred to the microscope chamber being exposed to ambient atmosphere no longer than 30 s.
Data availability
The data that support the findings of this study are enclosed in the supplementary material or are available from the corresponding author upon reasonable request.
Change history
09 April 2021
Open Access funding information has been added to this article.
References
Deng, D. Li-ion batteries: basics, progress, and challenges. Energy Sci. Eng. 3, 385–418 (2015).
Tarascon, J.-M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Hannan, M. A., Hoque, M. M., Mohamed, A. & Ayob, A. Review of energy storage systems for electric vehicle applications: Issues and challenges. Renew. Sust. Energy Rev. 69, 771–789 (2017).
Lin, D., Liu, Y. & Cui, Y. Reviving the lithium metal anode for high-energy batteries. Nat. Nanotechnol. 12, 194–206 (2017).
Xu, W. et al. Lithium metal anodes for rechargeable batteries. Energy Environ. Sci. 7, 513–537 (2014).
Liu, Y., Zhu, Y. & Cui, Y. Challenges and opportunities towards fast-charging battery materials. Nat. Energy 4, 540–550 (2019).
Bai, P., Li, J., Brushett, F. R. & Bazant, M. Z. Transition of lithium growth mechanisms in liquid electrolytes. Energy Environ. Sci. 9, 3221–3229 (2016).
Pei, A., Zheng, G., Shi, F., Li, Y. & Cui, Y. Nanoscale nucleation and growth of electrodeposited lithium metal. Nano Lett 17, 1132–1139 (2017).
Gireaud, L., Grugeon, S., Laruelle, S., Yrieix, B. & Tarascon, J. M. Lithium metal stripping/plating mechanisms studies: a metallurgical approach. Electrochem. Commun. 8, 1639–1649 (2006).
Hao, F., Verma, A. & Mukherjee, P. P. Mechanistic insight into dendrite-SEI interactions for lithium metal electrodes. J. Mater. Chem. A 6, 19664–19671 (2018).
Yoon, G., Moon, S., Ceder, G. & Kang, K. Deposition and stripping behavior of lithium metal in electrochemical system: continuum mechanics study. Chem. Mater. 30, 6769–6776 (2018).
Li, L. et al. Self-heating-induced healing of lithium dendrites. Science 359, 1513–1516 (2018).
Hundekar, P. et al. Exploiting self-heat in a lithium metal battery for dendrite healing. Energy Storage Mater 20, 291–298 (2019).
Wandt, J. et al. Operando electron paramagnetic resonance spectroscopy—formation of mossy lithium on lithium anodes during charge–discharge cycling. Energy Environ. Sci. 8, 1358–1367 (2015).
Bhattacharyya, R. et al. In situ NMR observation of the formation of metallic lithium microstructures in lithium batteries. Nat. Mater. 9, 504–510 (2010).
Chandrashekar, S. et al. 7Li MRI of Li batteries reveals location of microstructural lithium. Nat. Mater. 11, 311–315 (2012).
Feher, G. & Kip, A. F. Electron spin resonance absorption in metals. I. Experimental. Phys. Rev. 98, 337–348 (1955).
Dyson, F. J. Electron spin resonance absorption in metals. II. Theory of electron diffusion and the skin effect. Phys. Rev. 98, 349–359 (1955).
Taylor, D. R., Gillen, R. P. & Schmidt, P. H. Pulsed-microwave studies of conduction-electron spin resonance in lithium and sodium. Phys. Rev. 180, 427–431 (1969).
Wandt, J., Jakes, P., Granwehr, J., Eichel, R. A. & Gasteiger, H. A. Quantitative and time-resolved detection of lithium plating on graphite anodes in lithium ion batteries. Mater. Today 21, 231–240 (2018).
Niemöller, A., Jakes, P., Eichel, R. A. & Granwehr, J. EPR imaging of metallic lithium and its application to dendrite localisation in battery separators. Sci. Rep. 8, 14331 (2018).
Zhikharev, V. A. & Kessel’, A. R. Theory of spin induction and conduction-electron echo in bulky metallic samples. Sov. J. Exp. Theor. Phys. 40, 874 (1975).
Webb, R. H. Electron-spin-resonance line shape in spherical metal particles. Phys. Rev. 158, 225–233 (1967).
Johnston, D. C. Stretched exponential relaxation arising from a continuous sum of exponential decays. Phys. Rev. B Condens. Matter Mater. Phys. 74, 1–7 (2006).
Vashaee, S., Goora, F., Britton, M. M., Newling, B. & Balcom, B. J. Mapping B1-induced eddy current effects near metallic structures in MR images: a comparison of simulation and experiment. J. Magn. Reson. 250, 17–24 (2015).
Ilott, A. J. et al. Visualizing skin effects in conductors with MRI: 7Li MRI experiments and calculations. J. Magn. Reson. 245, 143–149 (2014).
Gaissmaier, D., Fantauzzi, D. & Jacob, T. First principles studies of self-diffusion processes on metallic lithium surfaces. J. Chem. Phys. 150, 041723 (2019).
Wang, X. et al. New insights on the structure of electrochemically deposited lithium metal and its solid electrolyte interphases via cryogenic TEM. Nano Lett. 17, 7606–7612 (2017).
Wang, X. et al. Glassy Li metal anode for high-performance rechargeable Li batteries. Nat. Mater. 19, 1339–1345 (2020).
Acknowledgements
J.A. gratefully acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy –Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832“. Funding for the Quanta FEG 650 by the Bundesministerium für Bildung und Forschung (BMBF, Federal Ministry of Education and Research) under the project SABLE, No 03EK3543 is thankfully acknowledged. We would like to thank Sven Jovanovic for assistance with 3D printing and Arvid Niemöller and Christian Hellenbrandt for the design of the electrochemical in operando cell.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
C.S. designed the in operando pEPR setup, carried out the experiments, and performed data processing. J.A. and C.S. recorded SEM images. C.S. and P.P.M.S. developed pEPR hardware modifications. C.S., J.A., P.P.M.S., P.J., R.-A.E., and J.G. analysed and discussed the results. C.S. and J.G. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary handling editors: Jie Xiao, John Plummer
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szczuka, C., Ackermann, J., Schleker, P.P.M. et al. Transient morphology of lithium anodes in batteries monitored by in operando pulse electron paramagnetic resonance. Commun Mater 2, 20 (2021). https://doi.org/10.1038/s43246-021-00126-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-021-00126-1
