
Abstract
Glycomimetics are structural mimics of naturally occurring carbohydrates and represent important therapeutic leads in several disease treatments. However, the structural and stereochemical complexity inherent to glycomimetics often challenges medicinal chemistry efforts and is incompatible with diversity-oriented synthesis approaches. Here, we describe a one-pot proline-catalyzed aldehyde α-functionalization/aldol reaction that produces an array of stereochemically well-defined glycomimetic building blocks containing fluoro, chloro, bromo, trifluoromethylthio and azodicarboxylate functional groups. Using density functional theory calculations, we demonstrate both steric and electrostatic interactions play key diastereodiscriminating roles in the dynamic kinetic resolution. The utility of this simple process for generating large and diverse libraries of glycomimetics is demonstrated in the rapid production of iminosugars, nucleoside analogues, carbasugars and carbohydrates from common intermediates.
Similar content being viewed by others

Introduction
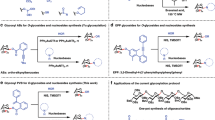
Carbohydrates are essential biomolecules that play critical roles in cell signaling, protein folding, and metabolism1. The recognition and regulation of carbohydrate structure are thus strictly controlled processes that rely on the fidelity of an array of carbohydrate-specific enzymes (e.g., glycosyl transferases and glycoside hydrolases) and binding proteins (e.g., lectins)2,3. While the use of carbohydrates as therapeutics is limited by generally poor pharmacokinetic properties, structural mimics of carbohydrates, also known as glycomimetics (1–6, Fig. 1), that interact in a similar manner with protein targets but have improved drug-like properties (e.g., affinity, stability and bioavailability) represent promising alternatives2,3,4,5,6,7,8. Common structural changes found in glycomimetics include replacement of the endocyclic oxygen with a carbon (carbasugars: 1)9 or nitrogen (iminosugar: 3 and 4)10 and substitution of a fluorine atom for hydroxyl groups (deoxyfluorosugars: 2, 5, and 6)11. For example, Oseltamivir (1)12 is a carbasugar mimic of sialic acid and a potent neuraminidase inhibitor that became the frontline antiviral drug used during the H1N1 pandemic in 2009. Likewise, Miglitol (3)13 is a glycomimetic that functions by inhibiting α-glucosidases and decreasing carbohydrate metabolism in patients with type II diabetes. Notably, iminosugars like 3 are typically protonated at physiological pH, which allows them to mimic the oxacarbenium ion transition state traversed during hydrolysis by glycoside hydrolases10.

Examples of glycomimetic drugs and DOS approaches to prepare DOS libraries of glycomimetics for screening purposes. Shaded regions highlight the glycomimetic structures.
The rational design of glycomimetic drugs often initiates with a structural analysis of the natural carbohydrate substrate bound to the protein target of interest, followed by iterative synthesis campaigns focused on stabilizing the bioactive conformation and removing unnecessary functional groups to improve drug-like properties5. Considering the vast array of biologically relevant carbohydrates, diversity-oriented synthesis (DOS)14,15 strategies that broadly sample this biologically relevant chemical space would also be well-suited to support drug discovery efforts16. However, the functional group density and stereochemical complexity inherent to carbohydrates often requires that DOS strategies rely on the derivatization of naturally occurring carbohydrates, ultimately limiting diversity16. For example, Wong has reported a library of potent and selective α-fucosidase inhibitors derived from 1-aminomethyl-fuconojirimycin (7) (Fig. 1)17. Here, late-stage amide coupling was used to modify the core scaffold 7, itself generated through a 12-step synthesis. A DOS approach has also been reported by Marcaurelle, where 2,3-unsaturated C-glycoside scaffolds were used to synthesize several new bicyclic carbohydrates18. Recently, Loh reported a strategy for the diversification of carbohydrates using hydrogen- and halogen-bond-catalyzed strain release glycosylation to produce complex O,N-glycoside analogs 11 (Fig. 1)19. Ultimately, this platform enabled the discovery of potential leads for treating acquired cancer resistance. While these limited examples highlight a clear role for DOS in the generation of glycomimetics, they also underscore the need for new synthetic strategies that enable rapid sampling of more diverse regions of carbohydrate-associated chemical space20.
As an alternative de novo approach to carbohydrates and glycomimetics, we have reported a one-pot α-chlorination/aldol reaction (αCAR, Fig. 2)21. As detailed in Fig. 2 (top panel), here proline catalyzes (i) the racemic α-chlorination of aldehydes, (ii) the interconversion of the resulting racemic α-chloroaldehydes 12, and (iii) their subsequent aldol reaction with dioxanone 13. To rationalize the preference for syn-chlorohydrins 14, we proposed that electrostatic repulsion between the Cl and O atoms in the transition structure (S)-TS (X = Cl) disfavors formation of anti-chlorohydrin 1521. The high degree of enantioselectivity results from H-bonding that directs the facial approach of the correctly configured α-chloroaldehyde to the proline-derived enamine through a Houk–List type transition structure22,23,24. Importantly, chlorohydrin scaffolds 14 produced via this dynamic kinetic resolution (DKR) can be readily converted into ribose analogs21 as well as other glycomimetics including carbasugars (e.g., 1625) and iminosugars26 (e.g. 1727). Recently, we reported a complimentary proline-catalyzed α-fluorination/aldol reaction (αFAR) that supports the rapid synthesis of nucleoside analogs (e.g., 18)28. While these two processes have provided access to a range of useful glycomimetics, both the αCARs and αFARs rely exclusively on dioxanone 13 as the ketone coupling partner24,29,30, which prevents wider application of these strategies to library synthesis and DOS pursuits.

a Proline-catalyzed α-chlorination- and α-fluorination/aldol reactions used for preparing glycomimetics. The fragments derived from aldehyde coupling partner are colored in blue. b Examples of glycomimetics that have been synthesized from α-chlorination- and α-fluorination/aldol reactions. c The discovery of new α-functionalization/aldol reactions to diversity-oriented synthesis of glycomimetics. The fragments derived from ketone coupling partner are colored in blue.
Results and discussion
α-Functionalization/aldol reactions
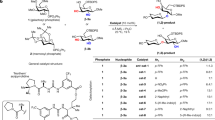
Based on the limitations noted above, we envisioned that new α-functionalization/aldol reactions31,32, involving a broader selection of (i) electrophiles and (ii) enolizable ketones, would support the construction of diverse collections of glycomimetics. Specifically, we aimed to exploit organocatalytic aldehyde α-functionalization reactions, including α-chlorination33,34,35, α-fluorination36,37,38, α-amination39,40, and α-trifluoromethylthiolation41 in combination with proline-catalyzed aldol reactions of various cyclic and acyclic ketones21. Importantly, these processes would avoid isolation27,28 of often unstable and configurationally labile α-functionalized aldehydes and expand their general utility. Toward this goal, we first investigated combinations of different proline-catalyzed α-functionalization reactions with proline-catalyzed aldol reaction of dioxanone 13. As summarized in Table 1, these reactions were performed as two-step-one-pot sequences (see Supplementary Table 1 for details). Specifically, l-proline-catalyzed α-functionalization using N-chlorosuccinimide34,42, N-bromosuccinimide43, N-fluorobenzenesulfonimide36,37,38, N-trifluoromethylthiophthalimide (PhthN-SCF3)44, or dibenzyl azodicarboxylate39,40 was followed by the direct addition of dioxanone 13. As summarized in Table 1, we found that these reactions preferentially afford syn-chlorohydrin 27, syn-fluorohydrin 28, syn-bromohydrin 29, syn-trifluoromethylthiohydrin 30, and syn-aminohydrin 31 with variable diastereoselectivity and in generally excellent enantioselectivity. Considering that the proline-catalyzed α-fluorination of aldehydes is not an enantioselective process (ee’s < 30%)45, the selective formation of fluorohydrin 28 was attributed to a DKR of the intermediate α-fluoroaldehyde as observed previously with α-chloroaldehydes21. In a separate experiment, involving the αFAR of hydrocinnamaldehyde, the diastereomeric ratio of products (>15:1) and enantiomeric excess of the intermediate α-fluoroaldehyde did not change over the course of the reaction even as the (R)-fluoroaldehyde was consumed, further confirming the role of a DKR in these processes (see Supplementary Table 2). Notably, the diastereoselectivity in the production of halohydrins (entries 2–4) correlates with increasing electronegativity of the halogen atom. Thus, despite a smaller van der Waals radius and shorter C–X bond length for X = F, the diastereoselective aldol reaction of the α-fluoroaldehyde was more greatly differentiated. The αCAR was also carried out in CH2Cl2 (rather than 9:1 CH2Cl2-DMF) and we observed a coincident increase in diastereoselectivity (2.2:1 to 6:1; entries 1 and 2) consistent with an increased influence of electrostatic interactions in the diastereodifferentiating step (Fig. 2). As summarized in entry 5, the α-trifluoromethylthioaldehyde derived from pentanal also underwent a diastero- and enantioselective aldol reaction with dioxanone 13. In the case of the α-aza aldehyde generated from the reaction of pentanal and dibenzyl azodicarboxylate (entry 6)40, steric hindrance precludes formation of a proline enamine required for racemization46. Thus, the ultimate diastereoselectivity (dr = 3:1) reflects the ratio of enantiomeric α-aza aldehydes generated in situ. Notably, when this reaction was repeated in CH2Cl2 or DMSO, the yield was significantly lower. The l-proline-catalyzed aldol reaction between (S)-2-Cbz-aminopentanal and dioxanone yielded the corresponding anti-aminohydrin as the sole product, and both l- and d-proline-catalyzed aldol reactions of (S)-N-Cbz prolinal (Supplementary Schemes 1 and 2) each gave single products without epimerization of the α-stereocenter, further confirming that α-aminoaldehydes do not racemize under these reaction conditions (krac << kaldol)47. We also examined the l-proline-catalyzed aldol reaction of (±)-2-phenylpropanal and dioxanone 13 (Supplementary Scheme 3), which afforded an equal mixture of syn- and anti- diastereomers again suggesting krac << kaldol.
Diversification of ketone coupling partners
We next looked to expand the scope of compatible ketones48. As summarized in Table 2, we began by examining the l-proline-catalyzed αCAR of tetrahydro-4H-thiopyranone (35)49 with isovaleraldehyde. Carrying out this reaction using our one-step-one-pot procedure in a variety of solvents afforded the desired syn-chlorohydrin 36, albeit in low yield (See Table 2, entries 1–3). Suspecting that competitive oxidation of tetrahydro-4H-thiopyran-4-one (35) was complicating this process, we adopted a two-step-one-pot sequence. Satisfyingly, when the l-proline-catalyzed α-chlorination of isovaleraldehyde was first carried out in CH2Cl2 at 0 °C followed by direct addition of thiopyranone 35 in DMSO, the desired aldol adduct 36 was produced in good diastereoselectivity (entry 4). Addition of catalytic amounts of water with the thiopyranone led to modest improvements in yield, and the chlorohydrin 36 was produced in excellent enantiomeric excess (94% ee). Using this straightforward process, several additional ketones (O-TBS-hydroxyacetone, cyclohexanone, and tetrahydro-4H-pyran-4-one)48 were engaged in αCARs giving syn-chlorohydrins 36 and 38–40 in good yield and excellent diastereo- and enantioselectivity (Fig. 3). Unfortunately, other simple aliphatic ketones such as acetone, cyclopentanone, and 3-pentanone were incompatible with this process (e.g., 41–45).

α-Chlorination/aldol reaction products from a range of ketones. Isolated yields for isolated diastereomer shown, diastereomeric ratio determined by 1H NMR spectroscopic analysis of crude reaction mixture.
Computational analysis of proline-catalyzed aldol reactions of α-haloaldehydes
To better understand the broad preference for the formation of syn-fluoro- and chlorohydrins (e.g., Tables 1 and 2) regardless of the ketone coupling partner, l-proline-catalyzed aldol reactions involving (R)- or (S)-2-fluoro- and 2-chloropentanal were examined using density functional theory (DFT) computations employing the program Gaussian 16 with the hybrid meta-GGA M06–2X functional of Truhlar (see Supplementary Data for DFT Calculations pp S146–S695)50,51. To start, we analyzed the l-proline-catalyzed aldol reaction between dioxanone 13 and (R)- or (S)-2-fluoropentanal resulting in the finding of low-energy transition states (R)-TS1O-F, (S)-TS2O-F, and (S)-TS3O-F (Fig. 4a). Notably, (S)-TS2O-F resembled an Evans–Cornforth52,53 type carbonyl addition mode (i.e., antiparallel alignment of carbonyl and α-F substituent) while (S)-TS3O-F resembled a Felkin–Anh54,55 addition mode (i.e., perpendicular orientation of carbonyl and α-F substituent). Further, each of these transition states possessed a characteristic Houk–List stabilizing O-H···O hydrogen-bonding interaction22. Among these structures, transition state (R)-TS1O-F leading to syn-fluorohydrin 28 was energetically favored over the diastereomeric transition states (S)-TS2O-F (ΔGrel = 3.5 kcal mol−1) and (S)-TS3O-F (ΔGrel = 2.5 kcal mol−1) affording anti-fluorohydrins, which is in agreement with experiment (Table 1, entry 3). The higher energy of the Evans–Cornforth-type transition state (S)-TS2O-F is attributed to a destabilizing interaction between an oxygen on the dioxanone-derived enamine and the aldehyde fluoride substituent with a distance measuring 2.74 Å. By comparison, the corresponding Felkin–Anh-type transition state (S)-TS3O-F is destabilized by unfavorable steric interactions as seen by a close hydrogen–hydrogen contact measuring 2.28 Å. Conversely, the lowest energy transition state (R)-TS1O-F benefited from stabilizing non-covalent interactions (NCIs) and fewer steric contacts as gauged by a NCI surface (see Supplementary Figure 7). Among these NCIs, interestingly, was a fluorine-hydrogen contact measuring 2.39 Å and, in part, deriving from fluorine atom lone pair (n) donation into a H–C(sp2) antibonding orbital (*) of the enamine (ηF → σ*(C–H)) contributing ~1.1 kcal mol−1 stability based upon natural bond orbital second-order perturbation theory.

a Transition state structures (R)-TS1O-F, (S)-TS2O-F and (S)-TS3O-F for l-proline-catalyzed aldol reactions of dioxanone 13 with (R)- or (S)-2-fluoropentanal. Oxygen atoms are colored in red, fluorine atoms are colored in green. b Transition state structures (R)-TS1O-Cl, (S)-TS2O-Cl, and (S)-TS3O-Cl for l-proline-catalyzed aldol reactions involving dioxanone 13 with (R)- or (S)-2-chloropentanal. DFT calculations were performed using IEFPCM(DCM)M06-2X/6-311++G(2d,2p)//IEFPCM(DCM)M06-2X/6-31+G(d,p) level of theory. The α-chloro and α-fluoroaldehydes are colored in blue.
The computed trends for reactions of (R)- and (S)-2-chloropentanal were similar to those discussed above. Namely, three low-energy transition states (R)-TS1O-Cl, (S)-TS2O-Cl, and (S)-TS3O-Cl were found with the latter two possessing structural attributes of prototypical Evans–Cornforth and Felkin–Anh carbonyl addition models (Fig. 4b). Among these structures, the transition state (R)-TS1O-Cl was lower in energy than both of the diastereomeric transition states (S)-TS2O-Cl and (S)-TS3O-Cl, in agreement with the preferential formation of syn-chlorohydrin 27 (Table 1, entry 1). In particular, the Evans–Cornforth-type transition state (S)-TS2O-Cl was destabilized by a repulsive interaction between an oxygen on the dioxanone-derived enamine and the aldehyde chlorine with a distance measuring 3.05 Å. In contrast, Felkin–Anh-type transition state (S)-TS3O-Cl suffered from steric interactions as seen from a close hydrogen–hydrogen contact with a distance of 2.21 Å. On the contrary, the energetically favored transition state (R)-TS1O-Cl contained fewer steric contacts and favorable NCIs, including a chlorine–hydrogen contact measuring 2.72 Å, visible from a NCI surface (see Supplementary Figure 3). Collectively, these studies revealed several conserved features irrespective of fluoro- or chloro-substitution and similar trends were found in the computed reactivity patterns of the cyclic ketones cyclohexanone, tetrahydropyranone, and thiopyranone 35 (Supplementary Tables 1–9). Notably, across this panel of ketones, the preferred transition state structures consistently shared geometries analogous to (R)-TS1O-F and (R)-TS1O-Cl. Moreover, these trends were adhered to with bromo-substitution (see Supplementary Table 6). In all cases, the minimization of repulsive steric interactions and favorable NCIs including a halogen–hydrogen (C–X···H) interaction were identified as key contributors to preferred formation of syn-halohydrin products.
Scope of α-functionalization/aldol reactions
With several new α-functionalization/aldol reactions in hand, we examined the reaction of a larger collection of aldehydes with one of the electrophiles demonstrated in Table 1 and ketones demonstrated in Table 2. As detailed in Fig. 5, this strategy allowed for the rapid construction of a collection of fluorohydrins (28, 47–58, and 64–70), chlorohydrins (59–62), trifluoromethylthiohydrins (30, 71–73), and aminohydrins (31 and 63) in excellent enantio- and diastereoselectivity. The functional group compatibility of the αFAR is highlighted in the reactions of TIPS-protected 3-hydroxypropanal and Cbz-protected 3-aminopropanal, which delivered the unusual fluorohydrins 54 and 58 bearing a heteroatom at each position in the carbon chain. Overall, the αFAR was tolerant to alkyl, alkene, alkyne, and heteroaryl substituents. Furthermore, simply using d-proline catalysis enables access to enantiomeric syn-fluorohydrins 49 and 54. Replacing dioxanone 13 with thiopyranone 35 or cyclohexanone provided anti-aldol-syn-fluorohydrin products (64–70) in good to excellent yield. We were particularly encouraged by productive reaction of α-fluoro-α-heteroaryl-acetaldehydes, which produced adducts 68–70 that should support the synthesis of nucleoside analogs28. Considering the SCF3 group is commonly used to increase lipophilicity of drug leads, we were pleased to find that syn-trifluoromethylthiohydrins containing an alkyl (30), alkenyl (71), aryl (72), or heteroaryl (73) group could all be prepared in excellent diastereo- and enantioselectivity. To the best of our knowledge, these are the first examples of enantioselective reactions involving α-trifluoromethylthioaldehydes. Finally, we explored the compatibility of a small collection of aldehydes in αCARs with O-TBS-hydroxyacetone, which afforded chlorohydrins 59–62, and demonstrated that α-amination/aldol reactions deliver enantiomerically enriched syn-aminohydrins 31 and 63.

Enantioselective synthesis of chlorohydrins (59–62), fluorohydrins (28, 47–58, 64–70), trifluoromethylthiohydrins (30, 71–73), and aminohydrins (31, 63). Yields are for the isolated diastereomer depicted. Diastereomeric ratios (dr) were determined by 1H NMR spectroscopic analysis of crude reaction mixtures. Ketones and the fragments of each product that are derived from the ketone are colored in blue. The atoms/groups used to functionalize the aldehyde are colored as follows: fluorine (green), chlorine (red), trifluoromethylthio (orange), and amino group (purple). [a] d-proline was used. [b] Selectfluor was used. [c] Yield over two steps following hydrogenation. [d] Stereochemistry at fluoromethine center not assigned.
Rapid synthesis of glycomimetics
To demonstrate the potential for this strategy to rapidly generate glycomimetics, several readily available chlorohydrins, fluorohydrins, trifluoromethylthiohydrins, and aminohydrins (Fig. 5) were converted into a diverse collection of iminosugars, furanose analogs, and bicyclic nucleoside analogs (Fig. 6). For example, hydrogenation of syn-aminohydrins allowed access to cyclic hydrazones 76 and 77 in excellent yield over two total steps (panel a). This route compares favorably with reported syntheses of the related azafagomines 74 and 75, inhibitors of α-fucosidase, that require 17 steps56. As depicted in panel b, a series of unusual furanose analogs 78–81 was prepared through 1,3-syn reduction of aldol adducts 36, 38, 40, and 61 with sodium borohydride followed by thermal cyclization21. Alternatively, reductive amination of aldol adducts 59, 60, and 62 with benzyl amine followed by reflux in toluene under basic conditions gave access to selectively protected 5′-deoxy-iminosugars 82–84 (panel c)26. The recent discovery of 85 as a potent and selective PRMT5 inhibitor for cancer treatment highlights bicyclic nucleoside analogs as a relatively unexplored scaffold in drug discovery57. Here, we prepared bicyclic nucleoside analogs 86–88 in two steps via a 1,3-syn reduction and indium chloride mediated cyclization28 from fluorohydrins 68a, 69, and 70 (panel d). Notably, reduction of 68a afforded preferentially a syn-diol intermediate that was readily cyclized to 86 without epimerization at the anomeric center28. Conversely, epimerization of 87 and 88 occurred following cyclization to give a mixture of α-d- and β-d- anomers as shown. Given the synthetic challenges associated with nucleoside analog synthesis, this short sequence provides new opportunities to explore structure activity relationships in this potentially important family of bicyclic nucleoside analogs.

Rapid diversification of α-functionalization/aldol adducts into glycomimetics including azasugars (a), furanose analogs (b), iminosugars (c), and bicyclic nucleoside analogs (d). [a] α:β = 2.5:1; [b] α:β = 3:1.
As summarized in Fig. 7, we also explored the utility of fluorinated aldol adducts to serve as precursors to fluorinated glycomimetics. Toward this goal, Julia–Kocienski olefination58 and subsequent ring closing metathesis59 of 51, 66, 67, and 71 afforded bicyclic fluorinated carbasugars 90–93 in good overall yield (panel a). To access 2-deoxy-2-fluoro sugars, a 1,3-syn or 1,3-anti-selective reduction of the fluorohydrin 58 was followed by removal of the silyl protecting group and oxidation of the resultant primary alcohol (panel b). This short sequence delivered the epimeric 2-deoxy-2-fluoropyranoses 94 and 95 and avoids the iterative alcohol protection–deprotection steps commonly required in fluorosugar synthesis. Hydrogenation of the Cbz-protected amino fluorohydrin 54 (made using d-proline catalysis) gave 96 directly and in excellent yield: a previously undescribed fluorinated analog of the drug migalastat (Galafold)60, which is a pharmacological chaperone used to treat Fabry disease (panel c). As an additional target of interest, we also prepared a fluorinated analog of D-ribo-phytosphingosine, a precursor to the potent natural killer T-cell stimulator α-galactosylceramide61, in a straightforward manner through the reductive amination of the fluorohydrin 49 with benzyl amine, followed by hydrogenolysis and acid deprotection (panel d).

Synthesis of fluorinated glycomimetics including carbasugars (a), 2-deoxy-2-fluoro sugars (b), a fluorinated iminosugar (c), and a fluorinated analog of phytosphingosine (d). Fluorine and fluorinated functional groups are highlighted in green. [c] Grela catalyst.
Conclusion
In summary, we have developed a flexible and robust DOS approach to glycomimetics that relies on achiral and readily accessible starting materials to access an array of highly relevant carbohydrate-like scaffolds. These processes are enabled by a family of new α-functionalization/aldol reactions that directly transform commercially available aldehydes and ketones into stereochemically rich and densely functionalized aldol adducts in good yield and excellent diastereo- and enantioselectivity. Notably, DFT analysis of aldol reactions of both α-chloro- and α-fluoroaldehydes identified a key stabilizing halogen–hydrogen (C–X···H) interaction in the lowest energy transition structure and support steric and electrostatic interactions playing key diastereodiscriminating roles in these reactions. Ultimately, the demonstration that a broad range of glycomimetics can be readily accessed using these strategies suggests these processes should inspire new efforts in drug discovery.
Methods
All experimental procedures for α-functionalization/aldol reactions and synthesis of glycomimetics are included in the Supplementary Information (Supplementary Methods).
Data availability
All characterization data including 1H and 13C NMR spectral data and sample spectra, IR data, [α]D, HRMS, and HPLC chromatograms used to determine enantiomeric purity are included in the Supplementary Information (Supplementary Figs. 1–80). The X-ray crystallographic coordinates for structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 1556397, 1556396, 1556395, 1556394, and 1556393. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The crystallographic information of compounds 28, 47, 56, 58, and 95 is available in Supplementary Data 2 and is summarized in Supplementary Figs. 81–85 and Supplementary Table 3. The supporting DFT calculations are summarized in Supplementary Fig. 86. Full details for DFT calcuations are available in Supplementary Data File 1 (Supplementary Figs. 87–104 and Supplementary Tables 4–24).
References
Overend, W. G. Detection of carbohydrate structures on isolated subcellular organelles of rat liver by heterophile agglutinins. Nature 242, 71–71 (1973).
Ernst, B. & Magnani, J. L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 8, 661–677 (2009).
Rojo, J., Sousa-Herves, A. & Mascaraque, A. in Comprehensive Medicinal Chemistry III (eds Chackalamannil, S., Rotella, D. & Ward, S. E.) 577–610 (Elsevier, 2017).
Zhang, Y. & Wang, F. Carbohydrate drugs: current status and development prospect. Drug Discov. Ther. 9, 79–87 (2015).
Magnani, J. L. & Ernst, B. Glycomimetic drugs-a new source of therapeutic opportunities. Discov. Med. 8, 247–252 (2009).
Pascolutti, M. & von Itzstein, M. Design and synthesis of carbohydrates and carbohydrate mimics as anti-influenza agents. in GlycochemIcal Synthesis, 455–481 (Glycochemical Synthesis: Strategies and Applications, John Wiley & Sons, Inc, 2016).
Hevey, R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals 12, 55 (2019).
Tamburrini, A., Colombo, C. & Bernardi, A. Design and synthesis of glycomimetics: recent advances. Med. Res. Rev. 40, 495–531 (2020).
Arjona, O., Gómez, A. M., López, J. C. & Plumet, J. Synthesis and conformational and biological aspects of carbasugars. Chem. Rev. 107, 1919–2036 (2007).
Compain, P. & Martin, O. R. Iminosugars: From Synthesis to Therapeutic Applications. 482 (Wiley, 2007).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Lew, W., Chen, X. W. & Kim, C. U. Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor. Curr. Med. Chem. 7, 663–672 (2000).
Scott, L. J. & Spencer, C. M. Miglitol: a review of its therapeutic potential in type 2 diabetes mellitus. Drugs 59, 521–549 (2000).
Schreiber, S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964 (2000).
Burke, M. D. & Schreiber, S. L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 43, 46–58 (2004).
Lenci, E., Menchi, G. & Trabocchi, A. Carbohydrates in diversity-oriented synthesis: challenges and opportunities. Org. Biomol. Chem. 14, 808–825 (2016).
Wu, C.-Y., Chang, C.-F., Chen, J. S.-Y., Wong, C.-H. & Lin, C.-H. Rapid diversity-oriented synthesis in microtiter plates for in situ screening: discovery of potent and selective α-fucosidase inhibitors. Angew. Chem. Int. Ed. 42, 4661–4664 (2003).
Gerard, B. et al. Synthesis of stereochemically and skeletally diverse fused ring systems from functionalized C-glycosides. J. Org. Chem. 78, 5160–5171 (2013).
Xu, C. & Loh, C. C. J. A multistage halogen bond catalyzed strain-release glycosylation unravels new hedgehog signaling inhibitors. J. Am. Chem. Soc. 141, 5381–5391 (2019).
Galloway, W. R. J. D., Isidro-Llobet, A. & Spring, D. R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 1, 80 (2010).
Bergeron-Brlek, M., Teoh, T. & Britton, R. A tandem organocatalytic α-chlorination-aldol reaction that proceeds with dynamic kinetic resolution: a powerful tool for carbohydrate synthesis. Org. Lett. 15, 3554–3557 (2013).
Bahmanyar, S., Houk, K. N., Martin, H. J. & List, B. Quantum mechanical predictions of the stereoselectivities of proline-catalyzed asymmetric intermolecular aldol reactions. J. Am. Chem. Soc. 125, 2475–2479 (2003).
Hoang, L., Bahmanyar, S., Houk, K. N. & List, B. Kinetic and stereochemical evidence for the involvement of only one proline molecule in the transition states of proline-catalyzed intra- and intermolecular aldol reactions. J. Am. Chem. Soc. 125, 16–17 (2003).
Enders, D., Grondal, C., Vrettou, M. & Raabe, G. Asymmetric synthesis of selectively protected amino sugars and derivatives by a direct organocatalytic mannich reaction. Angew. Chem. Int. Ed. 44, 4079–4083 (2005).
Adamson, C. et al. Structural snapshots for mechanism-based inactivation of a glycoside hydrolase by cyclopropyl carbasugars. Angew. Chem. Int. Ed. 55, 14978–14982 (2016).
Bergeron-Brlek, M., Meanwell, M. & Britton, R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 6, 6903 (2015).
Bergeron-Brlek, M. et al. A convenient approach to stereoisomeric iminocyclitols: generation of potent brain-permeable OGA inhibitors. Angew. Chem. Int. Ed. 54, 15429–15433 (2015).
Meanwell, M. et al. A short de novo synthesis of nucleoside analogs. Science 369, 725–730 (2020).
Grondal, C. & Enders, D. A direct organocatalytic entry to selectively protected aldopentoses and derivatives. Adv. Synth. Catal. 349, 694–702 (2007).
Mlynarski, J. & Gut, B. Organocatalytic synthesis of carbohydrates. Chem. Soc. Rev. 41, 587–596 (2012).
Quintard, A., Sperandio, C. & Rodriguez, J. Modular enantioselective synthesis of an advanced pentahydroxy intermediate of antimalarial bastimolide A and of fluorinated and chlorinated analogues. Org. Lett. 20, 5274–5277 (2018).
Quintard, A. & Rodriguez, J. Bicatalyzed three-component stereoselective decarboxylative fluoro-aldolization for the construction of elongated fluorohydrins. ACS Catal. 7, 5513–5517 (2017).
Brochu, M. P., Brown, S. P. & MacMillan, D. W. C. Direct and enantioselective organocatalytic α-chlorination of aldehydes. J. Am. Chem. Soc. 126, 4108–4109 (2004).
Halland, N., Braunton, A., Bachmann, S., Marigo, M. & Jorgensen, K. A. Direct organocatalytic asymmetric α-chlorination of aldehydes. J. Am. Chem. Soc. 126, 4790–4791 (2004).
Winter, P. et al. Transforming terpene-derived aldehydes into 1,2-epoxides via asymmetric α-chlorination: subsequent epoxide opening with carbon nucleophiles. Chem. Commun. 47, 12200–12202 (2011).
Steiner, D. D., Mase, N. & Barbas, C. F. III Direct asymmetric α-fluorination of aldehydes. Angew. Chem. Int. Ed. 44, 3706–3710 (2005).
Beeson, T. D. & MacMillan, D. W. C. Enantioselective organocatalytic α-fluorination of aldehydes. J. Am. Chem. Soc. 127, 8826–8828 (2005).
Marigo, M., Fielenbach, D., Braunton, A., Kjærsgaard, A. & Jørgensen, K. A. Enantioselective formation of stereogenic carbon–fluorine centers by a simple catalytic method. Angew. Chem. Int. Ed. 44, 3703–3706 (2005).
List, B. Direct catalytic asymmetric α-amination of aldehydes. J. Am. Chem. Soc. 124, 5656–5657 (2002).
Bøgevig, A., Juhl, K., Kumaragurubaran, N., Zhuang, W. & Jørgensen, K. A. Direct organo-catalytic asymmetric α-amination of aldehydes—a simple approach to optically active α-amino aldehydes, α-amino alcohols, and α-amino acids. Angew. Chem. Int. Ed. 41, 1790–1793 (2002).
Nagib, D. A., Scott, M. E. & MacMillan, D. W. C. Enantioselective α-trifluoromethylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 131, 10875–10877 (2009).
Ponath, S. et al. Mechanistic studies on the organocatalytic α-chlorination of aldehydes: the role and nature of off-cycle intermediates. Angew. Chem. Int. Ed. 57, 11683–11687 (2018).
Britton, R. & Kang, B. α-Haloaldehydes: versatile building blocks for natural product synthesis. Nat. Prod. Rep. 30, 227–236 (2013).
Hu, L. et al. Efficient catalytic α-trifluoromethylthiolation of aldehydes. N. J. Chem. 40, 6550–6553 (2016).
Enders, D. & Hüttl, M. R. M. Direct organocatalytic α-fluorination of aldehydes and ketones. Synlett 2005, 0991–0993 (2005).
Franzén, J. et al. A general organocatalyst for direct α-functionalization of aldehydes: stereoselective C−C, C−N, C−F, C−Br, and C−S bond-forming reactions. Scope and mechanistic insights. J. Am. Chem. Soc. 127, 18296–18304 (2005).
Marjanovic Trajkovic, J., Milanovic, V., Ferjancic, Z. & Saicic, R. N. On the asymmetric induction in proline-catalyzed aldol reactions: reagent-controlled addition reactions of 2,2-dimethyl-1,3-dioxane-5-one to acyclic chiral α-branched aldehydes. Eur. J. Org. Chem. 2017, 6146–6153 (2017).
Mukherjee, S., Yang, J. W., Hoffmann, S. & List, B. Asymmetric enamine catalysis. Chem. Rev. 107, 5471–5569 (2007).
Ward, D. E. The thiopyran route to polypropionates. Chem. Commun. 47, 11375–11393 (2011).
Frisch, M. J. et al. Gaussian 16, Revision C.01, (Gaussian, Inc., 2016).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Cornforth, J. W., Cornforth, R. H. & Mathew, K. K. 24. A general stereoselective synthesis of olefins. J. Chem. Soc. 112–127 (1959).
Cee, V. J., Cramer, C. J. & Evans, D. A. Theoretical investigation of enolborane addition to α-heteroatom-substituted aldehydes. Relevance of the Cornforth and Polar Felkin−Anh models for asymmetric induction. J. Am. Chem. Soc. 128, 2920–2930 (2006).
Chérest, M., Felkin, H. & Prudent, N. Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones. Tetrahedron Lett. 9, 2199–2204 (1968).
Anh, N. T. & Eisenstein, O. Theoretical interpretation of 1-2 asymmetric induction. The importance of antiperiplanarity. Nouv. J. Chim. 1, 61–70 (1977).
Moreno-Clavijo, E., Carmona, A. T., Moreno-Vargas, A. J., Rodríguez-Carvajal, M. A. & Robina, I. Synthesis and inhibitory activities of novel C-3 substituted azafagomines: a new type of selective inhibitors of α-l-fucosidases. Biorg. Med. Chem. 18, 4648–4660 (2010).
Machacek, M. et al. PRMT5 Inhibitors. (Merck Sharpe & Dohme Corp., Idenix Pharmaceuticals Llc., 2020). WO2020033288A1
Blakemore, P. R., Cole, W. J., Kocieński, P. J. & Morley, A. A stereoselective synthesis of trans-1,2-disubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett 1998, 26–28 (1998).
Ren, W. et al. Revealing the mechanism for covalent inhibition of glycoside hydrolases by carbasugars at an atomic level. Nat. Commun. 9, 3243 (2018).
Kato, A. et al. Biological properties of d- and l-1-deoxyazasugars. J. Med. Chem. 48, 2036–2044 (2005).
Gao, Y., He, X., Ding, F. & Zhang, Y. Recent progress in chemical syntheses of sphingosines and phytosphingosines. Synthesis 48, 4017–4037 (2016).
Acknowledgements
R.B. was supported by an NSERC Discovery Grant (2019-06368), research support from Merck & Co., a Strategic Initiative Grant from the Canadian Glycomics Network, and a Michael Smith Foundation for Health Research Career Scholar Award. M.M. was supported by a NSERC CGSD, M.B.-B. was supported by an NSERC PGSD, G.F., V.R., and C.A. were each supported by a NSERC CGSM. T.D. was supported by an NSERC Discovery Grant (2019-04205). Computations were carried out using facilities at SHARCNET (Shared Hierarchical Academic Research Computing Network: www.sharcnet.ca) and Compute/Calcul Canada. The authors acknowledge Bryton Varju and Declan McKearney for assistance with X-ray crystallographic analysis.
Author information
Authors and Affiliations
Contributions
M.M., G.F., W.R., B.A, V.R., J.L., S.M.S., C.A., M.B.-B., and V.R.C. assisted in the planning and optimization of reaction conditions, and execution of synthetic experiments. R.R., H.F., and T.D. designed and carried out computational studies. M.M., L.-C.C., and R.B. organized experiments, M.M., T.D., and R.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meanwell, M., Fehr, G., Ren, W. et al. Diversity-oriented synthesis of glycomimetics. Commun Chem 4, 96 (2021). https://doi.org/10.1038/s42004-021-00520-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-021-00520-3
This article is cited by
-
An α-chloroaldehyde-based formal synthesis of eribulin
Nature Communications (2023)
-
Fluorine-a small magic bullet atom in the drug development: perspective to FDA approved and COVID-19 recommended drugs
Chemical Papers (2023)
-
Practical and concise synthesis of nucleoside analogs
Nature Protocols (2022)
-
Multicomponent reaction derived small di- and tri-carbohydrate-based glycomimetics as tools for probing lectin specificity
Glycoconjugate Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
