
Abstract
Depression is a common mental disease, with some patients exhibiting ideas and behaviors such as self-harm and suicide. The drugs currently used to treat depression have not achieved good results. It has been reported that metabolites produced by intestinal microbiota affect the development of depression. In this study, core targets and core compounds were screened by specific algorithms in the database, and three-dimensional structures of these compounds and proteins were simulated by molecular docking and molecular dynamics software to further study the influence of intestinal microbiota metabolites on the pathogenesis of depression. By analyzing the RMSD gyration radius and RMSF, it was finally determined that NR1H4 had the best binding effect with genistein. Finally, according to Lipinski's five rules, equol, genistein, quercetin and glycocholic acid were identified as effective drugs for the treatment of depression. In conclusion, the intestinal microbiota can affect the development of depression through the metabolites equol, genistein and quercetin, which act on the critical targets of DPP4, CYP3A4, EP300, MGAM and NR1H4.
Similar content being viewed by others

Introduction
Depression is a common neuropsychiatric disorder with a high recurrence rate. Different types of depression affect approximately 350 million people worldwide, and the prevalence of depression is increasing1,2. Currently, the incidence of depression is increasing yearly, causing considerable losses to families and society. According to the World Health Organization, the disease burden of depression will rank first in Western countries, and depression will become the fourth most significant disease in the world by 20303,4. The etiology of depression is complex, the mechanism is still unclear, and the treatment response is insufficient, thereby reducing the efficiency of clinical diagnosis and treatment5,6. Therefore, it is critical to explore the etiologic mechanisms of depression. Selective serotonin reuptake inhibitors, serotonin, norepinephrine reuptake inhibitors, and other drugs are the primary treatment methods in modern medicine, but some patients do not achieve good efficacy and experience severe adverse reactions, such as cardiac toxicity7.
Ongoing research on this subject shows that the intestinal microbiota plays a crucial role in maintaining human health and preventing various diseases. The intestinal microbiota is also important for the normal development and maintenance of brain function and is related to various mental and neurological diseases8,9. The central nervous system can regulate the physiological activities of the intestine. In contrast, various physiological responses caused by changes in the composition of the intestinal microbiota can be transmitted to the brain and other central nerves to stimulate the intestinal microbiota, resulting in various physiological and pathological phenomena. Therefore, the “brain–gut” axis theory opens up a new avenue for treating depression10,11. Relevant studies have suggested that the intestinal microbiota of patients with depression differs significantly from that of people without depression and can affect brain function via the brain-gut axis, suggesting that the intestinal microbiota is closely related to the occurrence and progression of depression12,13,14. Many studies have shown that the microbiota–gut–brain (MGB) axis affects brain function and behavior; regulates stress, anxiety, and cognition; and causes depressive symptoms. Therefore, depression can be treated by regulating the intestinal microbiota15,16,17. Here, we explored the pathogenesis of depression based on the intestinal microbiota.
With the popularity of gene microarrays and RNA sequencing, bioinformatics has been widely used to analyze high-throughput sequencing data for various diseases. In this study, we used the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database and the GutMgene database (http://bio-annotation.cn/gutmgene/home.dhtml) to investigate the mechanism of depression and predict a promising drug for its treatment.
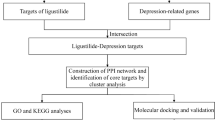
We obtained the intestinal microbiota and metabolite information from the human intestinal microbiota database. Then, we predicted the metabolite targets through two online target prediction websites, SEA and STP, and intersected the prediction results of the two websites to obtain the target genes of metabolites of intestinal microbiota and the potential effects on human diseases. Then, we selected three depression-related datasets from the GEO database, normalized them and used a volcano plot to show the differentially expressed genes in depression. The differentially expressed genes were intersected with the potential target genes of intestinal microbiota metabolites affecting human diseases, and the potential target genes of intestinal microbiota metabolites affecting depression were obtained. Subsequently, a protein‒protein interaction network and GO and KEGG enrichment analyses of these key differentially expressed genes were constructed, and the “intestinal microbiota–metabolites–target gene” network relationship was constructed. According to the network topology analysis by Cytoscape, we screened the key small-molecule compounds and proteins, carried out molecular docking and molecular dynamics simulation verification, and finally evaluated the potential small-molecule compounds for drug toxicity. The technology roadmap for this study is presented in Fig. 1.

Technology roadmap for this study.
Materials and methods
Acquisition of target genes of intestinal microbiota
Metabolite and target gene information for the intestinal microbiota and microflora were extracted from GutMgene, a database of intestinal microbiota. Using the similarity ensemble approach (SEA) (https://sea.bkslab.org/) and the SwissTargetPrediction (STP) (http://www.swisstargetprediction.ch/) database, we mined metabolites that may influence target genes and took the genes at the intersection of the two methods as intestinal microbiota target genes18,19,20.
Download and standardization of microarray data and identification of differentially expressed genes (DEGs)
Search strategies included “depression” [MeSH term] or “depression” [all fields]; “Homo sapiens” [porgn]; and “expression analysis by array” [filter].
According to the above retrieval strategies, the expression profiles of three gene sets, GSE19738, GSE76826, and GSE182193, were selected from the GEO database for analysis. After retrieving and standardizing the three datasets using the GEOquery package in the R language (version 4.1.3), boxplots of the gene expression data were designed using the ggplot2 package21,22. Genes with a threshold standard of |log FC > 0.5| and p value < 0.05 were selected as DEGs, and SAT samples were screened using a linear model of microarray data packets. The Pheat map, ggplot2, and RColorBrewer packages were used to create DEG volcano maps.
Identification of DEGs
The gene expression profiles of GSE19738, GSE76826, and GSE182193 were selected from the GEO database for analysis. DEGs were selected from the above three gene expression spectra, with threshold criteria of |logFC|> 0.5 and p < 0.05. After the duplicated DEGs were summarized and deleted, 44 differential genes were obtained from the intersection of DEGs and the target genes of intestinal microbiota. The 44 differentially expressed genes obtained through the intersection can be considered key genes that cause depression through the action of intestinal microbiota metabolites on target genes.
Enrichment analysis of gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathways
GO and KEGG enrichment analyses were performed on the selected 44 differentially expressed genes23. GO analysis annotates genetic information based on three aspects: molecular function, biological process, and cellular composition.
Establishment of protein‒protein interaction (PPI) networks and identification of hub genes
The 44 differentially expressed genes were imported into the STRING database (https://string-db.org/), and the protein interaction relationship was obtained after hiding the free nodes. The results were imported into Cytoscape 3.7.2 for visualization, and network topology analysis was performed using the CytoNCA function to identify the key target genes24,25.
Construction of the “intestinal microbiota–metabolites–target genes” network relationship
We used Cytoscape 3.7.2 to construct a "microbiota–metabolites–target genes" network to help determine the pharmacological mechanism. The degree value reflects the importance of nodes in the network. The higher the value, the more influential the node is. The core metabolites were identify by degree.
Molecular docking
The core metabolites were downloaded from the PubChem database. The small molecules were hydrogenated, and the charge was calculated using Autodock 4.2.6 software26. The core target structures were obtained using the AlphaFold Protein Structure Database27,28; they were then imported into Autodock, and an appropriate grid box was set for molecular docking. Finally, the conformation with the lowest docking binding energy was chosen as the final docking result, which was visualized using PyMOL. If the docking binding energy is less than − 5 kcal mol−1, the receptor and ligand can bind spontaneously. The lower the binding energy is, the greater the possibility of binding, the more stable the binding conformation, and the greater the possibility of reaction. We set the appropriate grid box and we presented the the values as follows: DPP4 (x = 40, y = 45, z = 40), CYP3A4 (x = 56, y = 43, z = 56), EP300 (x = 41, y = 45, z = 43), MGAM (x = 37, y = 37, z = 36), NR1H4 (x = 30, y = 40, z = 43).
Molecular dynamics simulation
Molecular dynamics (MD) simulation of the ligand‒receptor docked complex was carried out using GROMACS (version 2021.2)29. The protein topology file was generated using the AMBER99SB-ILDN force field30, whereas the ligand topology file was generated by the AnteChamber PYthon Parser interfacE (ACPYPE) script using the AMBER force field. MD simulations were carried out in a triclinic box filled with TIP3 water molecules and periodic bounding conditions. The system was neutralized with NaCl counter ions. Before MD simulation, the complex was minimized for 1000 steps and equilibrated by running canonical ensemble, constant-pressure (NVT) and constant-temperature (NPT) for 100 ps. Then, MD simulation was performed for 100 ns for each system under periodic boundary conditions at 310 K temperature and 1.0 bar pressure.
Drug similarity and toxicity profile assessment
Similarities and toxicity characteristics were determined using SwissADME31 (http://www.swissadme.ch/) and validation ADMETlab32 network tools (https://admetmesh.scbdd.com/). Because these two factors are critical in the promotion of new agents, we evaluated their physicochemical properties and side effects.
Results
Intersection of the Venn diagram
After obtaining human intestinal microbiota data from the GutMgene database, the target genes that metabolites could act on were predicted via the SEA and STP databases (Tables S1 and S2, Supplemental Digital Contents, which demonstrate the prediction results of SEA and STP). A total of 790 targets were obtained by the intersection of the genes obtained from the two databases (Fig. 2) (Table S3, Supplemental Digital Content, which demonstrates the result of the intersection of the STP and SEA results).

Acquisition of intestinal microbiota genes. There were 790 common targets between SEA (1251) and STP (1268).
Identification of DEGs in depression
After dataset standardization (Fig. 3), 1942 DEGs were selected (|logFC|> 0.5, p < 0.05) based on the methods described above. The results included 858 upregulated genes and 1084 downregulated genes. Next, 1908 DEGs were reconstructed by merging and removing duplicates (Fig. 4) (Table S4, Supplemental Digital Content, which demonstrates differential genes after GEO dataset processing).

Boxplot of expression profiles after merging and standardization. The x-axis represents the sample symbol, and the y-axis represents gene expression values. The black line in the boxplot shows the median gene expression. (a) Standardization of GSE19738. (b) Standardization of GSE76826. (c) Standardization of GSE182193.

Volcano map used to identify DEGs. (a) GSE19738, (b) GSE76826, and (c) GSE182193. The abscissa in the volcano plot is the log2 (fold change) value, and the ordinate is the log10 (p value). The red dots represent upregulated genes, and the blue dots represent downregulated genes.
Intersection between the target genes of the intestinal microbiota and depression in the GEO database
Seven hundred ninety metabolite target genes were intersected with 1908 depression-related differentially expressed genes screened in the GEO database. Accordingly, 44 differentially expressed genes were obtained, which can be considered the crucial genes of intestinal microbiota metabolites that affect depression (Fig. 5) (Table S5, Supplemental Digital Content, which demonstrates the intersection of metabolite targets and GEO data).

Intersection between target genes of the intestinal microbiota and depression in the GEO database.
GO enrichment analysis and KEGG analysis of core genes
The 44 differentially expressed genes were analyzed and visualized by GO and KEGG analyses based on the Metascape database (Fig. 6).

Enrichment analysis of GO and KEGG biological functions based on depression-related hub genes. (a) Molecular functions (MF); (b) biological processes (BP); (c) cellular components (CC); (d) KEGG.
Construction and analysis of the PPI network
The PPI network consisted of 28 nodes and 32 edges. Dipeptidyl peptidase 4 (DPP4), cytochrome P450 3A4 (CYP3A4), histone acetyltransferase p300 (EP300), maltase-glucoamylase (MGAM) and bile acid receptor (NR1H4) were identified as core targets according to degree values (Fig. 7) (Table S6, Supplemental Digital Content, which demonstrates the PPI network topology analysis results).

PPI network. Node size and color shade reflect the importance of the node.
Construction of the “intestinal microbiota–metabolites–target genes” network relationship
The network of "intestinal microbiota–metabolites–target genes" was constructed (Table S7, Supplemental Digital Content, which illustrates the intestinal microbiome–metabolite–substrate–target gene network). According to the degree values, equol, genistein, quercetin and glycocholic acid were considered the key metabolites affecting depression (Fig. 8).

“Intestinal microbiota–metabolites–substrate–genes” network. Green represents target genes; blue represents the intestinal microbiota; purple represents metabolites; and yellow represents the substrate.
Molecular docking results
The key metabolites screened were molecularly docked with depression-related targets. The binding energies of the main active constituents and main targets were all < − 7.0 kcal/mol (Table S8, Supplemental Digital Content, which demonstrates the molecular docking results). The smaller the binding energy is, the higher the binding activity is, and the easier the compound is to bind to the target. Our molecular docking results show that key genes and metabolites can form stable conformations. Among them, the key gene MGAM had the best molecular docking results with the metabolite glycocholic acid, and its value was − 9.7 kcal/mol (Table 1). All molecular docking results were visualized (Fig. 9).

Molecular docking demonstration of core metabolites and core targets.
Molecular dynamics simulation results
The root mean square deviation (RMSD) curve represents the fluctuation of protein conformation. It can be seen from the figure that in the beginning, RMSD increases because of the interactions between the complex and the solvent. Therefore, RMSD has certain fluctuations in the early stage. However, MGAM-glycocholic acid, NR1H4-equol, NR1H4-genistein and NR1H4-quercetin all increased briefly and tended to be stable, which indicated that the conformation of proteins would not change significantly after the combination of small molecular ligands with proteins, and the combination was relatively stable (Fig. 10a).

Molecular dynamics simulation results. (a) RMSD plot during molecular dynamics simulations. The black curve represents MGAM-glycocholic acid. The red curve represents NR1H4-equol. The blue curve represents NR1H4-genistein. The green curve represents NR1H4-quercetin. (b) Rog plot during molecular dynamics simulations. The black curve represents MGAM-glycocholic acid. The red curve represents NR1H4-equol. The blue curve represents NR1H4-genistein. The green curve represents NR1H4-quercetin. (c) RMSF plot during molecular dynamics simulations. The black curve represents MGAM-glycocholic acid-A. The red curve represents MGAM-glycocholic acid-B. The blue curve represents MGAM-glycocholic acid-C. The green curve represents NR1H4-equol. The purple curve represents NR1H4-genistein. The orange curve represents NR1H4-quercetin.
The gyration radius is often used to describe the change in the overall structure of a protein and to show the compactness of the overall structure. It can be seen from the figure that MGAM-glycocholic acid, NR1H4-equol, NR1H4-genistein and NR1H4-quercetin all have very stable gyration radii. This result is consistent with the RMSD curve reaction, which proves the stability of the protein conformation (Fig. 10b).
Root mean square fluctuation (RMSF) represents the fluctuation of the protein amino acid residues. This reflects the protein's flexibility in the molecular dynamics simulation process. Usually, after the drug is combined with the protein, the flexibility of the protein is reduced to stabilize the protein and play the role of enzyme. As seen from the figure, the simulation results of NR1H4 protein and equol drug, NR1H4 protein and genistein drug, and NR1H4 protein and quercetin drug show that the protein has good flexibility (Fig. 10c).
Evaluation of drug similarity and toxicity characteristics
Using the SwissADME and ADMETlab platforms and according to Lipinski's Five Laws, the similarity and toxicity of equol, genistein, quercetin and glycocholic acid were evaluated, including molecular weight (≤ 500), H-bond receptor (≤ 10), H-bond donor (≤ 5), MlogP (≤ 4.15), bioavailability score (> 0.1), and topological polar surface area (< 140). The results showed that equol, genistein, quercetin and glycocholic acid could be accepted as new drugs based on pharmacokinetic parameters (Table 2).
Despite an acceptable therapeutic value, a drug is still not acceptable as a final product if it exhibits unintended toxicity. Therefore, drug candidates should exceed toxicity limits for further validation. Therefore, equol, genistein, and quercetin were evaluated by the ADMETlab platform for hERG blockers, acute oral toxicity in rats, eye corrosion, and respiratory toxicity (including LD50 [5.238 mg/kg]). The results showed that these substances could play an important role in the treatment of depression (Table 3).
Discussion
Studies have shown that metabolites of intestinal microbes can play an essential role in a variety of psychiatric diseases via the “GMB” axis33,34.
In the PPI network, DPP4, CYP3A4, EP300 MGAM and NR1H4 showed higher degrees and were identified as essential genes of intestinal microbiota influencing the occurrence of depression through the brain–gut axis. The primary mechanism of action of DPP4 is the selective cleavage of cytokines and glucagon-like peptide-135. Previous studies have found that DPP4 inhibitors can improve cognitive function and mitochondrial function in the brain36. Clinical studies have also confirmed the hypothesis that low plasma DPP4 activity is a characteristic marker of major depression and that changes in DPP4 enzyme activity play a role in the pathophysiology of major depression37. CYP3A4 is a metabolic enzyme widely found in human tissues and organs, is involved in approximately 50% of drug metabolism and is an essential factor affecting drug metabolism and efficacy in vivo38. Recent studies have confirmed that the mechanism of action of most antidepressants is related to the regulation of CYP3A439,40,41,42,43. MGAM was associated with an increased risk of Alzheimer’s disease (AD) and major depressive disorder (MDD)44,45. NR1H4 is closely related to cholestasis, which can cause depression46,47. Cytochrome P450 oxidoreductase (POR) is involved in the biosynthesis of endogenous substances, such as bile acids and other steroids, as well as in the oxidative metabolism of xenobiotics, and POR knockdown resulted in the downregulation of NR1H4 (FXR) and the deregulation of bile acid and cholesterol biosynthesis48.
GO and KEGG enrichment analyses of hub genes showed that genes were primarily enriched in carbohydrate digestion and absorption, bile secretion, drug metabolism—cytochrome P450, and other pathways. Cytochrome P450 is one of the essential enzymes in drug oxidation metabolism because it can oxidize and metabolize many exogenous substances, including drugs. Secondary bile acids correspond to the metabolism of these products by the gut microbiome. The two primary BAs are cholic acid and deoxycholic acid, which are often secreted into bile in combination with taurine or glycine49. Activation of farnesoid X receptor (FXR) may play a central role in the onset of depression under pathological conditions50. During prenatal brain development, synapses form between neurons, resulting in neural circuits that support complex cognitive functions. Selective serotonin reuptake inhibitors are commonly used throughout pregnancy to treat depression51. Dysregulation of the serotonergic system has been reported to have a significant role in several neurological disorders, including depression, autism and substance abuse disorders52. Cholestasis can impair social motivation behavior and induce depression-like behavior. Cholestasis can also affect anxiety and pain behaviors in mice53. Pharmacotherapy for neuropsychiatric disorders, such as anxiety and depression, has been characterized by significant interindividual variability in drug response and the development of side effects. Pharmacogenetic research on depression and anxiety has focused on genetic polymorphisms affecting metabolism via cytochrome P450 (CYP)54. In antidepressant drug treatment, most drugs are metabolized via the polymorphic cytochrome P450 enzyme CYP2D655. Activating cAMP-PKA signaling could prevent the behavioral changes and hippocampal synaptic deficits elicited by posttraumatic stress disorder (PTSD)56. Restoring hippocampal cAMP and BDNF levels could be an antidepressant treatment57. Decreases in cAMP and ERK1/2 phosphorylation could reduce the immobility time of chronic restraint stress (CRS) mice in the FST58. Research has found that regulating the HIF-1 signaling pathway can improve LPS-induced depressive behavior59, and CPSP with comorbid anxiety and depression can be improved by increasing cerebral blood flow and inhibiting HIF-1α/NLRP3 inflammatory signaling60.
According to the network analysis of “intestinal microbiota–metabolites–substrate–target genes”, equol, genistein, quercetin and glycocholic acid were found to be the key metabolites affecting depression. Studies have shown that intestinal microbiota such as Enterococcus casseliflavus and Bacteroides sp. 45 can metabolize the flavonoid quercetin-3-glucoside to produce quercetin61,62. Quercetin acts as an antidepressant by regulating neurotransmitter levels, promoting hippocampal neuron regeneration, reducing inflammation and antioxidant stress, and increasing serotonin levels63,64. Equol, a key metabolite of isoflavone with estrogenic and antioxidant activities65, can decrease body weight, abdominal WAT, and depression-related behaviors66. Experimental studies have found that S-equol can help reduce depression and anxiety in individuals67. Genistein, which is produced by the strains “Hugonella massiliensis” DSM 101782T and “Senegalimassilia faecalis” KGMB04484T68, treats depression by suppressing the expression of miR-221/222 by targeting connexin69.
Molecular docking showed that the key metabolites had good binding activity with the hub genes, and the binding sites were hydrogen bonded to form a stable conformation, indicating that the combination of intestinal microbiota metabolites and depression targets may help in treating depression. Additionally, the molecular dynamics simulation results showed that MGAM-glycocholic acid, NR1H4-equol, NR1H4-genistein and NR1H4-quercetin bind stably.
Drug similarity and toxicity evaluations of equol, genistein, quercetin, and other metabolites revealed that they have antidepressant effects. Genistein can be found in Pueraria lobata, Tempeh tempeh, and Cistanche deserticola.
Conclusions
In this study, we developed a comprehensive strategy to analyze the metabolites of the intestinal microbiota and the target genes of the intestinal microbiota affecting depression through systems biology. We explored the potential targets and inhibitors of the intestinal microbiota in treating depression. We found that intestinal microbiota metabolites such as quercetin, equol, and glycocholic acid can affect the course of depression by acting on targets such as MGAM and NR1H4. The mechanisms of action are related to carbohydrate digestion and absorption, bile secretion, drug metabolism-cytochrome P450, and other pathways. The mechanism of action has multitarget and multipathway results. Subsequently, we further verified these results by molecular docking and molecular dynamics simulations. Finally, we also evaluated the critical metabolites' drug similarity and toxicity characteristics to further confirm their potential drug possibilities. However, accumulating microbiome information has certain limitations, and we plan to conduct further preclinical or clinical trials to provide more references for its clinical application and development.
Data availability
The depression data supporting this study's findings are available in the GEO database (http://www.ncbi.nlm.nih.gov/geo), reference numbers [GSE19738, GSE76826, and GSE182193], and the intestinal microbiota data can be found in the GutMgene database (http://bioannotation.cn/gutmgene/home.dhtml).
Abbreviations
- SEA:
-
Similarity ensemble approach
- STP:
-
SwissTargetPrediction
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein‒protein interaction
- MD:
-
Molecular dynamics
- ACPYPE:
-
AnteChamber PYthon Parser interface
- NVT:
-
Canonical ensemble, constant pressure
- NPT:
-
Constant temperature
- MF:
-
Molecular functions
- BP:
-
Biological processes
- CC:
-
Cellular components
- DPP4:
-
Dipeptidyl peptidase 4
- CYP3A4:
-
Cytochrome P450 3A4
- EP300:
-
Histone acetyltransferase p300
- MGAM:
-
Maltase-glucoamylase
- NR1H4:
-
Bile acid receptor
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- FXR:
-
Farnesoid X receptor
References
Dean, J. & Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatry 27, 101–111 (2017).
Ng, M. et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 384(9945), 766–781 (2014).
Moreno-Agostino, D. et al. Global trends in the prevalence and incidence of depression: A systematic review and meta-analysis. J. Affect. Disord. 281, 235–243 (2021).
Plana-Ripoll, O. et al. A comprehensive analysis of mortality-related health metrics associated with mental disorders: A nationwide, register-based cohort study. Lancet 394(10211), 1827–1835 (2019).
Tran, B. X. et al. Global mapping of interventions to improve quality of life of patients with depression during 1990–2018. Qual. Life Res. 29(9), 2333–2343 (2020).
Malhi, G. S. & Mann, J. J. Depression. Lancet 392(10161), 2299–2312 (2018).
Trivedi, M. H. et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: Implications for clinical practice. Am. J. Psychiatry 163(1), 28–40 (2006).
Li, Z., Zhu, H., Zhang, L. & Qin, C. The intestinal microbiome and Alzheimer’s disease: A review. Anim. Models Exp. Med. 1(3), 180–188 (2018).
Choi, H. H. & Cho, Y. S. Fecal microbiota transplantation: Current applications, effectiveness, and future perspectives. Clin. Endosc. 49(3), 257–265 (2016).
Wang, Y. & Kasper, L. H. The role of microbiome in central nervous system disorders. Brain Behav. Immun. 38, 1–12 (2014).
Farzi, A., Hassan, A. M., Zenz, G. & Holzer, P. Diabesity and mood disorders: Multiple links through the microbiota-gut-brain axis. Mol. Asp. Med. 66, 80–93 (2019).
Aizawa, E. et al. Possible association of Bifidobacterium and Lactobacillus in the gut microbiota of patients with major depressive disorder. J. Affect. Disord. 202, 254–257 (2016).
Luna, R. A. & Foster, J. A. Gut brain axis: Diet microbiota interactions and implications for modulation of anxiety and depression. Curr. Opin. Biotechnol. 32, 35–41 (2015).
Jiang, H. et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 48, 186–194 (2015).
Mayer, E. A., Knight, R., Mazmanian, S. K., Cryan, J. F. & Tillisch, K. Gut microbes and the brain: Paradigm shift in neuroscience. J. Neurosci. 34(46), 15490–15496 (2014).
Cruz-Pereira, J. S. et al. Depression’s unholy trinity: Dysregulated stress, immunity, and the microbiome. Annu. Rev. Psychol. 71, 49–78 (2020).
Kelly, J. R. et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 82, 109–118 (2016).
Cheng, L. et al. gutMGene: A comprehensive database for target genes of gut microbes and microbial metabolites. Nucleic Acids Res. 50(D1), D795–D800 (2022).
Keiser, M. J. et al. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25(2), 197–206 (2007).
Gfeller, D., Michielin, O. & Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 29(23), 3073–3079 (2013).
Davis, S. & Meltzer, P. S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23(14), 1846–1847 (2007).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis 1 (2016).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45(D1), D353–D361 (2017).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Otasek, D., Morris, J. H., Boucas, J., Pico, A. R. & Demchak, B. Cytoscape automation: Empowering workflow-based network analysis. Genome Biol. 20(1), 185 (2019).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31(2), 455–461 (2010).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589 (2021).
Varadi, M. et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50(D1), D439–D444 (2022).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4(3), 435–447 (2008).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78(8), 1950–1958 (2010).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Dong, J. et al. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 10(1), 29 (2018).
Hills, R. J. et al. Gut microbiome: Profound implications for diet and disease. Nutrients 11(7), 1613 (2019).
Asadi, A. et al. Obesity and gut-microbiota-brain axis: A narrative review. J. Clin. Lab. Anal. 36(5), e24420 (2022).
Taylor, S. I., Yazdi, Z. S. & Beitelshees, A. L. Pharmacological treatment of hyperglycemia in type 2 diabetes. J. Clin. Invest. https://doi.org/10.1172/JCI142243 (2021).
Pipatpiboon, N., Pintana, H., Pratchayasakul, W., Chattipakorn, N. & Chattipakorn, S. C. DPP4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur. J. Neurosci. 37(5), 839–849 (2013).
Maes, M. et al. Alterations in plasma dipeptidyl peptidase IV enzyme activity in depression and schizophrenia: Effects of antidepressants and antipsychotic drugs. Acta Psychiatr. Scand. 93(1), 1–8 (1996).
Liu, Y. T., Hao, H. P., Liu, C. X., Wang, G. J. & Xie, H. G. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab. Rev. 39(4), 699–721 (2007).
Kot, M., Haduch, A., Papp, M. & Daniel, W. A. The effect of chronic treatment with lurasidone on rat liver cytochrome P450 expression and activity in the chronic mild stress model of depression. Drug Metab. Dispos. 45(12), 1336–1344 (2017).
Andrade, C. Ketamine for depression, 5: Potential pharmacokinetic and pharmacodynamic drug interactions. J. Clin. Psychiatry 78(7), e858–e861 (2017).
Chen, L. et al. Evaluation of cytochrome P450 (CYP) 3A4-based interactions of levomilnacipran with ketoconazole, carbamazepine or alprazolam in healthy subjects. Clin. Drug Investig. 35(10), 601–612 (2015).
Ghosh, C. et al. Sertraline-induced potentiation of the CYP3A4-dependent neurotoxicity of carbamazepine: An in vitro study. Epilepsia 56(3), 439–449 (2015).
Menus, A. et al. Association of clozapine-related metabolic disturbances with CYP3A4 expression in patients with schizophrenia. Sci. Rep. 10(1), 21283 (2020).
Meng, L., Wang, Z., Ji, H. F. & Shen, L. Causal association evaluation of diabetes with Alzheimer’s disease and genetic analysis of antidiabetic drugs against Alzheimer’s disease. Cell Biosci. 12(1), 28 (2022).
Huang, S. S. et al. Investigating genetic variants for treatment response to selective serotonin reuptake inhibitors in syndromal factors and side effects among patients with depression in Taiwanese Han population. Pharmacogenom. J. 23(2–3), 50–59 (2023).
Khakpai, F., Rezaei, N., Issazadeh, Y. & Zarrindast, M. R. Modulation of social and depression behaviors in cholestatic and drug-dependent mice: Possible role of opioid receptors. J. Diabetes Metab. Disord. 22(1), 275–285 (2023).
Deng, F. et al. Multi-omics reveals 2-bromo-4,6-dinitroaniline (BDNA)-induced hepatotoxicity and the role of the gut-liver axis in rats. J. Hazard. Mater. 457, 131760 (2023).
Heintze, T. et al. Effects of diminished NADPH: Cytochrome P450 reductase in human hepatocytes on lipid and bile acid homeostasis. Front. Pharmacol. 12, 769703 (2021).
Chiang, J. Y. Bile acid metabolism and signaling. Compr. Physiol. 3(3), 1191–1212 (2013).
Trzeciak, P. & Herbet, M. Role of the intestinal microbiome, intestinal barrier and psychobiotics in depression. Nutrients 13(3), 927 (2021).
Tate, K., Kirk, B., Tseng, A., Ulffers, A. & Litwa, K. Effects of the selective serotonin reuptake inhibitor fluoxetine on developing neural circuits in a model of the human fetal cortex. Int. J. Mol. Sci. 22(19), 10457 (2021).
Chaji, D., Venkatesh, V. S., Shirao, T., Day, D. J. & Ellenbroek, B. A. Genetic knockout of the serotonin reuptake transporter results in the reduction of dendritic spines in in vitro rat cortical neuronal culture. J. Mol. Neurosci. 71(11), 2210–2218 (2021).
Khakpai, F., Issazadeh, Y., Rezaei, N. & Zarrindast, M. R. Enhanced anxiolytic and analgesic effectiveness or a better safety profile of morphine and tramadol combination in cholestatic and addicted mice. NeuroReport 33(16), 681–689 (2022).
Radosavljevic, M., Svob, S. D., Jancic, J. & Samardzic, J. The role of pharmacogenetics in personalizing the antidepressant and anxiolytic therapy. Genes (Basel) 14(5), 1095 (2023).
Kirchheiner, J. & Seeringer, A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim. Biophys. Acta 1770(3), 489–494 (2007).
Ji, M. et al. Curculigoside rescues hippocampal synaptic deficits elicited by PTSD through activating cAMP-PKA signaling. Phytother. Res. 37(2), 759–773 (2023).
Zaki, E. S., Sayed, R. H., Saad, M. A. & El-Yamany, M. F. Roflumilast ameliorates ovariectomy-induced depressive-like behavior in rats via activation of AMPK/mTOR/ULK1-dependent autophagy pathway. Life Sci. 327, 121806 (2023).
Oh, D. R. et al. Antidepressant effects of p-coumaric acid isolated from Vaccinium bracteatum leaves extract on chronic restraint stress mouse model and antagonism of serotonin 6 receptor in vitro. Phytomedicine 116, 154871 (2023).
Li, A. et al. Roxadustat (FG-4592) abated lipopolysaccharides-induced depressive-like symptoms via PI3K signaling. Front. Mol. Neurosci. 16, 1048985 (2023).
Shi, Z. M. et al. Stellate ganglion block ameliorated central post-stroke pain with comorbid anxiety and depression through inhibiting HIF-1alpha/NLRP3 signaling following thalamic hemorrhagic stroke. J. Neuroinflamm. 20(1), 82 (2023).
Chen, S. et al. Antidepressant potential of quercetin and its glycoside derivatives: A comprehensive review and update. Front. Pharmacol. 13, 865376 (2022).
Shen, Z. et al. Avicularin relieves depressive-like behaviors induced by chronic unpredictable mild stress in mice. Med. Sci. Monit. 25, 2777–2784 (2019).
Bax, E. N., Cochran, K. E., Mao, J., Wiedmeyer, C. E. & Rosenfeld, C. S. Opposing effects of S-equol supplementation on metabolic and behavioral parameters in mice fed a high-fat diet. Nutr. Res. 64, 39–48 (2019).
Schneider, H., Simmering, R., Hartmann, L., Pforte, H. & Blaut, M. Degradation of quercetin-3-glucoside in gnotobiotic rats associated with human intestinal bacteria. J. Appl. Microbiol. 89(6), 1027–1037 (2000).
Yang, J. et al. Identification of rutin deglycosylated metabolites produced by human intestinal bacteria using UPLC-Q-TOF/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 898, 95–100 (2012).
Mayo, B., Vazquez, L. & Florez, A. B. Equol: A bacterial metabolite from the daidzein isoflavone and its presumed beneficial health effects. Nutrients 11(9), 2231 (2019).
Blake, C., Fabick, K. M., Setchell, K. D., Lund, T. D. & Lephart, E. D. Neuromodulation by soy diets or equol: Anti-depressive & anti-obesity-like influences, age- & hormone-dependent effects. BMC Neurosci. 12, 28 (2011).
Soukup, S. T. et al. Metabolism of daidzein and genistein by gut bacteria of the class Coriobacteriia. Foods 10(11), 2741 (2021).
Shen, F. et al. Genistein improves the major depression through suppressing the expression of miR-221/222 by targeting connexin 43. Psychiatry Investig. 15(10), 919–925 (2018).
Acknowledgements
This research was funded by the Famous Old Traditional Chinese Medicine Experts Inheritance Studio Construction Project (202275). We would also like to thank Bullet Edits (www.bulletedits.cn/Email: info abulletedits) and Springer Nature Author Services (https://secure.authorservices.springernature.com) for help with postediting proofreading. FT would like to thank his friends and his girlfriend FG; even though youth will eventually end, we at least fought for the dream of scientific research in our hearts.
Funding
This work was supported by funding from the Famous Old Traditional Chinese Medicine Experts Inheritance Studio Construction Project (202275).
Author information
Authors and Affiliations
Contributions
Conceptualization, J.W.; methodology, investigation, F.T., Z.L., and G.F.; data curation, F.T., Z.L. and F.G.; writing-original draft preparation, F.G., J.L., X.T., J.L., X.W. and H.G.; validation, X.T.; writing-review and editing, J.W.; investigation, J.W.; formal analysis, J.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teng, F., Lu, Z., Gao, F. et al. Systems biology approaches to identify potential targets and inhibitors of the intestinal microbiota to treat depression. Sci Rep 13, 11225 (2023). https://doi.org/10.1038/s41598-023-38444-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38444-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
