
Abstract
Multiple roles of reactive oxygen species (ROS) and their consequences for health and disease are emerging throughout biological sciences. This development has led researchers unfamiliar with the complexities of ROS and their reactions to employ commercial kits and probes to measure ROS and oxidative damage inappropriately, treating ROS (a generic abbreviation) as if it were a discrete molecular entity. Unfortunately, the application and interpretation of these measurements are fraught with challenges and limitations. This can lead to misleading claims entering the literature and impeding progress, despite a well-established body of knowledge on how best to assess individual ROS, their reactions, role as signalling molecules and the oxidative damage that they can cause. In this consensus statement we illuminate problems that can arise with many commonly used approaches for measurement of ROS and oxidative damage, and propose guidelines for best practice. We hope that these strategies will be useful to those who find their research requiring assessment of ROS, oxidative damage and redox signalling in cells and in vivo.
Similar content being viewed by others

Main
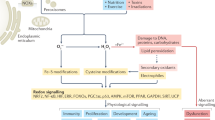
Reactive oxygen species (ROS) (Box 1) are intimately involved in redox signalling but in some situations can also lead to oxidative damage. Hence, they have both physiological and pathophysiological roles in biology1,2,3,4. Consequently, researchers from diverse fields often need to measure ROS, to assess oxidative events and to investigate their biological importance using antioxidants (Box 1) or inhibitors to modulate the phenomena observed. There are many assays and commercial kits available, but their use and interpretation are challenging and open to artefacts. There is a well-established field of biophysics/biochemistry/chemistry focusing on the identification of ROS, their chemical reactions and products of oxidative damage. However, as with many specialized fields, this literature can be hard to interpret by those working outside the area. Frequently problems arise due to reliance on commercial kits that claim to measure ‘ROS’ or ‘oxidative damage’, or from the use of ‘antioxidants’ in general terms, when progress requires understanding of specific molecular mechanisms.
To address these points, this international group has set out guidelines on the nomenclature and measurement of ROS, oxidative reactions and oxidative damage. Our focus is on the techniques used to measure ROS and oxidative damage. These can be applied to their role in pathology, but it is also important to note that changes in the levels of ROS and consequent changes in the activity of redox-sensitive cellular processes are central to the field of redox signalling1,2,3,4. We hope that these guidelines will be useful for researchers who find themselves carrying out experiments in this area. These topics, and indeed the approaches we advocate, have been covered by many reviews in the past1,2,3,4,5,6,7,8,9,10,11 and which researchers are strongly encouraged to read. Here we distill the key points underlying this consensus statement.
What are ROS, antioxidants and oxidative damage?
One problem that underlies the measurement of ROS and oxidative damage and the use of ‘antioxidants’ is the lack of precision in the use of these terms. ROS is an abbreviation that covers a wide range of chemical species with different properties, reactivities and interactions (Box 1, Table 1). For example, one important reactive species found in biology, the superoxide radical anion (O2•−), is formed by the one-electron reduction of oxygen (O2). In itself, O2•− is not very reactive, except with another radical nitric oxide (•NO) to form peroxynitrite11, or with Fe–S clusters in proteins12. Similarly, hydrogen peroxide (H2O2), formed by various oxidase enzymes1,4 and by the action of superoxide dismutase (SOD), is poorly reactive, which allows its use as an important signalling molecule in vivo2,4. Nevertheless, in the presence of ferrous or cuprous ions, H2O2 forms the extremely reactive hydroxyl radical (•OH) by Fenton chemistry: •OH reacts non-specifically and essentially instantaneously with any nearby biomolecule (Table 1)1,13. The availability of transition metal ions to catalyse Fenton chemistry is carefully controlled in vivo1, but these can be released by tissue injury or when certain proteins with Fe–S clusters encounter O2•− (refs. 1,12,14). Their importance in vivo has recently been underscored by the growing literature on ferroptosis, a form of cell death involving ‘catalytic’ iron ions15. H2O2 is a substrate for haem peroxidases such as myeloperoxidase, generating further reactive species such as HOCl (Table 1). Despite its poor overall reactivity, H2O2 can selectively oxidize some methionine (Met) and cysteine (Cys) residues16,17 in certain proteins.
A (far from complete) list of the physicochemical properties of the most common ROS encountered in biology is given in Table 1, which provides insights into what reactions might be plausible in vivo when these species are generated. What should also be evident is that ‘reactive’ is highly context dependent, because the reactivity of different ROS varies over a wide scale, as do their lifespans, ability to diffuse and potential to generate further downstream reactive species. In short, not all ROS are the same. The generalization ‘ROS’, although widely used (including in this paper!) does not give information about the actual chemical species causing the observed effect. Recommendation 1: wherever possible, the actual chemical species involved in a biological process should be stated and consideration given to whether the observed effect is compatible with its reactivity, lifespan, products generated and fate in vivo. If this is not possible, caveats about use of the term ‘ROS’ should be discussed.
A wide range of antioxidants is present in biology. These include enzymes and small molecules that react with individual ROS to decrease oxidative damage and/or modulate redox signalling1,2. As with ‘ROS’, the use of ‘antioxidant’ as a general term can be imprecise and misleading (Box 1). Often the effect of a putative antioxidant on a biological outcome is used to infer a role for a ROS, as if all antioxidants were equivalent. However, each antioxidant has its own specific chemistry and reactivity with different ROS. Furthermore, the major antioxidants in vivo are enzymatic systems such as SOD for O2•−, peroxidases for H2O2, and metal ion sequestration1,14. Most low-molecular-mass compounds commonly employed as ‘antioxidants’ are stoichiometric scavengers of certain ROS and often have modest (if any) reactivity with O2•− or H2O2. For example, N-acetylcysteine (NAC) is a widely used ‘antioxidant’ but it has other (and sometimes more important18) modes of action. NAC can indeed scavenge some ROS in vitro, but not others, most notably not H2O2 (ref. 18). It can also increase the cellular Cys pool and thereby enhance glutathione (GSH) levels, generate H2S, and directly cleave protein disulphides18. Low-molecular-mass compounds that do act as antioxidants in vivo include vitamin E, which scavenges lipid peroxyl radicals19. Sometimes ‘•OH scavengers’ are used to infer a role for this ROS yet they can rarely, if ever, achieve a sufficiently high concentration to prevent the effectively instantaneous reaction of •OH with biomolecules1,7,13. Consequently, many of the biological effects assigned to ‘antioxidants’, especially NAC, are due to other effects. Other agents often used as ‘antioxidants’, such as TEMPO/TEMPOL, mito-TEMPO and porphyrin-based ‘SOD mimetics’, undergo complex redox reactions in vivo and are better described as ‘redox modulators’ rather than ‘antioxidants’ or ‘O2•− scavengers’1,20,21. Recommendation 2: for an intervention to be attributed to an antioxidant activity, the particular chemical species targeted by the ‘antioxidant’ needs to be made explicit. It should be recognized that low-molecular-mass ‘antioxidants’ are unlikely to act by scavenging H2O2. The specificity, rate constant, location and concentration of the antioxidant within the cell should render an antioxidant effect chemically plausible. Wherever possible the activity of the antioxidant should be confirmed by measuring a decrease in oxidative damage.
A key procedure to attributing oxidative damage, or activation of a redox signalling pathway, to a particular ROS can be by selective generation of the ROS in a biological context. This can be done by using redox cycling compounds such as paraquat (PQ) or quinones to generate O2•−, or MitoPQ to generate O2•− within mitochondria1,22. Of course, increased O2•− generation will also increase H2O2 production by O2•− dismutation. Glucose oxidase can be used to generate H2O2 directly in vitro, while the regulated generation of H2O2 within cells can be achieved using genetically expressed d-amino acid oxidase, an enzyme that generates H2O2 as it oxidizes d-amino acids23. It can be targeted to different sites in the cell, and the flux regulated by varying the added concentration of its substrate, d-alanine23. NADPH oxidase (NOX) enzymes are important sources of O2•−and H2O2 for redox signalling, as well as oxidative damage9,24, and modulation of their activity is an important approach to understanding these processes. A number of fairly specific inhibitors of NOX enzymes have been described24. However, the use of compounds such as apocynin and diphenyleneiodonium as ‘NOX inhibitors’ is still widespread, even though their lack of specificity is well established1,24. Recommendation 3: we recommend the use of PQ, quinones and MitoPQ for selective generation of O2•− and the cellular expression of d-amino acid oxidase for controlled generation of H2O2. Avoid the use of inhibition of a phenomenon by apocynin or diphenyleneiodonium as sole evidence for a role of NOX enzymes, or at least discuss their lack of specificity. Specific inhibitors24 or deletion or knockdown of NOX components should be used to identify their roles.
General principles of measurement of ROS and oxidative damage
When investigating ROS in biological systems, it is important to detect and quantify the ROS of interest. This can be done using electron paramagnetic (spin) resonance (EPR/ESR), various probe molecules or by measuring oxidative modifications (‘oxidative damage’, Box 1) caused by the ROS1. Most ROS probes capture only a small percentage of any ROS formed. Indeed, if the probe reacted with most of the ROS generated this would perturb the system and affect experimental results (for example, inhibition of oxidative damage or interference with redox signalling). However, it is important that the percentage capture remains approximately constant over different rates of production of the ROS in question.
Oxidative damage can take many forms; the chemical processes by which it arises from a particular ROS and how it is assessed and quantified are complex. Furthermore, the final level of any oxidative damage biomarker measured is the difference between its rate of production and its removal by repair, degradation, excretion or diffusion. Recommendation 4: when oxidative damage levels to any biomolecule are presented, the chemical processes by which they arise and the methods used to quantify them should be made explicit. The impact of repair and clearance on the final levels measured should be considered and discussed.
Guidelines and limitations of the detection and measurement of ROS
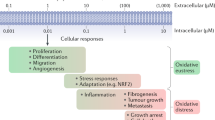
Consideration of ROS, antioxidants and oxidative damage as monolithic concepts limits the precision and interpretation of experiments and glosses over the need to establish precise molecular mechanisms. To put these precepts into practice requires measurement of specific ROS and/or oxidative damage products, as well as the effects of antioxidants. This is a major practical challenge, because most ROS are short lived (lifespans of milliseconds or less) and their steady-state levels are low (picomolar to low micromolar) and alter rapidly, because they are affected by continuously varying rates of generation, chemical reaction and diffusion.
In simple in vitro systems it is possible to detect several ROS (Table 2). For example, O2•− production can be monitored by the reduction of cytochrome c, and its selectivity assessed by inhibition by added SOD. However, even such a ‘simple’ system can be surprisingly complex. For example, semiquinones can reduce cytochrome c in a reaction inhibited by SOD25. The bottom line is that all methods used to assess ROS are susceptible to artefact, and appropriate controls are required to be certain of the species and amounts measured. Hence, it is important to corroborate measurements with ‘orthogonal techniques’ that rely on an alternative approach using a different detection method to avoid method-specific artefacts. These complexities are magnified when one attempts to measure ROS in cells. Commonly used cell culture conditions promote oxidative damage due to both limited antioxidants in the medium and high O2 concentrations relative to those in vivo26. Consequently, cultured cells generate more ROS than these cells would in vivo.
Recommendation 5: use commercial kits only if the actual species being measured and the method of detection are explained in the kit materials, are chemically plausible and the limitations are understood. The use of commercial kits without such information is strongly discouraged. To avoid method-specific artefacts, confirm results using techniques that rely on different principles of detection.
Small-molecule fluorescent probes are frequently used to assess ROS within cells. In some cases, often involving kits, a lack of description of the chemical reactivity or structures of these probes makes it difficult to interpret results and therefore such probes should be avoided. Even for probes of known structure there can be concerns. Consider the widely used fluorescent probe 2',7'-dichlorodihydrofluorescein (DCFH), usually administered in its diacetate (DCFH-DA) form, which enters cells readily. DCFH is oxidized to the fluorescent product 2',7'-dichlorofluorescein (DCF) by several ROS, and so it is not specific for any particular ROS6,7. DCFH is not oxidized directly by H2O2 (which it is often claimed to detect), but only after H2O2 is converted to more reactive species by redox-active metals or by haem proteins such as cytochrome c or peroxidases. Furthermore, the oxidation of DCFH and fluorescence of DCF are sensitive to local O2 levels and pH, and fluorescence yield may not be linear with increased ROS levels27,28,29. This is not to say that DCFH, and other non-specific fluorescent probes such as dihydrorhodamine, should never be used, but their limitations (selectivity, problems of quantification, linearity of response and susceptibility to artefact) should be understood and results interpreted cautiously28. In particular, their response should not be attributed to a specific ROS without detailed controls to validate this, and their use should be restricted to an initial assessment of a change in cellular redox state, to be followed up by a more detailed investigation into mechanism. While many small-molecule and protein fluorescent probes are more selective than DCF, it is always important to validate data by a number of simple controls: does the response change over time and with the amount of biological sample in a plausible manner? Can the effect be replicated by generating the ROS of interest (for example, using PQ for O2•− or d-amino acid oxidase for H2O2)? Do negative controls that should abolish the ROS-generating process (for example, gene knockouts, knockdowns, inhibitors, radical scavengers) respond as expected? Recommendation 6: when using fluorescent ROS probes (especially DCFH-DA), the chemistry involved, the selectivity for particular chemical species and potential artefacts should be made clear and discussed. Wherever possible, controls to show that the response is due to the proposed species should be carried out and orthogonal techniques used to corroborate the conclusion.
Extending measurements of ROS from cells in culture to tissues in vivo or ex vivo is vital. However, in some cases this gap has been addressed by the addition of ‘ROS probes’ to fresh or previously frozen tissue slices or homogenates ex vivo. These measurements may be meaningless, because the very short lifespan of ROS means that any present in vivo will have long disappeared by the time the material is assayed. Furthermore, freezing or homogenization disrupts membranes and alters substrate and ion concentrations (for example, raising levels of Ca2+ or ‘catalytic’ Fe2+)1, such that any ROS production in the tissue slice or homogenate bears no relation to the levels that would have been generated in vivo. There are valid methods available to assess ROS in vivo or in perfused organs, but in these situations the process is either monitored in vivo (for example, see Table 2 for the use of catalase compound I to measure H2O2) or the system is quenched to stabilize the probe for analysis ex vivo. Recommendation 7: measurements of ROS should be carried out in cells, tissues or organs under physiologically relevant conditions in vivo or ex vivo. ROS should not be ‘measured’ in tissue homogenates or cryosections, unless the probe or sensor employed is able to irreversibly capture the reactive species when the cells/tissues/organs are under biologically relevant conditions.
Direct measurement of ROS
Here we outline what we consider to be, currently, the best approaches to the measurement of commonly encountered ROS.
Superoxide
In simple systems O2•− can be measured in a number of ways, such as by the SOD-inhibitable reduction of cytochrome c (ref. 25). The generation of O2•− can also be assessed by spin trapping followed by EPR, which has the benefit of direct detection of the radical1. The Fe–S cluster in aconitase is inactivated by O2•−, and by other ROS, but its interaction with O2•− is fast, reasonably specific and reversible, making it a good indicator for O2•− in mitochondria30. The chemiluminescent ‘superoxide probes’ luminol and lucigenin are widely used to ‘detect O2•−’, but interpretation of such data is difficult because these probes generate radicals that produce O2•− themselves; they do not react with O2•− directly31,32.
Recommendation 8: the use of luminol and lucigenin to ‘detect O2•−’ should be discouraged, but they can be used as general indicators of increased ROS production. SOD-sensitive reduction of cytochrome c in vitro and aconitase inactivation within mitochondria are better strategies.
In cells, O2•− is often detected by measuring the fluorescence arising from oxidation of dihydroethidium (sometimes called hydroethidine (HE)), or mitochondria-targeted HE (MitoSOX). Unfortunately, detection by fluorescence is misleading because these probes form both ethidium (E+), a non-specific oxidation product, and the O2•−-specific product 2-hydroxyethidium. Because these two products have overlapping fluorescence spectra, it is hard to differentiate the contribution of non-specific oxidation and O2•−-dependent oxidation (if any) to the overall fluorescence33. Accurate quantification of the 2-hydroxyethidium product can be achieved using liquid chromatography–mass spectrometry (LC–MS)33. Another factor that should be considered is the extent of cellular uptake of HE/MitoSOX and the intracellular concentrations of these and their multiple products. Furthermore, HE oxidation products intercalate into DNA, greatly enhancing their fluorescence and creating another artefact. NeoD and MitoNeoD contain a modified HE that does not intercalate into DNA34.
Mitochondrially accumulated O2•−probes, such as MitoSOX, are often used to ‘detect O2•−’ within mitochondria. When using these probes, and others that have positive charges or generate positively charged species (including 2-hydroxyethidium and ethidium), it is important to remember that probe accumulation is dependent on plasma and mitochondrial membrane potentials and mitochondrial size, shape and mass35. Furthermore, fluorescence can be quenched when these are present at high concentrations in mitochondria36.
Recommendation 9: use only HE or MitoSOX probes to detect O2•−by simple fluorescence measurements when the product has been independently validated as 2-hydroxyethidium. Fluorescence measurements with probes such as dihydroethidium and MitoSOX33 should be conducted using the lowest probe concentration possible, and must include controls for changes in plasma and mitochondrial membrane potentials and mitochondrial mass and morphology, such as normalization to a similar membrane-potential-responsive, but redox-insensitive, probe. LC–MS methods, which measure all modified species33, should be performed when possible.
Hydrogen peroxide
In simple systems, H2O2 can be measured by horseradish peroxidase (HRP)-oxidizing substrates, one frequently used being Amplex Red. These methods can be interfered with by other HRP substrates (for example, ascorbate and NAC)1 and by O2•− (which can inactivate HRP), the latter preventable by the addition of SOD37. Since H2O2 can cross membranes directly or via aquaporins, this system can also be used to measure H2O2 release from cells. However, please be aware that this release reflects the balance between H2O2 production, removal by intracellular enzymes and the rate of diffusion out of the cell.
Within cells, H2O2 detection by phenylboronate-based probes is more reliable38 although these may lack sufficient sensitivity because they react only slowly with H2O2, which can make it difficult to detect small or localized changes in H2O2 levels39. However, recent studies suggest that borinic acids, which react more rapidly with H2O2, may be more sensitive detectors40. The mechanism of oxidation of phenylboronates to phenols requires a two-electron oxidant, such as H2O2. Because H2O2 is typically generated at higher concentrations than other ROS, boronate probes can be selective for H2O2 detection subject to proper controls39,40. However, boronate probes react with ONOO−/ONOOH or HOCl much more rapidly than they do with H2O2 which can sometimes complicate measurements, and orthogonal approaches or the use of inhibitors can aid validation41. For example, H2O2- and peroxynitrite-dependent signals can be distinguished using nitric oxide synthase (NOS) inhibitors and catalase38,39,41,42.
Genetically encoded fluorescent protein sensors have provided major advances in cellular H2O2 detection43,44,45,46. These probes contain a dithiol switch that changes the overall fluorescence of the probe depending on its oxidation status. High sensitivity and specificity for H2O2 have been achieved by coupling a redox-sensitive green fluorescent protein (GFP) mutant to a H2O2-sensitive thiol protein, such as oxyR (HyPer series), or to a peroxidase such as Orp1 or TSA2 (roGFP2-based probes). HyPer7 and roGFP2 coupled to a peroxiredoxin provide the highest sensitivity44,45. While HyPer7- and roGFP2-based probes are pH stable, earlier versions of HyPer are not and require expression of a control probe (SypHer) to control for signal changes due to variation in pH43. Imaging analysis by fluorescence microscopy is normally employed, but fluorescence plate readers can also be used. The redox status measured represents a balance between the rate of oxidation and re-reduction of the probes by cellular reductants, including glutaredoxin/GSH and thioredoxin, permitting real-time, live-cell assessments of redox state. Because excitation wavelengths of both reduced and oxidized probes are used, the probes are ratiometric and the output is not dependent on the level of protein probe expression. By incorporation of appropriate targeting gene sequences, these probes can be directed to different cell compartments, including mitochondria, microtubules, endoplasmic reticulum, nucleus and cytoplasm43,44,45,46. Hence, subcellular regions of interest can be studied and the probe then calibrated at the end by full reduction (2 mM dithiothreitol), washout and full oxidation (2 mM t-butylhydroperoxide)44,45. This calibration yields a measure of oxidation percentage, permitting comparisons across experiments and among subcellular compartments44,45. These probes have been expressed in transgenic animals to provide useful assessments of in vivo H2O2 generations46,47. Plasmid transfection of viral vectors can be used with cultured cells, and targeted roGFP2 probes are available commercially (www.addgene.com).
In most experiments the H2O2 probes are expressed as free proteins that distribute within the cell. Nevertheless, given uncertainties about intracellular H2O2 diffusion distances, it is still unclear what resolution is needed to understand subcellular H2O2 distribution. Therefore, tethering H2O2 probes to sub-compartmental locations such as protein complexes or organelle contact sites is an important approach.
Recommendation 10: genetically encoded fluorescent probes (some of which are commercially available) are currently the most sensitive detectors of H2O2 and we recommend their use in cells and animals if expression is possible. Boronate probes (some of which are also commercially available) are the preferred small-molecule probes, but controls to determine specificity for H2O2 are required and sensitivity is limited for physiological H2O2 levels. Amplex Red with HRP can measure H2O2 release from cells if other reducing agents or peroxidase substrates are absent.
Peroxynitrite
Peroxynitrite (ONOO−) exhibits complex chemistry42,48,49 and itself can oxidize certain biomolecules. A major physiological reaction is with CO2 (Table 1), and hence the CO2/ \({{{\mathrm{HCO}}}}_3^{- }\) content of biological systems plays a role in determination of the biological impact50 of ONOO−. Products of this reaction include reactive species such as the carbonate radical anion (CO3•−) and the nitrating agent nitrogen dioxide (NO2•) (Table 1), both of which react with many of the general ‘ROS probes’. Peroxynitrite oxidizes boronate-based probes nearly a million times more rapidly than H2O2 and, under the right conditions, these probes can be used to assess ONOO−/ONOOH production42,49. Peroxynitrite has been measured in tissues ex vivo using boronate probes51.
Hypochlorous acid and other reactive halogen species
HOCl, hypobromous acid (HOBr) and some of the chloramines and bromamines derived from them (Table 1) react with most of the general probes used to detect ROS, including DCFH and luminol. However, many of these probes are also substrates of the peroxidases that generate HOCl or HOBr, confounding their use. More specific fluorescent probes for reactive halogen species have been reported and some are commercially available52. A genetically encoded probe for reactive halogen species has been developed, enabling dynamic monitoring of these species both in cell culture and in vivo53.
Measurement of oxidative damage
The presence of ROS can be inferred by their effects on protein, carbohydrates, nucleic acids and lipids to generate specific compounds which, so long as they cannot be formed by other mechanisms, can be used as ‘biomarkers’ of oxidative damage (Box 1)1,54,55. However, do note that the measured levels of biomarkers represent a balance between the generation and removal of the biomarker (for example, by degradation, diffusion or excretion), plus any artefactual increased levels caused by oxidative damage during isolation or analysis.
Lipid peroxidation
Polyunsaturated fatty acids (PUFAs), are readily oxidized, and hence lipid peroxidation products are widely used to characterize oxidative damage56,57,58. Lipid peroxidation can be initiated by certain ROS and proceed as a random, non-enzymatic (often chain) radical process. However, there are also enzymatic mechanisms (for example, lipoxygenases) available for peroxidation of free PUFAs or PUFA-phospholipids that produce specific signalling products with biological roles. Thus, when measuring lipid peroxidation the focus might be placed on either (1) establishment of increased lipid peroxidation as an example of oxidative damage or (2) identification of individual oxidatively modified lipid molecules acting as signals by selective interaction with certain cellular targets.
In PUFAs, the presence of a double bond adjacent to a methylene group makes the methylene C–H bond weaker and therefore the bis-allylic hydrogen is more susceptible to H• abstraction56,57,58. The carbon-centred radical (L) generated by H• abstraction is stabilized by delocalization over the double bonds. Subsequent reaction with O2 gives a peroxyl radical (LOO) with the formation of a conjugated diene system and a range of peroxides (LOOH). LOO• can react further to yield highly oxidized secondary products, including epoxy-, oxo- or cyclic peroxides56,57,58. Hence, there are multiple end products of lipid peroxidation that show vast chemical heterogeneity and variable stability and polarity, and thus measurement of only a single oxidation product in no way represents the whole process of lipid peroxidation.
Several methods are available to assess ‘general’ lipid peroxidation. In simple model systems (for example, isolated lipoproteins), diene conjugation can be measured by ultraviolet (UV) absorbance but this method is not suitable for use in cells or body fluids because of the presence of interfering UV-absorbing molecules that do not result from lipid peroxidation1. In cells, ‘lipid peroxidation’ can be assessed by changes in the fluorescence of BODIPY conjugated to a peroxidation-sensitive undecanoic acid moiety59. This assay is technically simple but should be interpreted cautiously because BODIPY’s rate of reaction with peroxyl radicals is slower than that of radical-scavenging antioxidants, and hence suppression of BODIPY fluorescence by antioxidants need not always reflect their ability to suppress lipid peroxidation59. Another fluorometric assay for lipid peroxidation employs cis-parinaric acid (PnA), a fatty acid with four conjugated double bonds. Oxidation of PnA disrupts its conjugated system and hence fluorescence. Because PnA may be incorporated into different classes of phospholipid, high-performance liquid chromatography (HPLC) separation provides information on the oxidation of different phospholipids60. However, extrapolation of PnA-based results to endogenous phospholipid oxidation is difficult due to the higher oxidation rate of PnA, its vulnerability to photobleaching and variable metabolic incorporation of PnA into different phospholipids60.
Lipid peroxidation is frequently assessed by the measurement of end products such as α,β-unsaturated hydroxyalkenals61, ideally by MS-based techniques. In particular, 4-hydroxynonenal (HNE) formation has been widely used. Antibodies against the protein adducts formed by HNE are widely available and frequently used in immunostaining of tissues, but it should be realized that different antibodies can detect different epitopes and so give different answers, depending on what amino acid residues the HNE binds to in proteins61,62,63,64.
One minor end product of lipid peroxidation is malondialdehyde (MDA)61, which can also be a useful biomarker if measured by MS techniques. However, the widely used ‘MDA assays’ utilizing thiobarbituric acid-reactive substances (TBARS) are unspecific since TBA generates chromogens from many biomolecules other than MDA1,65. Use of HPLC to separate the ‘real’ TBA–MDA adduct from false chromogens increases specificity but does not eliminate all problems1.
Recommendation 11: application of the simple TBA test (TBARS), or kits based on its use, to cells, tissues or body fluids is not recommended as the only test used for evaluation of oxidative lipid damage because of the low specificity that can result in false-positive results. HPLC-based TBA tests are less prone to artefacts.
The detection of lipid oxidation products has been revolutionized by the development of LC–MS for detailed analysis of oxidized lipid mixtures66. Collection and storage of samples to avoid artefactual peroxidation is key to any lipid peroxidation study, and samples for later analysis should be immediately frozen in liquid nitrogen. Biofluids may require the addition of chemicals (for example, butylated hydroxytoluene) to prevent auto-oxidation during storage1,67. The internal standards used for quantification should be added to samples before solvent extraction. Such LC–MS-based methods have the advantages of high sensitivity, small sample volume requirements and the ability to detect multiple end products of lipid peroxidation. This makes LC–MS protocols the methods of choice for assessment of general lipid peroxidation and identification of individual products, including those with specific signalling functions. However, limitations of available standards may sometimes preclude quantitative analysis of certain products. Scrupulous attention to methodology is required in such studies68.
Prominent among lipid oxidation products that have been quantified by MS-based approaches are the F2-isoprostanes (F2-IsoPs)69. Sixty-four F2-IsoP stereoisomers can be generated from the free-radical-induced, non-enzymatic oxidation of arachidonic acid and can be separated from those that arise from the enzymatic oxidation of arachidonic acid by cyclo-oxygenase enzymes (COX-1/-2). F3- and F4-isoprostanes arise from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), respectively, but have been less well characterized than the F2-isoprostanes. ELISA methods have been developed to quantify one F2-IsoP isomer, 8-iso-PGF2α (also referred to as 15-F2t-IsoP or iPF2α-III), and compared with gas chromatography–MS and LC–tandem MS (LC–MS/MS) methods69,70,71,72,73. In all these studies there was poor agreement between commercially available ELISA kits and MS methods; 8-iso-PGF2α is one of 64 different F2-IsoP isomers generated during arachidonic acid peroxidation, and antibody cross-reactivity between 8-iso-PGF2α and related isomers is challenging. Pre-analysis sample clean-up may allow for more precise measurement of 8-iso-PGF2α by ELISA72,73, but by far the most accurate method for quantification of F2-IsoPs is by LC–MS/MS and is very strongly recommended.
Recommendation 12: F2-IsoPs are a generally accepted biomarker of lipid peroxidation, but it should be realized that they are one of many end products and that the levels of various types can be affected by experimental conditions69. Quantification using ELISA is susceptible to artefact, but sample clean-up may allow measurement of 8-iso-PGF2α by ELISA73. LC–MS/MS with appropriate internal standards is the preferred approach.
Protein damage
Amino acid residues in proteins are sensitive to oxidative modification, some forms of which provide useful biomarkers74,75. Detailed protocols for measurement of multiple products can be found in refs. 76,77. A common protein modification is the formation of ‘protein carbonyls’ due to oxidation of specific amino acid residues to carbonyl group-bearing products; carbonyls can also be formed by the reaction of aldehydes with nucleophilic sites on proteins or by glycation75,77. Many assays involve derivatization of the carbonyl group with 2,4-dinitrophenylhydrazine to form a dinitrophenylhydrazone (DNP). This product can be detected spectrophotometrically, although this approach can suffer from high background and low reproducibility; to circumvent this, DNP adducts can be separated by LC before measurement. Alternatively, carbonyls can be detected using an antibody against DNP products by ELISA or immunoblotting78. Changes in protein carbonyls can be measured in tissue homogenates treated with fluorescein-5-thiosemicarbazide (FTC) to generate fluorophore-labelled proteins that can be separated by gel electrophoresis79. Enrichment methods using biotin-tagged derivatization coupled with LC–MS detection have been developed80. Protein carbonyls, α-aminoadipic semialdehyde and glutamic semialdehyde have also been assayed individually by stable isotopic dilution analysis LC–MS/MS77. Of course, data from a single time point reflect the difference between the rates of formation and removal (for example, by repair or proteolysis) of these products.
Protein analysis using MS allows detection and identification of modifications with characteristic mass increases (for example, hydroxylation, nitration and chlorination)75,76. This has been particularly useful in studies of oxidative damage to brain proteins in patients with dementia by ‘redox proteomics’81. Peptide-level mapping after proteolytic cleavage allows detection of the nature of the modification, its location within the protein sequence and concomitant loss of the parent peptide, allowing relative quantification. Amino acid analysis after complete digestion allows determination of types and absolute concentrations of particular species (determined by the use of isotope-labelled standards) together with the parent species, allowing determination of a ‘mass balance’75,76,77,82. Care must be taken in sample handling to prevent artefactual oxidation of Cys or Met, and also during protein hydrolysis, because some products of oxidative protein damage are labile. LC–MS analyses have many advantages including high specificity, high sensitivity, the capacity to detect many different modifications and parent species concurrently, as well as the capacity to detect products that are diagnostic of the ROS involved, such as chlorination from HOCl83 and nitrated species arising from the action of myeloperoxidase in the presence of NO2− and/or by reactions of ONOO−/ONOOH49,84). These LC–MS approaches can be carried out on materials ranging from isolated proteins to tissue samples. Quantification relative to non-modified amino acids or peptides, and preferably against added heavy-isotope-labelled materials, is recommended to overcome potential artefacts arising from sample handling and preparation. However, do bear in mind that plasma and urinary levels of oxidized amino acids might have contributions from absorption of oxidized amino acids from proteins in food or from increased tissue proteolysis as a result of pathology. Neither of these have yet been studied in detail.
Cysteine is a major target for modification due to its ease of oxidation (particularly in its thiolate form, RS−) and its nucleophilicity, which results in ready adduct formation with electrophiles. Oxidation can be irreversible—for example, to a sulphinic or sulphonic acid, which can be useful biomarkers of oxidative protein damage4,85. Reversible oxidation of Cys residues in proteins is a prominent mechanism of redox signalling2,4. Reversible products include disulphides, sulphenic acids, S-nitrosothiol and persulphide species2,4,85,86,87. These modifications can be reversed by the GSH/glutaredoxin or thioredoxin systems87. A common approach in detection of the pool of reversibly modified Cys residues is to first block reduced thiols with a reactive reagent and then reduce and derivatize the previously oxidized residues with a tag that can be identified by LC–MS of tryptic digests88. These approaches can be extended to the use of modification-specific chemistry to tag only a particular oxidation product, such as an S-nitrosothiol, sulphenic acid or persulphide88. Until recently, major limitations to these approaches were low coverage of the total Cys pool and lack of quantitation of the modification at individual residues87,88,89. The latter is of particular importance when interpreting the biological importance of reversible modifications. Substantial improvements in quantification have been achieved by isobaric tagging, in which reduced Cys residues are first labelled with one tag, reversibly modified residues are reduced and then labelled with a chemically identical, but heavy-isotope-modified, tag that enables quantification of the proportion oxidized for each particular Cys. These methods have been extended to tags that incorporate moieties such as biotin, which enable enrichment of the labelled peptides, greatly enhancing Cys coverage. The most recent iteration of this approach, as exemplified by the OxiMOUSe study89, has superseded previous methods.
Methionine is also a major site of redox post-translational modifications. Oxidation to methionine sulphoxide can be reversed enzymatically by methionine sulphoxide reductase enzymes, potentially facilitating redox signalling by the installation/removal of a single oxygen atom17. Reagents have been developed for methionine bioconjugation that can identify and characterize redox-sensitive methionine sites in proteomes90.
Recommendation 13: ELISA, FTC and immunoblotting are useful tools in the detection of protein carbonyls as a biomarker of general oxidative protein damage, although it must be realized that not all protein oxidation products contain carbonyls. LC–MS approaches, using carefully prepared samples, are the best available techniques for assessment of protein oxidation due to the sensitivity, selectivity and quantitation available with these methods. The use of orthogonal approaches, such as specific and validated antibodies (see below) against individual oxidation products, is also encouraged.
Nucleic acids
Oxidative modifications of DNA and RNA are often used as biomarkers of oxidative damage1,13,91. One method used to assess ‘general’ oxidative damage to DNA in cells is the comet assay, which detects DNA strand breaks. Such breaks can arise by several mechanisms, not necessarily via oxidative damage, but the use of repair enzymes that ‘nick’ DNA at the sites of oxidation increases the specificity for oxidative DNA damage. The simplest measurement is the length of the DNA ‘ghost’ following electrophoresis of cells embedded on a gel on a microscope slide92.
Oxidative damage to DNA usually focuses on oxidation of guanine to 8-oxo-7,8-dihydro-2′-deoxyguanosine (8OHdG, or 8-oxodG). Data on modifications at other bases are limited, although these are likely to be biologically important1,13. These measurements require the isolation of the DNA and its digestion to release modified bases, and there can be spurious oxidation during sample handling and analysis. Multi-laboratory initiatives93 have established protocols to avoid this and have determined ‘normal’ levels of 8OHdG. The amount of 8OHdG (or any other product of oxidative DNA damage) measured in DNA is the balance between the rate of oxidation and that of repair. The best methodology is ultra-performance LC–MS/MS (UPLC–MS/MS)94. Caution should be exercised in using ELISA methods, which lack sensitivity and specificity and can give variable results between batches, and there is sometimes cross-reaction between 8-hydroxyguanosine (8OHG) and 8OHdG. However, immunohistochemistry can be useful in identification of cells that have higher amounts of 8-OHdG in vivo, if applied appropriately95.
Oxidized nucleosides from both DNA and RNA can be detected in various body fluids. Originally they were believed to arise from DNA repair, particularly nucleotide excision repair. However, they also arise from oxidation of the DNA and RNA nucleotide precursor pools ‘sanitized’ by removal of oxidized products94. The relative contributions of DNA repair and nucleotide pool sanitization to the levels of oxidized nucleosides detected in body fluids are currently unclear. Urine collected over 24 h will represent the number of guanines in DNA/RNA and/or the respective nucleotide precursor pools oxidized during that period96. Urine sampling represents formation within the entire body, and is best suited to situations where all tissues are assumed to be affected, but it could be inadequate in the detection of changes that occur only in certain organs. Measurement in specific tissues will be a snapshot of the balance between generation and repair, and may not represent processes in other organs.
Recommendation 14: when measuring oxidative modifications of nucleic acids from extracted cells or tissue samples, great care must be taken to avoid spurious oxidation in the preparative and analytical steps. Methods such as the comet assay (using DNA repair enzymes) on isolated cells and UPLC–MS/MS for 8OHdG and 8OHG determination in body fluids or nucleic acids extracted from tissues are presently the best available. ELISA-based methods, especially in kit form, are usually insufficiently validated and their use is not recommended.
Some general comments on antibodies
As discussed above, antibodies have been widely used to detect oxidation products (and also adducts) formed on proteins (for example, carbonyls and 3-nitro- and 3-chlorotyrosine), DNA (for example, 8-oxodG) and lipids (F2-Isoprostanes). They have been used, for example, in ELISA, immunohistochemistry and immune precipitation formats, but often suffer from background reactivity, cross-reactivity and lack of specificity. To address this, the epitope used to generate the antibody should be documented (for example, as for HNE)62,63,64 and controls to eliminate background should be included. Blocking by authentic samples of the epitope is recommended to determine selectivity. Relative quantification is possible but absolute quantification can be difficult—for example, due to poor epitope accessibility (for example, in proteins the oxidation product may be buried). In addition, antibodies are typically generated against unstructured, chemically modified peptides and the epitope(s) recognized may not always have been determined.
Recommendation 15: well-validated antibodies against specific products are useful detection tools when used with appropriate care and controls, including those for non-specific interactions. Competition data with authentic epitopes should be included whenever possible.
Measurement of ROS and oxidative damage in vivo
Measurement of ROS in vivo is a challenge. EPR methods have been developed but are not yet widely used. Bioluminescent approaches to ROS detection include peroxy-caged luciferin-1 which, upon oxidation, forms luciferin in situ that is oxidized in luciferase-transfected systems to generate bioluminescence97. As noted earlier, genetically encoded redox biosensors have been used in animal studies. With the development of improved sensitivity and detection modalities, positron emission tomography is now being used to image ROS in vivo98 but is still in its infancy. In mitochondria of cells and tissues, changes in H2O2 can be assessed using the mitochondria-targeted boronate MitoB, which accumulates in these organelles and is converted by H2O2 into MitoP. The ratio of MitoP to MitoB can then be determined by MS99.
Measurement of ROS and oxidative damage in clinical trials
Because oxidative damage plays a central role in many human pathologies, there is considerable interest in developing therapeutic interventions to decrease this damage1,2,3. A corollary is that in clinical trials we should be able to demonstrate how these interventions affect oxidative damage. For example, many double-blinded randomized clinical trials have been conducted using ‘antioxidants’ such as beta-carotene, vitamin C and vitamin E. These generally failed to influence disease activity. Unfortunately, in most cases the effect of the intervention on oxidative damage was not measured, making it uncertain whether the putative therapy was actually effective at decreasing oxidative damage: if it was not, lack of effect is predictable1,55.
To address this, it is essential to assess the impact of these interventions on levels of oxidative damage in the patients in clinical trials. Currently, methods are limited to measuring end points of oxidative damage in either biopsies (for example, skin or muscle) or clinically accessible body fluids such as plasma, saliva, sputum or urine, and sometimes cerebrospinal fluid. These biomarkers have included those for oxidation of nucleic acids such as 8OHG and 8OHdG100 and F2-isoprostanes as a biomarker of lipid peroxidation69,101. To date, limited use has been made of biomarkers of protein oxidation in clinical trials. However, there is evidence for strong associations of alterations in protein thiol/disulphide ratios, and increased protein carbonyls and other modifications with pathologies75,76,77.
More generally, clinical trials should include internationally validated biomarkers: the biomarker should ideally have undergone interlaboratory comparison. Many biomarkers rely on concentration measurement in body fluids such as plasma, but these reflect only the balance between formation and elimination rates and therefore cannot readily be interpreted as ‘oxidative stress’. However, models have been developed to estimate the 24 h production of certain biomarkers100. Ideally a panel of biomarkers should be used54,55 since end products of oxidative damage to lipids, proteins and nucleic acids do not necessarily correlate with each other, nor would we expect them to since they are different molecular targets of different ROS.
Recommendation 16: if intervening with antioxidants, first use biomarkers in preliminary dose-ranging studies to determine whether the intervention does indeed decrease oxidative damage to the relevant biomolecules. They should include well-defined biomarkers analysed with a validated methodology and/or orthogonal approaches. We do not recommend in clinical (or other!) studies the use of the d-ROMS assay (for reasons explained in ref. 1), TBARS, determinations of total antioxidant activity1,102 or kit-based methods where the methodology behind the kit is not clear and/or has not been validated.
Concluding remarks
The goal of this consensus statement is to generate a useful resource for researchers from diverse fields who find themselves needing to measure ROS and to assess oxidative events to investigate their biological importance. We have discussed the limitations of many of the procedures currently used and suggested the best currently available approaches. Inevitably, new techniques will be developed and applied in the future, but the principles of our cautious philosophy, illustrated by our 16 recommendations (summarized in Fig. 1), will remain valid.

Here we have summarized and abbreviated the recommendations for best practices developed in this manuscript.
References
Halliwell, B. & Gutteridge J. M. C. Free Radicals in Biology and Medicine 5th edn (Oxford Univ. Press, 2015).
Sies, H. et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-022-00456-z (2022).
Forman, H. J. & Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20, 689–709 (2021).
Lennicke, C. & Cochemé, H. M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 81, 3691–3707 (2021).
Winterbourn, C. C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 1840, 730–738 (2014).
Zielonka, J. et al. Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analyses. J. Biol. Chem. 287, 2984–2995 (2012).
Forman, H. J. et al. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 78, 233–235 (2015).
Brewer, T. F., Garcia, F. J., Onak, C. S., Carroll, K. S. & Chang, C. J. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 84, 765–790 (2015).
Janssen-Heininger, Y. M. et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 (2008).
Murphy, M. P. et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 13, 361–366 (2011).
Möller, M. N. et al. Detection and quantification of nitric oxide-derived oxidants in biological systems. J. Biol. Chem. 294, 14776–14802 (2019).
Varghese, S., Tang, Y. & Imlay, J. A. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J. Bacteriol. 185, 221–230 (2003).
Halliwell, B., Adhikary, A., Dingfelder, M. & Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 50, 8355–8360 (2021).
Halliwell, B. Reflections of an aging free radical. Free Radic. Biol. Med. 161, 234–245 (2020).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Winterbourn, C. C. Hydrogen peroxide reactivity and specificity in thiol-based cell signalling. Biochem. Soc. Trans. 48, 745–754 (2020).
Lim, J. M., Kim, G. & Levine, R. L. Methionine in proteins: it’s not just for protein initiation anymore. Neurochem. Res. 44, 247–257 (2019).
Pedre, B., Barayeu, U., Ezeriņa, D. & Dick, T. P. The mechanism of action of N-acetylcysteine (NAC): the emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 228, 107916 (2021).
Blaner, W. S., Shmarakov, I. O. & Traber, M. G. Vitamin A and vitamin E: will the real antioxidant please stand up? Annu. Rev. Nutr. 41, 105–131 (2021).
Policar, C., Bouvet, J., Bertrand, H. C. & Delsuc, N. SOD mimics: from the tool box of the chemists to cellular studies. Curr. Opin. Chem. Biol. 67, 102109 (2022).
Samuni, U., Samuni, A. & Goldstein, S. Cyclic hydroxylamines as monitors of peroxynitrite and superoxide–revisited. Antioxidants (Basel) 11, 40 (2021).
Dhar, S. K., Scott, T., Wang, C., Fan, T. W. M. & St Clair, D. K. Mitochondrial superoxide targets energy metabolism to modulate epigenetic regulation of NRF2-mediated transcription. Free Radic. Biol. Med. 179, 181–189 (2022).
Steinhorn, B. et al. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun. 9, 4044 (2018).
Herb, M., Gluschko, A. & Schramm, M. Reactive oxygen species: not omnipresent but important in many locations. Front. Cell Dev. Biol. 9, 716406 (2021).
Nauseef, W. Detection of superoxide and H2O2 produced by NADPH oxidases. Biochim. Biophys. Acta 1840, 757–767 (2014).
Halliwell, B. Cell culture, oxidative stress, and antioxidants: avoiding pitfalls. Biomed. J. 37, 99–105 (2014).
Kowaltowski, A. J. Strategies to detect mitochondrial oxidants. Redox Biol. 21, 101065 (2019).
Kalyanaraman, B. et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6 (2012).
Brandes, R. P., Rezende, F. & Schröder, K. Redox regulation beyond ROS: why ROS should not be measured as often. Circ. Res. 123, 326–328 (2018).
Gardner, P. R. Superoxide-driven aconitase Fe-S center cycling. Biosci. Rep. 17, 33–42 (1997).
Vásquez-Vivar, J., Hogg, N., Pritchard, K. A. Jr, Martasek, P. & Kalyanaraman, B. Superoxide anion formation from lucigenin: an electron spin resonance spin-trapping study. FEBS Lett. 403, 127–130 (1997).
Zielonka, J., Lambeth, J. D. & Kalyanaraman, B. On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: a reevaluation. Free Radic. Biol. Med. 65, 1310–1314 (2013).
Zielonka, J., Vasquez-Vivar, J. & Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat. Protoc. 3, 8–21 (2008).
Shchepinova et al. MitoNeoD: a mitochondria-targeted superoxide probe. Cell Chem. Biol. 24, 1285–1298 (2017).
Kowaltowski, A. J. et al. Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. FASEB J. 33, 13176–13188 (2019).
Nicholls, D. G. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods Mol. Biol. 1782, 121–135 (2018).
Kettle, A. J., Carr, A. C. & Winterbourn, C. C. Assays using horseradish peroxidase and phenolic substrates require superoxide dismutase for accurate determination of hydrogen peroxide production by neutrophils. Free Radic. Biol. Med. 17, 161–164 (1994).
Lippert, A. R., Van de Bittner, G. C. & Chang, C. J. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc. Chem. Res. 44, 793–804 (2011).
Winterbourn, C. C. Biological production, detection, and fate of hydrogen peroxide. Antioxid. Redox Signal. 29, 541–551 (2018).
Gatin-Fraudet, B. et al. Evaluation of borinic acids as new, fast hydrogen peroxide-responsive triggers. Proc. Natl Acad. Sci. USA 118, e2107503118 (2021).
Miller, E. W., Tulyathan, O., Isacoff, E. Y. & Chang, C. J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 3, 263–267 (2007).
Zielonka, J. et al. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 25, 1793–1799 (2012).
Bilan, D. S. & Belousov, V. V. In vivo imaging of hydrogen peroxide with HyPer probes. Antioxid. Redox Signal. 29, 569–584 (2018).
Morgan, B. et al. Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat. Chem. Biol. 12, 437–443 (2016).
Pak, V. V. et al. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 31, 642–653 (2020).
Guzman, J. N. et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700 (2010).
Breckwoldt, M. O. et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 20, 555–560 (2014).
Augusto, O. et al. Carbon dioxide-catalyzed peroxynitrite reactivity – the resilience of the radical mechanism after two decades of research. Free Radic. Biol. Med. 135, 210–215 (2019).
Ferrer-Sueta, G. et al. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 118, 1338–1408 (2018).
Augusto, O. & Truzzi, D. R. Carbon dioxide redox metabolites in oxidative eustress and oxidative distress. Biophys. Rev. 13, 889–891 (2021).
Kameritsch, P. et al. The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc. Natl Acad. Sci. USA 118, e1921828118 (2021).
Ma, C. et al. Recent development of synthetic probes for detection of hypochlorous acid/hypochlorite. Spectrochim. Acta A Mol. Biomol. Spectrosc. 240, 118545 (2020).
Kostyuk, A. I. et al. Hypocrates is a genetically encoded fluorescent biosensor for (pseudo)hypohalous acids and their derivatives. Nat. Commun. 13, 171 (2022).
Frijhoff, J. et al. Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 23, 1144–1170 (2015).
Halliwell, B. Establishing the significance and optimal intake of dietary antioxidants: the biomarker concept. Nutr. Rev. 57, 104–113 (1999).
Kagan, V. E. Lipid Peroxidation in Biomembranes (CRC Press, 1988).
Niki, E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic. Biol. Med. 47, 469–484 (2009).
Yin, H., Xu, L. & Porter, N. A. Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 111, 5944–5972 (2011).
MacDonald, M. L., Murray, I. V. & Axelsen, P. H. Mass spectrometric analysis demonstrates that BODIPY 581/591 C11 overestimates and inhibits oxidative lipid damage. Free Radic. Biol. Med. 42, 1392–1397 (2007).
Kuypers, F. A., van den Berg, J. J., Schalkwijk, C., Roelofsen, B. & Op den Kamp, J. A. Parinaric acid as a sensitive fluorescent probe for the determination of lipid peroxidation. Biochim. Biophys. Acta 921, 266–274 (1987).
Esterbauer, H., Schaur, R. J. & Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 (1991).
Waeg, G., Dimsity, G. & Esterbauer, H. Monoclonal antibodies for detection of 4-hydroxynonenal modified proteins. Free Radic. Res. 25, 149–159 (1996).
Toyokuni, S. et al. The monoclonal antibody specific for the 4-hydroxy-2-nonenal histidine adduct. FEBS Lett. 359, 189–191 (1995).
Ozeki, M. et al. Susceptibility of actin to modification by 4-hydroxy-2-nonenal. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 827, 119–126 (2005).
Yin, H. & Porter, N. A. Specificity of the ferrous oxidation of xylenol orange assay: analysis of autoxidation products of cholesteryl arachidonate. Anal. Biochem. 313, 319–326 (2003).
Li, L. et al. Recent development on liquid chromatography-mass spectrometry analysis of oxidized lipids. Free Radic. Biol. Med. 144, 16–34 (2019).
Liu, W. et al. Ex vivo oxidation in tissue and plasma assays of hydroxyoctadecadienoates: Z,E/E,E stereoisomer ratios. Chem. Res. Toxicol. 23, 986–995 (2010).
O’Donnell V. B. et al. Failure to apply standard limit-of-detection or limit-of-quantitation criteria to specialized pro-resolving mediator analysis incorrectly characterizes their presence in biological samples. Zenodo https://zenodo.org/record/5766267#.Yp4YzvnMLIU (2021).
Milne, G. L., Musiek, E. S. & Morrow, J. D. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers 10, S10–S23 (2005).
L’yasova, D., Morrow, J. D., Ivanova, A. & Wagenknecht, L. E. Epidemiological marker for oxidant status: comparison of the ELISA and the gas chromatography/mass spectrometry assay for urine 2,3-dinor-5,6-dihydro-15-F2t-isoprostane. Ann. Epidemiol. 14, 793–797 (2004).
Soffler, C., Campbell, V. L. & Hassel, D. M. Measurement of urinary F2-isoprostanes as markers of in vivo lipid peroxidation: a comparison of enzyme immunoassays with gas chromatography-mass spectrometry in domestic animal species. J. Vet. Diagn. Invest. 22, 200–209 (2010).
Tsikas, D. & Suchy, M.-T. Assessment of urinary F(2)-isoprostanes in experimental and clinical studies: mass spectrometry versus ELISA. Hypertension 60, e14 (2012).
Tsikas, D. Quantitative analysis of biomarkers, drugs and toxins in biological samples by immunoaffinity chromatography coupled to mass spectrometry or tandem mass spectrometry: a focused review of recent applications. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 133–148 (2010).
Davies, K. J. Protein damage and degradation by oxygen radicals: I general aspects. J. Biol. Chem. 262, 9895–9901 (1987).
Hawkins, C. L. & Davies, M. J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 294, 19683–19708 (2019).
Hawkins, C. L., Morgan, P. E. & Davies, M. J. Quantification of protein oxidation by oxidants. Free Radic. Biol. Med. 46, 965–988 (2009).
Rabbani, N. & Thornalley, P. J. Reading patterns of proteome damage by glycation, oxidation and nitration: quantitation by stable isotopic dilution analysis LC-MS/MS. Essays Biochem. 64, 169–183 (2020).
Winterbourn, C. C. & Buss, I. H. Protein carbonyl measurement by enzyme-linked immunosorbent assay. Methods Enzymol. 300, 106–111 (1999).
Chaudhuri, A. R. et al. Detection of protein carbonyls in aging liver tissue: a fluorescence-based proteomic approach. Mech. Ageing Dev. 127, 849–861 (2006).
Havelund, J. F. et al. A biotin enrichment strategy identifies novel carbonylated amino acids in proteins from human plasma. J. Proteom. 156, 40–51 (2017).
Butterfield, D. A. & Boyd-Kimball, D. Redox proteomics and amyloid β peptide: insights into Alzheimer’s disease. J. Neurochem. 151, 459–487 (2019).
Gamon, L. F. et al. Absolute quantitative analysis of intact and oxidized amino acids by LC-MS without prior derivatization. Redox Biol. 36, 101586 (2020).
Nybo, T., Davies, M. J. & Rogowska-Wrzesinska, A. Analysis of protein chlorination by mass spectrometry. Redox Biol. 26, 101236 (2019).
Halliwell, B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett. 411, 157–160 (1997).
Poole, L. B. et al. Introduction to approaches and tools for the evaluation of protein cysteine oxidation. Essays Biochem. 64, 1–17 (2020).
Shi, Y. & Carroll, K. S. Activity-based sensing for site-specific proteomic analysis of cysteine oxidation. Acc. Chem. Res. 53, 20–31 (2020).
Murphy, M. P. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 (2012).
Yang, J., Carroll, K. S. & Liebler, D. C. The expanding landscape of the Thiol Redox Proteome. Mol. Cell Proteom. 15, 1–11 (2016).
Xiao, H. et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 180, 968–983 (2020).
Lin, S. et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2017).
Poulsen, H. E. et al. RNA modifications by oxidation: a novel disease mechanism? Free Radic. Biol. Med. 52, 1353–1361 (2012).
Muruzabal, D., Collins, A. & Azqueta, A. The enzyme-modified comet assay: past, present and future. Food Chem. Toxicol. 147, 111865 (2021).
Gedik, C. M. & Collins, A. & ESCODD (European Standards Committee on Oxidative DNA Damage).Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 19, 82–84 (2005).
Henriksen, T., Weimann, A., Larsen, E. L. & Poulsen, H. E. Quantification of 8-oxo-7,8-dihydro-2′-deoxyguanosine and 8-oxo-7,8-dihydro-guanosine concentrations in urine and plasma for estimating 24-h urinary output. Free Radic. Biol. Med. 172, 350–357 (2021).
Toyokuni, S. et al. Quantitative immunohistochemical determination of 8-hydroxy-2’-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab. Invest. 76, 365–374 (1997).
Jorgensen, A., Thygesen, M. B., Kristiansen, U. & Poulsen, H. E. An in silico kinetic model of 8-oxo-7,8-dihydro-2-deoxyguanosine and 8-oxo-7,8-dihydroguanosine metabolism from intracellular formation to urinary excretion. Scand. J. Clin. Lab. Invest. 81, 540–545 (2021).
Van de Bittner, G. C., Dubikovskaya, E. A., Bertozzi, C. R. & Chang, C. J. In vivo imaging of hydrogen peroxide production in a murine tumor model with a chemoselective bioluminescent reporter. Proc. Natl Acad. Sci. USA 107, 21316–21321 (2010).
Boutagy, N. E. et al. In vivo reactive oxygen species detection with a novel positron emission tomography tracer, 18F-DHMT, allows for early detection of anthracycline-induced cardiotoxicity in rodents. JACC Basic Transl. Sci. 3, 378–390 (2018).
Cairns, A. G., McQuaker, S. J., Murphy, M. P. & Hartley, R. C. Insights on targeting small molecules to the mitochondrial matrix and the preparation of MitoB and MitoP as exomarkers of mitochondrial hydrogen peroxide. Methods Mol. Biol. 2275, 87–117 (2021).
Larsen, E. L., Weimann, A. & Poulsen, H. E. Interventions targeted at oxidatively generated modifications of nucleic acids focused on urine and plasma markers. Free Radic. Biol. Med. 145, 256–283 (2019).
Ahmed, O. S. et al. Moving forward with isoprostanes, neuroprostanes and phytoprostanes: where are we now? Essays Biochem. 64, 463–484 (2020).
Sies, H. et al. The use of total antioxidant capacity as surrogate marker for food quality and its effect on health is to be discouraged. Nutrition 30, 791–793 (2014).
Sies, H. & Chance, B. The steady state level of catalase compound I in isolated hemoglobin-free perfused rat liver. FEBS Lett. 11, 172–176 (1970).
Acknowledgements
We apologise to our colleagues for the many key papers we were unable to cite due to space limitations, combined with the breadth of coverage required in a Consensus Statement. We thank A. J. Kowaltowski for helpful comments on the manuscript. Work in the laboratory of M.P.M. is supported by a grant from the Medical Research Council UK (no. MC_UU_00015/3) and by a Wellcome Trust Investigator award (no. 220257/Z/20/Z). Work in the laboratories of H.B. and V.E.K. is supported by National Institutes of Health (NIH), USA (grant nos. AI145406, CA165065, CA266342, HL114453, AI156924, NS076511, AI156923, NS061817 and NS117000). K.J.A.D. was supported by grant no. ES003598 from the National Institute of Environmental Health Sciences of the US NIH, and by grant no. AG052374 from the National Institute on Aging of the US NIH. R.R. was supported by grants from Universidad de la República, Uruguay (nos. CSIC_2018 and EI_2020). Research in the laboratory of B.H. is supported by grants from the National Medical Research Council of Singapore, National University of Singapore, National Research Foundation, Singapore and the Tan Chin Tuan Foundation. C.J.C. is supported by NIH (nos. GM 79465, GM 139245 and ES 28096) and is a CIFAR Scholar. Work in the laboratory of S.T. is supported by JST CREST (grant no. JPMJCR19H4), JSPS Kakenhi (grant nos. JP16H06276[AdAMS], JP19H05462 and JP20H05502) and a Research Grant of the Princess Takamatsu Cancer Research Fund (no. 19-25126). Work in the laboratory of T.P.D. is supported by grants from the German Research Council (nos. DFG, TRR184 and SPP2306) and the European Research Council (no. 742039). Work in the laboratory of H.Y. is supported by grants from the National Natural Science Foundation of China (nos. 32030053 and 32150710522). Work in the laboratory of M.J.D. is supported by the Novo Nordisk Foundation (grant nos. NNF13OC0004294, NNF19OC0058493 and NNF20SA0064214). Work in the laboratory of N.-G.L. is supported by the Swedish Research Council (2015-00418), Swedish Cancer Foundation, the Knut and Alice Wallenberg foundation, European Research Council (Advanced Grant 2016-741366) and the Novo Nordisk Foundation. B.H. is grateful to Alvin Loo for raising the issue of dubious kits to measure ROS.
Author information
Authors and Affiliations
Contributions
The initial impetus for this consensus statement came from B.H. and M.P.M., who wrote both initial and final versions. H.B., V.B., C.J.C., K.J.A.D., M.J.D., T.P.D., T.F., H.J.F., Y.J.-H., D.G., V.E.K., B.K., N.-G.L., G.L.M., T.N., H.E.P., R.R., H.V.R., P.T.S., P.J.T., S.T., C.C.W. and H.Y. wrote parts of the manuscript and edited and approved the entire manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Liron Bar-Peled, Kathy Griendling and Pietro Ghezzi for their contribution to the peer review of this work. Primary Handling Editor: Christoph Schmitt, in collaboration with the Nature Metabolism team.
Rights and permissions
About this article

Cite this article
Murphy, M.P., Bayir, H., Belousov, V. et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab 4, 651–662 (2022). https://doi.org/10.1038/s42255-022-00591-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-022-00591-z
This article is cited by
-
Cellular zinc metabolism and zinc signaling: from biological functions to diseases and therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Effect of very long-term storage and multiple freeze and thaw cycles on 11-dehydro-thromboxane-B2 and 8-iso-prostaglandin F2α, levels in human urine samples by validated enzyme immunoassays
Scientific Reports (2024)
-
A strategy of novel molecular hydrogen-producing antioxidative auxiliary system improves virus production in cell bioreactor
Scientific Reports (2024)
-
Pro-ferroptotic signaling promotes arterial aging via vascular smooth muscle cell senescence
Nature Communications (2024)
-
Optimization of pacing parameters to entrain slow wave activity in the pig jejunum
Scientific Reports (2024)
