
Abstract
Gut microbiota, or the collection of diverse microorganisms in a specific ecological niche, are known to significantly impact human health. Decreased gut microbiota production of short-chain fatty acids (SCFAs) has been implicated in type 2 diabetes mellitus (T2DM) disease progression. Most microbiome studies focus on ethnic majorities. This study aims to understand how the microbiome differs between an ethnic majority (the Dutch) and minority (the South-Asian Surinamese (SAS)) group with a lower and higher prevalence of T2DM, respectively. Microbiome data from the Healthy Life in an Urban Setting (HELIUS) cohort were used. Two age- and gender-matched groups were compared: the Dutch (n = 41) and SAS (n = 43). Microbial community compositions were generated via DADA2. Metrics of microbial diversity and similarity between groups were computed. Biomarker analyses were performed to determine discriminating taxa. Bacterial co-occurrence networks were constructed to examine ecological patterns. A tight microbiota cluster was observed in the Dutch women, which overlapped with some of the SAS microbiota. The Dutch gut contained a more interconnected microbial ecology, whereas the SAS network was dispersed, i.e., contained fewer inter-taxonomic correlational relationships. Bacteroides caccae, Butyricicoccus, Alistipes putredinis, Coprococcus comes, Odoribacter splanchnicus, and Lachnospira were enriched in the Dutch gut. Haemophilus, Bifidobacterium, and Anaerostipes hadrus discriminated the SAS gut. All but Lachnospira and certain strains of Haemophilus are known to produce SCFAs. The Dutch gut microbiome was distinguished from the SAS by diverse, differentially abundant SCFA-producing taxa with significant cooperation. The dynamic ecology observed in the Dutch was not detected in the SAS. Among several potential gut microbial biomarkers, Haemophilus parainfluenzae likely best characterizes the ethnic minority group, which is more predisposed to T2DM. The higher prevalence of T2DM in the SAS may be associated with the gut dysbiosis observed.
Similar content being viewed by others


Introduction
A microbiome is a community of phylogenetically diverse microorganisms and their multi-omic content that inhabit a specific ecological niche, such as the human gut1. The microbiome and the host are embedded in a mutually dependent relationship that impacts host behavior and microbial community structure and function2. As host-microbe interactions shape reciprocal fitness, phenotype, and metabolism, the host and the microbiome coevolve2,3. Dysbiosis, or pathologic alteration of the baseline microbial milieu, may drive and/or result from disease progression4,5. Our resident microbial flora has been widely recognized as a key but not yet fully understood mediator in the pathophysiology of many communicable and noncommunicable diseases5,6,7,8,9,10,11, including type 2 diabetes mellitus (T2DM)12,13,14,15,16.
Diabetes is a chronic, metabolic disease in which hyperglycemia leads to multi-organ damage over time17. Since the 1990s, the global prevalence of T2DM has increased, and the age-adjusted prevalence is expected to rise from 6.3% in 2019 to 7.8% in 2045 across Europe18. Disproportionately high rates of diabetes and related complications affect migrant and ethnic minority groups living in Western societies19. Diet, genetic predisposition, body weight, and sedentary lifestyle are key factors in the multifaceted pathophysiology of T2DM20. Recently, the gut microbiome of diabetics has been shown to be distinctly different from that of normoglycemic, insulin-sensitive individuals14,15,16,21. Most agree that the abundances of Ruminococcus, Fusobacterium, and Blautia are positively correlated with T2DM while Bifidobacterium, Bacteroides, Faecalibacterium, Akkermansia, and Roseburia are negatively correlated with T2DM13,22,23,24. However, a large knowledge gap still exists: how do the quantitative presences of the many other gut inhabitants vary with hyperglycemia and insulin resistance? For example, many disagree on the nature of the correlative relationship between T2DM and the abundance of Lactobacillus23.
Short-chain fatty acids (SCFAs), namely, acetate, butyrate, and propionate, and the microbes that produce them have been of particular interest in the diabetic gut because of the favorable effect that these molecules have on host function25,26,27. Most of the beneficial effects of SCFAs on glucose metabolism and insulin signaling are mediated via the GPR41 and GPR43 receptors27,28,29. These molecules can activate intestinal gluconeogenesis27,30, potentiate glucose-stimulated insulin secretion through glucagon like peptide-1 (GLP-1) dependent and independent pathways28,29,31, and attenuate the chronic release of pro-inflammatory cytokines that worsens insulin resistance32,33,34. SCFAs have many other important functions on host metabolism, which are well described by recent reviews26,27. Ultimately, a decrease in SCFA-producing taxa is thought to be comorbid with T2DM and may cause or worsen the disease. Of note, cross-feeding between SCFA-producing taxa plays a major role in the gut microbial functional ecology. For example, Bacteroides thetaiotaomicron, Blautia obeum, Roseburia inulinivorans, Listeria sp., and Clostridium sphenoides can all create a major intermediate metabolite (1,2-propanediol) which can be used by Lactobacillus reuteri to make propionate. Roseburia can then take up acetate, the most widely produced SCFA, and make butyrate from glucose35.
Ethnicity and place of habitation are thought to play an even larger role than metabolic health in shaping gut microbiome composition36,37,38. Ethnicity is a particularly important factor as it connotes similar diet, shared genetics, and migration patterns, all of which have their own variable impact on gut microbial flora. The multi-ethnic Healthy Life in an Urban Setting (HELIUS) prospective cohort study estimated that the impact of ethnicity on gut microbiome composition is ~ 6%37. The HELIUS cohort included participants from the six major ethnic groups living in Amsterdam, the Netherlands at the time of sample collection. Of these ethnicities, the Dutch, Ghanaian, and South-Asian Surinamese (SAS) were found to have the most discriminant gut microbiomes37. In the Dutch population, the ethnic majority of Amsterdam, nine core gut bacterial species were identified: Subdoligranulum sp., Alistipes onderdonkii, Alistipes putredinis, Alistipes shahii, Bacteroides uniformis, Bacteroides vulgatus, Eubacterium rectale, Faecalibacterium prausnitzii and Oscillibacter sp. These were also consistently found across many other populations38. Of these, several were shown to be significantly depleted, e.g., Faecalibacterium prausnitzii, and enriched, e.g., Bacteroides sp., in the SAS as compared to the Dutch37.
The aim of our study is to compare the gut microbiome ecology between the Dutch and SAS groups, as these had the lowest and highest prevalence of T2DM among the HELIUS ethnicities, respectively. Our objective is to characterize and replicate, as microbiome studies are notoriously challenged by reproducibility, the bacterial biomarkers and inter-taxa relationships between these two groups. Network analysis was performed to compare the gut microbiomes of the two ethnicities because it can elucidate co-occurrence patterns, which can identify relationships between different bacterial groups. We demonstrate differences in the SCFA-producing taxa and correlate these with primarily ethnicity and secondarily metabolic health.
Methods
Genomic data source
Our dataset is from a multi-ethnic prospective cohort study, the HELIUS study. This cohort is composed of five major ethnic groups (Surinamese, Dutch, Ghanaian, Moroccan, and Turkish) aged 18–70 years old. Participants were randomly invited, and then stratified by ethnicity. All were living in Amsterdam, the Netherlands at the time of sample collection (2011–2015). A total of 2170 stool samples, each from a different individual, were collected, and the metagenomes were sent for sequencing of the 16S rRNA V4 hypervariable region. This was done on a 2 × 250 base-pair (bp) MiSeq system with use of the 515F and 806R primers. The 2170 sequenced fecal samples yielded a total raw read count of 177,089,775. Further detail about cohort composition, data collection, and gene sequencing protocols have been previously described37,39. The HELIUS study was approved by the medical ethics committee of the Amsterdam University Medical Center, and all participants provided informed consent prior to enrollment in the study. Experiments were performed in accordance with all relevant regulations and guidelines, as approved by the Amsterdam University Medical Center and Declaration of Helsinki (6th, 7th revisions).
Ethnicity
A person was defined as of non-Dutch ethnic origin if he/she fulfilled one of two criteria: (1) he/she was born outside of the Netherlands and had at least one parent born outside the Netherlands (first generation) or (2) he/she was born in the Netherlands but both parents were born outside of the Netherlands (second generation). For the Dutch samples, people who were born in the Netherlands and whose parents were born in the Netherlands were invited. The country of birth indicator for ethnicity was limited in that people who were born in the same country might be of different ethnic background, which in the Dutch context was applicable to the Surinamese population. Therefore, after data collection, participants of Surinamese ethnic origin were further classified by self-reported ethnic origin (obtained by questionnaire) as ‘African’, ‘South-Asian’, ‘Javanese’, or ‘other’.
Microbial community composition
Paired-end reads were input into a DADA2 (v1.16)40 workflow to generate microbial compositions. All parameters were kept at default values except those described here. First, primers were removed from sequence reads. Then, reads were quality filtered and truncated. Two bp errors were allowed per 250 bp read, and read-ends were trimmed down to a quality score threshold of 3041. Then, reads were dereplicated and merged. A minimum overlap of 30 bp was required for merging to occur. After merging, chimeric sequences were removed from the amplicon sequence variants (ASVs). Lastly, taxonomy was assigned via queries to the SILVA 16S rRNA gene reference database (v138.1)42. Species-level phylogenetic classification was attained for 44.8% of the ASVs, genus-level for 47.8%, and family-level or higher (or unassigned) for the remaining 7.4%. A Mann–Whitney U test, or Wilcoxon rank-sum test, was calculated between the relative abundances (RAs) of each taxon to determine if significant differences in a particular taxon existed between the two ethnic groups.
Principal coordinate analysis
To estimate the degree of differentiation between the common core microbiota43 of the Dutch and SAS samples, Principal Coordinate Analysis (PCoA)44 was applied using Bray–Curtis distance45 based on the RAs of the taxa. First, a prevalence threshold of 50% was applied within each group. Then, the first two principal components were plotted in a two-dimensional space. The PCoA was supported by a PERMANOVA analysis46.
Microbiota diversity
To measure the microbial diversity within each sample, the following alpha diversity indices were computed: Chao richness47, Shannon48, Fisher49, and inverse Simpson50. Weighted UniFrac distances51,52 were computed to estimate beta diversity, or how different the samples within each ethnic group were from one another in terms of phylogeny and abundance.
Biomarker analyses
To identify potential biomarkers that could distinguish the Dutch and SAS gut microbiota, a linear discriminant analysis of effect size, or LEfSe analysis53, was performed (p < 0.05 and LDA effect size > 1). LEfSe proposes microbial biomarkers based on RA, effect size, and biological consistency. It can also rank the significance of the biomarkers, i.e., taxa, because it calculates the effect size of each. This rank of significance is provided via the LDA score. LEfSe is especially useful for determining significant differences in taxa with low RAs, which is difficult to do using pure abundance data alone.
Additionally, a differential expression analysis for sequence count data, or DESeq254,55, algorithm was used to calculate if significant differences existed between the bacterial abundances of the two ethnic groups. Lastly, to determine likely species- and strain-level taxonomic identities of the proposed biomarkers, the nucleotide sequences of the corresponding ASVs were input into a BLAST56 search for similar sequences contained only in rRNA/ITS databases.
Network analysis
To estimate two-way ecological relationships in the Dutch and SAS microbiota57, we built microbial co-occurrence (social) networks58 using RA data at the lowest possible taxonomic classification level. We computed SparCC59 correlations between each pair of taxa (p < 0.01). Results were displayed as a network, with nodes representing taxa (size proportional to RA), and edges representing correlation (green = positive, estimating cooperation; red = negative, estimating competition). Networks were visualized using the Fruchterman-Reingold algorithm60 to clarify community structure.
Ethics approval and consent to participate
The HELIUS study was approved by the medical ethics committee of the Amsterdam University Medical Centre, and all participants provided informed consent prior to enrollment in the study.
Results
Cohort analysis
This study analyzed a strictly age- (53 to 55 years-old) and gender-matched subset of the HELIUS gut microbiome cohort to account for any potential confounding effects of these variables on gut microbiota61,62. Only the Dutch and SAS ethnicities were analyzed because these were reported to have the lowest (five %) and highest (21.5%) prevalence of T2DM, respectively, among the ethnic groups of the HELIUS study63. After matching, 41 Dutch and 43 SAS subjects were analyzed. 34.9% of the SAS and 4.9% of the Dutch individuals included in our study were afflicted by T2DM (Table 1).
Diversity analyses
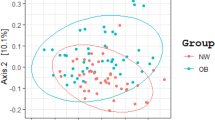
To estimate the degree of differentiation between the common core microbiota of each ethnicity, PCoA44 was applied based on the RAs of the top 25 most abundant taxa (Fig. 1a). The Dutch had a more similar core gut microbiota to each other as far as abundance goes. Contrarily, the SAS varied much more from person to person. Qualitatively, the SAS abundances formed two distinct clusters while the Dutch formed a single, tight cluster. Within the Dutch cluster, the Dutch females formed an even tighter cluster. There was some overlap between both ethnicities and genders. These qualitative observations were well-supported by a PERMANOVA analysis46 (Table 2).

Microbial diversity between samples: (a) To estimate the degree of differentiation between each ethnicity’s common core microbiota, which was approximated by the top 25 most abundant taxa, PCoA44 was applied based on the relative abundances. First, a prevalence threshold of 50%111 was applied. Then, the first two principal components were plotted in a two-dimensional space. The PCoA showed that the Dutch had more similar gut microbiomes regarding the quantitative presence of the common core taxa. Contrarily, the SAS varied much more from person to person. Qualitatively, the SAS formed two clusters (A and B), and the Dutch formed a single cluster (C). Microbiota from the Dutch women formed the tightest cluster (D). There is some overlap between all the groups. (b) To estimate beta diversity, or how different the samples within each ethnic group were from one another for phylogeny and abundance, weighted UniFrac distances51,52 were computed. On average, the Dutch had a significantly more consistent gut microbiota between samples than did the SAS (p = 8.88 × 10–208).
Four different metrics of alpha diversity were computed (Table 3) to achieve consensus on the overall trend in microbial diversity across the samples, as each measurement of alpha diversity can be biased because this index encompasses both species richness and evenness. Chao1 estimates species richness. The Shannon and Simpson indices measure both richness and evenness, with Shannon focusing more on richness and Simpson more on evenness. Though Shannon has been shown to underestimate alpha diversity64. All metrics indicated that the Dutch gut harbored a significantly more diverse microbial milieu (p < 0.001). Weighted UniFrac distances51,52 between samples were computed to approximate beta diversity (Fig. 1b). A lower UniFrac distance indicates a greater degree of similarity between two sets of taxa in terms of phylogeny and abundance. The Dutch had significantly more similar gut bacterial communities to each other than did individuals of SAS origin (p = 8.88 × 10–208).
Microbial abundances
DADA240 was used to infer gut microbiome compositions. The common core gut microbiota of the Dutch and SAS were represented by the top 25 most abundant genera (Fig. 2), or otherwise lowest possible phylogenetic rank, because of the limitations of identifying finer phylogenetic resolutions with short-read, targeted 16S sequencing43,65. Several tests of differential abundance (DA) between the two ethnicities were performed because methods for approximating DA largely vary in output. So, it is best to perform several to attain consensus on the differentially abundant taxa, or potential biomarkers66.

Relative abundances of common core gut microbiota: the common core gut microbiota of the Dutch and South-Asian Surinamese ethnic groups were represented by each ethnicity’s 25 most abundant genera, or otherwise lowest possible phylogenetic rank. Relative abundances were computed by DADA2 (v1.16)40. The blue triangles denote the taxa detected as significantly differentially abundant by a DESeq2 analysis54,55, the orange circles denote those identified as potential biomarkers by LEfSe, and the pink stars represent taxa found to be in significantly different abundance by a Mann–Whitney U test. Taxa highlighted by more than one test of differential abundance are more likely to truly differentiate the two groups.
First, a Mann–Whitney U test between each of the shared common core taxa was calculated (p < 0.05 with Benjamini–Hochberg correction). The following taxa had significantly higher average RAs in the Dutch guts: Subdoligranulum, Ruminococcus, a group of unclassified Lachnospiraceae genera, Christensenellaceae R7 group, Alistipes, Closridium sensu stricto 1, Odoribacter, Butyricicoccus, and Akkermansia. The following taxa had significantly higher average RAs in the SAS guts: Streptococcus, Romboutsia, Haemophilus, and Bifidobacterium.
The second test of DA performed was a DESeq2 analysis54,55. In the Dutch samples, Clostridium sensu stricto 1 was found to be in significantly higher abundance while Bifidobacterium, Haemophilus, and Anaerostipes were found to be in significantly higher abundances in the SAS samples (p-values < 0.05).
Lastly, a LEfSe53 biomarker analysis was computed (Fig. 3). The LDA scores assigned by the LEfSe estimate the individual contribution of each bacterium to the overall uniqueness of the community. Per the LEfSe, the discriminating bacterial biomarkers of the Dutch gut included Odoribacter splanchnicus, Lachnospira, Bacteroides caccae, Alistipes putredinis, Coprococcus comes, and Butyricicoccus. Haemophilus and its ascending phylogenetic ranks characterized the SAS gut.

LEfSe biomarker analysis: to identify potential microbial biomarkers that could distinguish the Dutch and SAS gut microbiomes, a LEfSe analysis53 was performed (p < 0.05 and LDA effect size > 1). The LEfSe was set to propose discriminating taxa at the lowest possible phylogenetic level. The LDA score for each potential biomarker is displayed as a histogram, with all scores falling between |2.5—3|. The proposed biomarkers of the Dutch gut microbiome included Odoribacter splanchnicus, Coprococcus comes, Bacteroides caccae, Alistipes putredinis, Lachnospira, and Butyricicoccus. Haemophilus and its ascending phylogenetic ranks discriminated the SAS gut.
The full phylogenetic lineage of each differentiating taxon proposed by the DESeq2 and LEfSe analyses is provided (Fig. 4). We also inferred the most likely species- and strain-level identities of the differentially abundant taxa by performing a BLAST56 search of the nucleotide sequence of each corresponding ASV uncovered in our sequencing (Table 4). There was notably more species and strain diversity in Clostridium sensu stricto 1 and Bifidobacterium as compared to all the other discriminating taxa. Size factors for DESeq2 ranged from 0.23 to 3.08 for genus level and 0.25–3.47 for species level.

Phylogenetic tree of the proposed gut microbial biomarkers: the phylogenetic lineages of the taxa that were found to distinguish the gut microbiomes of the Dutch and SAS ethnic groups, as per the LEfSe53 or DESeq254,55 analyses, are shown. The blue circles represent microbes that are potential biomarkers of the metabolically healthier Dutch, and the pink circles represent those of the more T2DM-afflicted SAS.
The per sample distributions of the RAs of the differentially abundant taxa, as determined by either a Mann–Whitney U test, DESeq2 analysis, and/or LEfSe biomarker analysis were then plotted (Fig. 5). Clostridium sensu stricto 1 was entirely absent from the SAS guts while Streptococcus, Akkermansia, and Romboustia were totally missing from the Dutch guts. Some bacteria significantly varied in RA between individuals of one ethnicity. Bacteroides, one of the most abundant taxa, showed the most person-to-person variation in both ethnic groups, but this was more pronounced in the SAS. Subdoligranulum was widely distributed across the SAS. The RAs of Streptococcus in the SAS gut had the most outliers. The middle quartiles of the RAs for a group of unclassified Lachnospiraceae genera were almost completely non-overlapping between the two ethnic groups.

Per sample distribution of relative abundances of differentiating taxa: the per sample distributions of the RAs of the differentially abundant genera (p-values < 0.05), as determined by either a Mann–Whitney U test, DESeq2 analysis, and/or LEfSe biomarker analysis were plotted as a box and whisker plot. The Mann–Whitney U test highlighted Subdoligranulum, Ruminococcus, a group of unclassified Lachnospiraceae genera, Christensenellaceae R7 group, Alistipes, Clostridium sensu stricto 1, Odoribacter, Butyricicoccus, and Akkermansia as being significantly enriched RAs in the Dutch while Streptococcus, Romboutsia, Haemophilus, and Bifidobacterium as being significantly enriched in the SAS guts. DESeq2 identified Clostridium sensu stricto 1 as significantly differentially abundant in the Dutch and Bifidobacterium, Haemophilus, and Anaerostipes as significantly differentially abundant in the SAS. Per the LEfSe, the biomarkers of the Dutch gut included Odoribacter splanchnicus, Lachnospira, Bacteroides caccae, Alistipes putredinis, Coprococcus comes, and Butyricicoccus while Haemophilus characterized the SAS gut.
Co-occurrence network analyses
To depict the ecological framework of each ethnic group’s gut microbiome, bacterial co-occurrence networks were constructed (Fig. 6), as previously described58. The thickness of the lines, or edges, is indicative of correlative strength. Green lines represent positive and red lines represent negative correlations. The number next to some of the organisms is a centrality score67, which indicates how important that taxon is to the overall community structure. At a macroscopic view, the community is tightly knit with many positive correlational relationships between taxa in the Dutch. This interconnectedness was undetectable in the SAS.

Bacterial co-occurrence networks: bacterial co-occurrence (social) networks of the Dutch and SAS gut microbiomes are shown. RA data was used at the species or next lowest possible taxonomic classification level. These networks were constructed from pairwise correlation (SparCC59, p < 0.01) matrices using Cytoscape (v3.10.1), and visualized using the Fruchterman-Reingold algorithm60 to clarify community structure. Each node is a distinct ASV, and the node colors represent different phyla as shown in the key. The thickness of the lines, or edges, indicates correlative strength, with green lines representing positive and red lines representing negative correlations. The numbers next to many of the organisms are centrality scores, which indicate how important that taxon is to the overall community.
Blautia appears to be the most integral group for the gut of both ethnicities, since this genus was ranked as the most central taxon in both networks68. Subsequent Affinity Propagation69 analysis revealed the sixth ranked node in the Dutch network, Blautia massiliensis, as the centroid of this large cluster.
Coprococcus comes was ranked as the second most central node in SAS and was unranked in the Dutch network. In the SAS, C. comes negatively correlated with two other taxa (Anaerostipes hadrus and Clostridium sensu stricto 1). In the Dutch, this bacterium positively correlated with eight other taxa (Agathobacter, Bacteroides, Blautia massiliensis, Blautia, Anaerostipes hadrus, Dorea formicigenerans, Lachnospira, and UCG-002) and negatively correlated with one other taxon (Erysipelotrichaceae UCG-003). Oscillibacter was another group that changed its direction of correlation with other bacteria between the two ethnicities, as it held a positive correlation with Blautia massiliensis in the Dutch and a negative correlation with the Christensenellaceae R7 group in the SAS.
Haemophilus and Bifidobacterium shared several positive edges with multiple other highly ranked nodes in the SAS network, and, in the Dutch, these genera were completely disconnected from the other nodes. In the SAS, Haemophilus shared positive edges with Streptococcus (ranked third) and Anaerostipes hadrus (ranked eleventh) while Bifidobacterium shared positive edges with Blautia (ranked first), Blautia massiliensis (ranked fourth), and Agathobacter (ranked 15th). A similar phenomenon occurred with Bacteroides and Erysipelotrichaceae UCG-003.
In the Dutch network, Erysipelotrichaceae UCG-003 (ranked fourth) was negatively correlated with eight other taxa (Agathobacter, Bacteroides, Oscillibacter, Blautia, Dorea formicigenerans, Lachnospira, and UCG-002), and positively correlated with one other taxon (Marvinbryantia). Also in the Dutch, Bacteroides (ranked third) held eight positive edges (Agathobacter, Coprococcus comes, UCG-002, Anaerostipes hadrus, Lachnospira, Dorea formicigenerans, Blautia, and Blautia massiliensis) and two negative edges (Marvinbryantia and Erysipelotrichaceae UCG-003). In the SAS, Bacteroides and Erysipelotrichaceae UCG-003 were not correlated with any other nodes.
Discussion
The T2DM gut microbiome has been shown to be distinctly different from that of normoglycemic, insulin-sensitive individuals14,15,16,21. However, consensus on which taxa are enriched versus depleted as compared to healthy controls has yet to be reached23. Higher microbial production of SCFAs in the gut lumen is protective against T2DM26,27. The dynamics of SCFA-producing taxa have not been well-documented in minority populations because many microbiome studies are largely based on cohorts consisting of ethnic majority groups. It is important to understand how these taxa change with insulin resistance across all populations to develop effective and equitable microbiome-based therapeutics. In this study, the differential bacteria and ecological framework of the gut microbiota from two ethnic groups, the Dutch and South-Asian Surinamese (SAS), living in the same city (Amsterdam, the Netherlands) were analyzed. These two groups, both from the HELIUS cohort37, were compared as the SAS had a much greater prevalence of T2DM. Microbiota compositions were generated via DADA2 (v1.16)40. Abundances were clustered using PCoA44. Metrics of alpha- and beta-diversity were computed. A Mann–Whitney U test and two algorithms, DESeq254,55 and LEfSe53, were used to estimate differentially abundant taxa, or potential biomarkers. Lastly, bacterial co-occurrence networks, which can extract simple patterns from complex microbial community data58, were constructed to compare gut microbial ecology.
Distinct SCFA-producing gut microbial milieus between ethnicities
The DESeq2 identified several taxa that are capable of SCFA production to be significantly enriched in the SAS gut. These included Bifidobacterium, Anaerostipes, and certain strains of Haemophilus70,71,72,73,74,75. Clostridium sensu stricto 1 was detected as significantly enriched in the Dutch. It is unclear if this is a SCFA-producing taxon76,77,78. Contrarily, the LEfSe (Fig. 3) discriminated the Dutch gut from that of the SAS by multiple SCFA-producers: Butyricicoccus, Coprococcus comes, Lachnospira, and Odoribacter splanchnicus79,80,81,82,83,84. Alistipes putredinis was also proposed as a biomarker of the Dutch gut, but this species produces SCFAs in small quantities85. Other than Haemophilus, of which only certain strains produce SCFAs75, no SCFA-producing taxa were proposed as biomarkers of the SAS gut by the LEfSe.
It is unclear from these different results if the overall abundances (Fig. 5) of SCFA-producing taxa truly vary between the two ethnicities. Though, of the potential biomarkers identified, Haemophilus parainfluenzae ATCC 33392 (Table 4) would likely serve as the best discriminating bacterium between these two ethnicities. A consensus approach using various DA methods has been proposed as the best way to identify truly differential taxa. Compared to other DA tests, LEfSe has been shown to have a higher false discovery rate and predisposition to identify more abundant taxa as differential66,86. It has also been demonstrated that the Mann–Whitney U test has a comparatively higher false positive rate66. Contrarily, DESeq2 has a lower type I error rate while maintaining moderate sensitivity87,88.
Given these biases, Haemophilus is likely the most robust biomarker because of the concordance among our DA tests on its significance and it being of relatively low RA in both groups. Odoribacter is also likely a robust biomarker given its low RA across both ethnicities. Bacteroides may be a false call by LEfSe because it was one of the most abundant taxa in both ethnicities. Finally, Akkermansia, enriched in the Dutch, and Streptococcus, enriched in the SAS, might be notable biomarkers even though they were only called by the Mann–Whitney U test. This is because of their complete absence across most of the subjects of each respective ethnicity, as LEfSe and DESeq2 are unable to model zeros88. In future work, it would be beneficial to add more specific methods, such as ALDEx289 or ANCOM-BC90, to identify differential taxa with greater certainty. While SCFA-producers may not be present in significantly different quantities between the Dutch and SAS, the Dutch gut contained a more phylogenetically diverse SCFA-producing microbial milieu.
Haemophilus parainfluenzae, a known respiratory pathobiont, is also a gut pathobiont, as it plays a pro-inflammatory role in Crohn’s Disease91,92. A higher abundance of Haemophilus parainfluenzae has been positively correlated with obesity and cardiometabolic disease by some93, but negatively associated with similar entities by others94,95. To the best of our knowledge, Haemophilus parainfluenzae has not been clearly shown to produce SCFAs in a significant quantity. Our work indicates that this bacterium could be significantly associated, or even a pathobiont, in the guts of those with risk factors for metabolic disease, especially in ethnic minorities. Overall, our results indirectly indicate that normo- and hyperglycemic patients are unable to be distinguished based on microbial SCFA production.
A more complex gut microbiome ecological framework in the ethnic majority
Our PCoA (Fig. 1a) displayed that RAs and phylogeny were more similar across the Dutch samples as compared to the SAS. Sex was not observed to significantly impact gut microbiota because each PCoA cluster contained a roughly equal proportion of both sexes. This is consistent with previous work that has failed to demonstrate a clear relationship between sex and the gut microbiome62. Additionally, the overlap between the Dutch and SAS abundances may be explained by how long ago the SAS individuals had immigrated to the Netherlands, as over time they likely increasingly incorporated Dutch foods and were exposed to the Dutch environmental microbiome.
Bacterial co-occurrence, or social, networks of the Dutch and SAS gut microbiota were computed (Fig. 6). Network analyses can help determine the ecological core microbiota, i.e., which taxa are key for community structure and, possibly, function96. Compared to the SAS, the intra-taxonomic relationships in the Dutch gut were more interconnected, with many more positive correlational relationships. The Dutch network also contained a large cluster of SCFA-producing taxa (Coprococcus comes, Lachnospira, Blautia, and Haemophilus). This positively interconnected ecological framework correlates with the significantly higher average alpha diversity (Table 3), lower beta diversity, and more stable community composition and phylogenetic distribution across the Dutch samples (Fig. 1). Previous studies have associated the SAS and diabetics with a less diverse gut microbiota23,97.
Our networks support the notion that the role played (cooperation versus competition) by a taxon may change with ethnicity and is possibly a function of host glycemic phenotype. In the Dutch network, Erysipelotrichaceae UCG-003 was estimated to be the fourth most important taxon for the community structure and had several negative relationships with other taxa. However, in the SAS network, this taxon was unranked and lacked any correlations with other community members. Erysieplotrichaceae UCG-003 has previously been positively associated with insulin resistance and obesity98,99. Erysieplotrichaceae UCG-003 has also been proposed as a marker of healthy aging100. This taxon may be negatively correlated with others in the Dutch network because there is competition for a mutual resource. In the Dutch, the other likely beneficial taxa that Erysieplotrichaceae UCG-003 is correlated with may be better able to acquire that resource and use it to produce a metabolite that is beneficial for the host, such as a SCFA. This competition was not observed in the SAS.
The Dutch, less afflicted by T2DM, may have a more interconnected gut microbial ecology because of greater bacterial cross-feeding interactions. Cross-feeding is dependent on microbiota spatial organization and is critical for SCFA production26,27. Several taxa that were either SCFA producers or negatively associated with T2DM were members of the large, interconnected Dutch network. Bacteroides, which has a strong negative association with T2DM22,24,101, was disconnected and unranked in the SAS network, but was ranked as the third most integral taxon in the Dutch network. So, the metabolically beneficial role of this bacterium is likely a reproducible and accurate finding. Oscillibacter, ranked sixth in the Dutch and ninth in the SAS network, has been shown to be positively associated with T2DM102,103. Bifidobacterium, which has been reported to be protective against T2DM13,22, had more positive relationships with other bacteria in the SAS group, but was unranked in both networks. So, the role of a particular taxon in relation to metabolic fitness may depend on host ethnicity.
The main limitation of our study was that we were unable to stratify per-sample the two ethnicities by diabetic status. Although subjects were strictly age- and gender-matched, other factors, such as ethnically variable genetic predispositions104,105, epigenetics106,107, diet61,108,109, and socioeconomic status110 likely confounded our findings. The ethnic minorities of the HELIUS cohort were of lower socioeconomic status, which significantly impacts health equity39. Differences in dietary and exercise patterns between the ethnicities were also observed in the HELIUS study39. Additionally, most of the SAS were first-generation immigrants, so they had a shorter length of residence in the Netherlands37. More work is needed to understand gut microbiota in the context of each ethnicity to drive forward personalized medicine for metabolic diseases.
Data availability
The HELIUS data are owned by the Amsterdam University Medical Centers, location AMC, in Amsterdam, the Netherlands. Any researcher can request the data by submitting a proposal to the HELIUS Executive Board, as outlined at http://www.heliusstudy.nl/en/researchers/collaboration, by email to heliuscoordinator@amsterdamumc.nl. The HELIUS Executive Board will check proposals for compatibility with the general objectives, ethical approvals, and informed consent forms of the HELIUS study. There are no other restrictions to obtaining the data and all data requests will be processed in the same manner. The microbial genomic sequences from the HELIUS cohort, which were used for this study, are stored under protected access on the European Genome-Phenome Archive (https://ega-archive.org/datasets/EGAD00001004106).
Code availability
Our entire downstream analysis through the open-source PluMA11 initiative is available at http://biorg.cs.fiu.edu/pluma/pipelines.html.
Abbreviations
- ASV:
-
Amplicon sequence variant
- BP:
-
Base-pair
- DA:
-
Differential abundance
- DESeq:
-
Differential expression analysis for sequence count data
- FPG:
-
Fasting plasma glucose
- GLP-1:
-
Glucagon-like peptide 1
- HELIUS:
-
Healthy life in an urban setting
- PCoA:
-
Principal coordinate analysis
- RA:
-
Relative abundance
- SAS:
-
South-Asian Surinamese
- SCFA:
-
Short-chain fatty acid
- T2DM:
-
Type two diabetes mellitus
References
Berg, G. et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome 8, 103 (2020).
Münger, E., Montiel-Castro, A. J., Langhans, W. & Pacheco-López, G. Reciprocal interactions between gut microbiota and host social behavior. Front. Integr. Neurosci. 12, (2018).
Ogunrinola, G. A., Oyewale, J. O., Oshamika, O. O. & Olasehinde, G. I. The human microbiome and its impacts on health. Int. J. Microbiol. 2020, e8045646 (2020).
Hooks, K. B. & O’Malley, M. A. Dysbiosis and its discontents. mBio 8, e01492-e1517 (2017).
Foster, J. A. & McVey Neufeld, K.-A. Gut–brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 36, 305–312 (2013).
Scher, J. U. & Abramson, S. B. The microbiome and rheumatoid arthritis. Nat. Rev. Rheumatol. 7, 569–578 (2011).
Wong, S. H. & Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 16, 690–704 (2019).
Khan, I. et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 8, 126 (2019).
Romano, S. et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 7, 1–13 (2021).
Kang, D.-W. et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 5, 10 (2017).
Mathee, K., Cickovski, T., Deoraj, A., Stollstorff, M. & Narasimhan, G. The gut microbiome and neuropsychiatric disorders: Implications for attention deficit hyperactivity disorder (ADHD). J. Med. Microbiol. 69, 14–24 (2020).
Devaraj, S., Hemarajata, P. & Versalovic, J. The human gut microbiome and body metabolism: Implications for obesity and diabetes. Clin. Chem. 59, 617–628 (2013).
Bielka, W., Przezak, A. & Pawlik, A. The role of the gut microbiota in the pathogenesis of diabetes. Int. J. Mol. Sci. 23, 480 (2022).
Vals-Delgado, C. et al. An altered microbiota pattern precedes Type 2 diabetes mellitus development: From the CORDIOPREV study. J. Adv. Res. 35, 99–108 (2022).
Fang, Y. et al. Characteristics of the gut microbiota and metabolism in patients with latent autoimmune diabetes in adults: A case-control study. Diabetes Care 44, 2738–2746 (2021).
Zhou, W. et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature 569, 663–671 (2019).
National Diabetes Statistics Report | Diabetes | CDC. https://www.cdc.gov/diabetes/data/statistics-report/index.html (2022).
International Diabetes Federation (2019) IDF Diabetes Atlas: Ninth edition 2019. Available from https://diabetesatlas.org/upload/resources/material/20200302_133351_IDFATLAS9e-final-web.pdf. Accessed Sept 7th, 2023.
Agyemang, C., van der Linden, E. L. & Bennet, L. Type 2 diabetes burden among migrants in Europe: Unravelling the causal pathways. Diabetologia 64, 2665–2675 (2021).
DeFronzo, R. A. et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 1, 15019 (2015).
Cui, J. et al. Butyrate-producing bacteria and insulin homeostasis: The microbiome and insulin longitudinal evaluation study (MILES). Diabetes 71, 2438–2446 (2022).
Gurung, M. et al. Role of gut microbiota in type 2 diabetes pathophysiology. eBioMedicine 51, (2020).
Letchumanan, G. et al. Gut microbiota composition in prediabetes and newly diagnosed type 2 diabetes: A systematic review of observational studies. Front. Cell. Infect. Microbiol. 12, (2022).
Guo, Z., Pan, J., Zhu, H. & Chen, Z.-Y. Metabolites of gut microbiota and possible implication in development of diabetes mellitus. J. Agric. Food Chem. 70, 5945–5960 (2022).
Sanna, S. et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51, 600–605 (2019).
Morrison, D. J. & Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7, 189–200 (2016).
Blaak, E. et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 11, 411–455 (2020).
McNelis, J. C. et al. GPR43 potentiates β-cell function in obesity. Diabetes 64, 3203–3217 (2015).
Priyadarshini, M. et al. An acetate-specific GPCR, FFAR2, regulates insulin secretion. Mol. Endocrinol. 29, 1055–1066 (2015).
Reddy, M. A. et al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes 63, 4249–4261 (2014).
Pingitore, A. et al. The diet-derived short chain fatty acid propionate improves beta-cell function in humans and stimulates insulin secretion from human islets in vitro. Diabetes Obes. Metab. 19, 257–265 (2017).
Canfora, E. E. et al. Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: A randomized crossover trial. Sci. Rep. 7, 2360 (2017).
van der Beek, C. M. et al. Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin. Sci. 130, 2073–2082 (2016).
Freeland, K. R. & Wolever, T. M. S. Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br. J. Nutr. 103, 460–466 (2010).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41 (2017).
Gaulke, C. A. & Sharpton, T. J. The influence of ethnicity and geography on human gut microbiome composition. Nat. Med. 24, 1495–1496 (2018).
Deschasaux, M. et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531 (2018).
Gacesa, R. et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 604, 732–739 (2022).
Snijder, M. B. et al. Cohort profile: The healthy life in an urban setting (HELIUS) study in Amsterdam, The Netherlands. BMJ Open 7, e017873 (2017).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Nayman, E. I. et al. Microbiome depiction through user-adapted bioinformatic pipelines and parameters. J. Med. Microbiol. 72, 001756 (2023).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Neu, A. T., Allen, E. E. & Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. 118, e2104429118 (2021).
Pearson, K. L. I. I. I. On lines and planes of closest fit to systems of points in space. Lond. Edinburgh Dublin Philos. Mag. J. Sci. 2, 559–572 (1901).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). in Wiley StatsRef: Statistics Reference Online 1–15 (John Wiley & Sons, Ltd, 2017). https://doi.org/10.1002/9781118445112.stat07841.
Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270 (1984).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423 (1948).
Fisher, R. A., Corbet, A. S. & Williams, C. B. The relation between the number of species and the number of individuals in a random sample of an animal population. J. Anim. Ecol. 12, 42–58 (1943).
Simpson, E. H. Measurement of diversity. Nature 163, 688–688 (1949).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. & Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 5, 169–172 (2011).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Schoch, C. L. et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database (Oxford) 2020, baaa062 (2020).
Faust, K. et al. Microbial co-occurrence relationships in the human microbiome. PLOS Comput. Biol. 8, e1002606 (2012).
Fernandez, M., Riveros, J. D., Campos, M., Mathee, K. & Narasimhan, G. Microbial ‘social networks’. BMC Genomics 16, S6 (2015).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLOS Comput. Biol. 8, e1002687 (2012).
Fruchterman, T. M. J. & Reingold, E. M. Graph drawing by force-directed placement. Softw. Pract. Exp. 21, 1129–1164 (1991).
Nagpal, R. et al. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 4, 267–285 (2018).
Kim, Y. S., Unno, T., Kim, B.-Y. & Park, M.-S. Sex differences in gut microbiota. World J. Mens. Health 38, 48–60 (2020).
Snijder, M. B. et al. Case finding and medical treatment of type 2 diabetes among different ethnic minority groups: The HELIUS study. J. Diabetes Res. 2017, 9896849 (2017).
Willis, A. D. Rarefaction, Alpha Diversity, and Statistics. Front. Microbiol. 10, (2019).
Janda, J. M. & Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764 (2007).
Nearing, J. T. et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 13, 342 (2022).
Cickovski, T., Aguiar-Pulido, V. & Narasimhan, G. MATria: A unified centrality algorithm. BMC Bioinf. 20, 278 (2019).
Freeman, L. C. Centrality in social networks conceptual clarification. Soc. Netw. 1, 215–239 (1978).
Frey, B. J. & Dueck, D. Clustering by passing messages between data points. Science 315, 972–976 (2007).
Belenguer, A. et al. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 72, 3593–3599 (2006).
Duncan, S. H., Louis, P. & Flint, H. J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 70, 5810–5817 (2004).
Lee, J.-Y. et al. Anaerostipes hominis sp. nov., a novel butyrate-producing bacteria isolated from faeces of a patient with Crohn’s disease. Int. J. Syst. Evol. Microbiol. 71, 1 (2021).
Sato, T. et al. Isolation of lactate-utilizing butyrate-producing bacteria from human feces and in vivo administration of Anaerostipes caccae strain L2 and galacto-oligosaccharides in a rat model. FEMS Microbiol. Ecol. 66, 528–536 (2008).
Schwiertz, A. et al. Anaerostipes caccae gen. nov., sp. nov., a new saccharolytic, acetate-utilising, butyrate-producing bacterium from human faeces. Syst. Appl. Microbiol. 25, 46–51 (2002).
López-López, N. et al. Haemophilus influenzae glucose catabolism leading to production of the immunometabolite acetate has a key contribution to the host airway-pathogen interplay. ACS Infect. Dis. 6, 406–421 (2020).
Huart, J. et al. Gut microbiota and fecal levels of short-chain fatty acids differ upon 24-hour blood pressure levels in men. Hypertension 74, 1005–1013 (2019).
Wang, Y. et al. Opportunistic bacteria confer the ability to ferment prebiotic starch in the adult cystic fibrosis gut. Gut Microbes 10, 367–381 (2018).
Hu, C. et al. A comprehensive analysis of the colonic flora diversity, short chain fatty acid metabolism, transcripts, and biochemical indexes in heat-stressed pigs. Front. Immunol. 12, 1 (2021).
Amiri, P. et al. Role of butyrate, a gut microbiota derived metabolite, in cardiovascular diseases: A comprehensive narrative review. Front. Pharmacol. 12, 1 (2022).
Eeckhaut, V. et al. Butyricicoccus pullicaecorum gen. nov., sp. nov., an anaerobic, butyrate-producing bacterium isolated from the caecal content of a broiler chicken. Int. J. Syst. Evol. Microbiol. 58, 2799–2802 (2008).
Nogal, A. et al. Circulating levels of the short-chain fatty acid acetate mediate the effect of the gut microbiome on visceral fat. Front. Microbiol. 12, (2021).
Abdugheni, R. et al. Metabolite profiling of human-originated Lachnospiraceae at the strain level. iMeta 1, e58 (2022).
Hiippala, K. et al. Novel odoribacter splanchnicus strain and its outer membrane vesicles exert immunoregulatory effects in vitro. Front. Microbiol. 11, 1 (2020).
Werner, H., Rintelen, G. & Kunstek-Santos, H. A new butyric acid-producing bacteroides species: B. splanchnicus n. sp. (author’s transl). Zentralbl Bakteriol Orig A 231, 133–144 (1975).
Parker, B. J., Wearsch, P. A., Veloo, A. C. M. & Rodriguez-Palacios, A. The genus alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 11, (2020).
Wallen, Z. D. Comparison study of differential abundance testing methods using two large Parkinson disease gut microbiome datasets derived from 16S amplicon sequencing. BMC Bioinf. 22, 265 (2021).
Calgaro, M., Romualdi, C., Waldron, L., Risso, D. & Vitulo, N. Assessment of statistical methods from single cell, bulk RNA-seq, and metagenomics applied to microbiome data. Genome Biol. 21, 191 (2020).
Cho, H. et al. Comprehensive evaluation of methods for differential expression analysis of metatranscriptomics data. Brief. Bioinf. 24, bbad279 (2023).
Fernandes, A. D., Macklaim, J. M., Linn, T. G., Reid, G. & Gloor, G. B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLOS ONE 8, e67019 (2013).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514 (2020).
Sohn, J. et al. Periodontal disease is associated with increased gut colonization of pathogenic Haemophilus parainfluenzae in patients with Crohn’s disease. Cell Rep. 42, 112120 (2023).
Fitzgerald, R. S., Sanderson, I. R. & Claesson, M. J. Paediatric inflammatory bowel disease and its relationship with the microbiome. Microb. Ecol. 82, 833–844 (2021).
Chen, J. et al. Gut microbiome alterations in patients with carotid atherosclerosis. Front. Cardiovasc. Med. 8, 1 (2021).
Palmnäs-Bédard, M. S. A. et al. The human gut microbiota and glucose metabolism: A scoping review of key bacteria and the potential role of SCFAs. Am. J. Clin. Nutr. 116, 862–874 (2022).
Makki, K., Deehan, E. C., Walter, J. & Bäckhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 23, 705–715 (2018).
Atzeni, A. et al. Taxonomic and functional fecal microbiota signatures associated with insulin resistance in non-diabetic subjects with overweight/obesity within the frame of the PREDIMED-plus study. Front. Endocrinol. (Lausanne) 13, 804455 (2022).
Roelofsen, H., Priebe, M. G. & Vonk, R. J. The interaction of short-chain fatty acids with adipose tissue: Relevance for prevention of type 2 diabetes. Benef. Microbes 1, 433–437 (2010).
Li, Y. et al. Effects of Konjaku flour on the gut microbiota of obese patients. Front. Cell. Infect. Microbiol. 12 (2022).
Singh, H. et al. Gastro-intestinal and oral microbiome signatures associated with healthy aging. GeroScience 41, 907–921 (2019).
Wu, X. & Park, S. Fecal bacterial community and metagenome function in Asians with type 2 diabetes according to enterotypes. Biomedicines 10, 2998 (2022).
Zeevi, D. et al. Structural variation in the gut microbiome associates with host health. Nature 568, 43–48 (2019).
Thingholm, L. B. et al. Obese individuals with and without Type 2 diabetes show different gut microbial functional capacity and composition. Cell Host Microbe 26, 252-264.e10 (2019).
Langenberg, C. & Lotta, L. A. Genomic insights into the causes of type 2 diabetes. Lancet 391, 2463–2474 (2018).
Sirdah, M. M. & Reading, N. S. Genetic predisposition in type 2 diabetes: A promising approach toward a personalized management of diabetes. Clin. Genet. 98, 525–547 (2020).
Stronks, K. et al. Unravelling the impact of ethnicity on health in Europe: The HELIUS study. BMC Public Health 13, 402 (2013).
Shojima, N. & Yamauchi, T. Progress in genetics of type 2 diabetes and diabetic complications. J. Diabetes Investig. 14, 503–515 (2023).
Stephenson, E. J., Smiles, W. & Hawley, J. A. The relationship between exercise, nutrition and type 2 diabetes. Med. Sport Sci. 60, 1–10 (2014).
Delpino, F. M. et al. Ultra-processed food and risk of type 2 diabetes: A systematic review and meta-analysis of longitudinal studies. Int. J. Epidemiol. 51, 1120–1141 (2022).
Canedo, J. R., Miller, S. T., Schlundt, D., Fadden, M. K. & Sanderson, M. Racial/ethnic disparities in diabetes quality of care: The role of healthcare access and socioeconomic status. J. Racial Ethn. Health Disparities 5, 7–14 (2018).
Espelt, A. et al. Socioeconomic position and type 2 diabetes mellitus in Europe 1999–2009: A panorama of inequalities. Curr. Diabetes Rev. 7, 148–158 (2011).
Custer, G. F., Gans, M., van Diepen, L. T. A., Dini-Andreote, F. & Buerkle, C. A. Comparative analysis of core microbiome assignments: Implications for ecological synthesis. mSystems 8, e01066–22 (2023).
Acknowledgements
The HELIUS study was conducted by the Amsterdam University Medical Centers, location AMC and the Public Health Service of Amsterdam. Both organizations provided core support for HELIUS. We are most grateful to the participants of the HELIUS study and the management team, research nurses, interviewers, research assistants, and other staff who have taken part in gathering the data of this study. We would like to thank the members of the FIU Bioinformatics Research Group for their valuable feedback throughout several discussions of this work, especially Dr. Vitalii Stebliankin for his statistical support.
Funding
The HELIUS study was funded by the Dutch Heart Foundation, the Netherlands Organization for Health Research and Development (ZonMw), the European Union (FP-7), and the European Fund for the Integration of non-EU immigrants (EIF). M. Nieuwdorp was supported by a personal ZONMW-VICI grant 2020 (09150182010020).
Author information
Authors and Affiliations
Contributions
Conceptualization: E.I.N., B.A.S, M.P., A.C.G, and K.M.; methodology: E.I.N., B.A.S, M.N., and T.C.; data analysis: E.I.N., B.A.S., M.P., A.C.G., M.N., T.C., and K.M.; writing and editing: E.I.N., B.A.S., M.N., T.C., and K.M.; supervision: T.C. and K.M. All authors have read and agreed to the published version of the manuscript. All authors consent for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nayman, E.I., Schwartz, B.A., Polmann, M. et al. Differences in gut microbiota between Dutch and South-Asian Surinamese: potential implications for type 2 diabetes mellitus. Sci Rep 14, 4585 (2024). https://doi.org/10.1038/s41598-024-54769-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54769-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
