
Abstract
Proximal spinal muscular atrophy (SMA) is a leading genetic cause for infant death in the world and results from the selective loss of motor neurons in the spinal cord. SMA is a consequence of low levels of SMN protein and small molecules that can increase SMN expression are of considerable interest as potential therapeutics. Previous studies have shown that both 4-phenylbutyrate (4PBA) and trichostatin A (TSA) increase SMN expression in dermal fibroblasts derived from SMA patients. AR42 is a 4PBA-tethered TSA derivative that is a very potent histone deacetylase inhibitor. SMA patient fibroblasts were treated with either AR42, AR19 (a related analogue), 4PBA, TSA or vehicle for 5 days and then immunostained for SMN localization. AR42 as well as 4PBA and TSA increased the number of SMN-positive nuclear gems in a dose-dependent manner while AR19 did not show marked changes in gem numbers. While gem number was increased in AR42-treated SMA fibroblasts, there were no significant changes in FL-SMN mRNA or SMN protein. The neuroprotective effect of this compound was then assessed in SMNΔ7 SMA (SMN2+/+;SMNΔ7+/+;mSmn−/−) mice. Oral administration of AR42 prior to disease onset increased the average lifespan of SMNΔ7 SMA mice by ~ 27% (20.1 ± 1.6 days for AR42-treated mice vs. 15.8 ± 0.4 days for vehicle-treated mice). AR42 treatment also improved motor function in these mice. AR42 treatment inhibited histone deacetylase (HDAC) activity in treated spinal cord although it did not affect SMN protein expression in these mice. AKT and GSK3β phosphorylation were both significantly increased in SMNΔ7 SMA mouse spinal cords. In conclusion, presymptomatic administration of the HDAC inhibitor AR42 ameliorates the disease phenotype in SMNΔ7 SMA mice in a SMN-independent manner possibly by increasing AKT neuroprotective signaling.
Similar content being viewed by others


Introduction
Proximal spinal muscular atrophy (SMA) is an autosomal recessive, early-onset neurodegenerative disease characterized by selective loss of α motor neurons in the anterior horn of the spinal cord1,2.This loss of α motor neurons leads to atrophy of limb and trunk muscles. SMA results from the loss or mutation of the SMN1 (survival motor neuron) gene3. In humans, SMN1 is duplicated and the two genes, SMN1 and SMN2, functionally differ by a single cytosine to thymine transition within exon 74,5. While translation of transcripts from SMN1 produce full-length SMN (FL-SMN) protein, most of the transcripts from SMN2 lack exon 7 (SMNΔ7) and produce a truncated, unstable SMNΔ7 protein. About 10–20% of SMN2 transcripts are alternatively spliced to include exon 7 which then results in the production of FL-SMN protein4,5. Although SMA results from the loss of a single gene, it has a wide clinical spectrum of disease severity1. The severity of SMA depends on the copy number of SMN2 and the consequent levels of the SMN protein (reviewed in6). SMN2 is, therefore, a genetic modifier of disease severity in SMA.
Unlike humans, mice carry only one SMN gene (mSmn or Smn1) which is orthologous to SMN17,8. Loss of mSmn results in embryonic lethality in the mouse suggesting that the mSmn gene product is essential for cell function and survival9. Transgenic insertion of SMN2 into mSmn null mice rescues the embryonic lethality phenotype10,11. However, mice with low copy numbers (i.e. 1–2) of SMN2 develop severe SMA and die at 6–8 days10,11,12. Increasing SMN2 copy number results in a milder phenotype in the SMA mice12. When the SMN2 copy number is high, the resultant SMA mice are phenotypically indistinguishable from non-SMA littermates11. Introducing SMN lacking exon 7 (SMNΔ7)—the predominant mRNA produced by SMN2—into a severe SMA mouse genetic background partially ameliorates the SMA phenotype and these mice die at 14–15 days13. This suggests that SMNΔ7 might be partially functional but not enough to completely rescue SMA-like motor neuron degeneration. Collectively, these studies demonstrate that increasing SMN2 expression ameliorates the SMA phenotype in mouse model as it does in humans, making SMN2 a desirable target for the development of therapeutic agents. Accordingly, there are currently three different FDA-approved SMN-inducing therapies for SMA which either modulate SMN2 splicing or aim to replace SMN1: nusinersen (Spinraza)14,15, risdiplam (Evrysdi)16 and onasemnogene abeparvovec (Zolgensma)17.
SMN2 expression can be increased by inhibiting histone deacetylase (HDAC) activity which remodels the chromatin containing this gene18. Many different HDAC inhibitors of different classes increase SMN2 expression in cultured cells. These SMN2-inducing HDAC inhibitors include butyric acid (BA;19), 4-phenylbutyric acid (4PBA;20,21), valproic acid (VPA;22,23,24,25), trichostatin A (TSA;26), suberoylanilide hydroxamic acid (SAHA;27,28,29), M34427,29, MS-27527, m-carboxycinnamic acid bis-hydroxamide (CBHA;27), LBH58930 and JNJ-2648158531. It is thought that HDAC inhibitors increase SMN2 promoter activity as a result of increased acetylation of histones32. Accordingly, using isoform-specific short hairpin RNAs, knockdown of all HDAC isoforms, except for HDAC5, increases SMN2 promoter activity33.
TSA improves the survival and phenotype of SMNΔ7 SMA mice following repeated intraperitoneal injections26,34,35. We have previously demonstrated that short-chain fatty acid derivative 4PBA significantly improves the survival of SMNΔ7 SMA mice when administered orally36. AR42 is a novel, orally bioavailable HDAC inhibitor that is 4PBA-tethered derivative of TSA37,38. As TSA and 4PBA both increase SMN2 expression in SMA cells and exert protective effects on mouse models for SMA, we examined the effect of AR42 on the expression of SMN2 in human SMA fibroblasts as well on the survival and phenotype of SMNΔ7 SMA mice.
Results
Effects of AR42 and AR19 on SMN expression in cultured cells
SMN localizes to the cytosol and discreet nuclear bodies called gems39. Subnuclear gems can be detected in cell using immunohistochemistry. The number of nuclei with gems and the number of gems per cell is reduced in SMA fibroblasts40. As TSA and 4PBA increase the number of gems in SMA fibroblasts20,21,26, we examined the effect of AR42 and its less potent analogue AR19 (Fig. 1) on SMN localization to gems in GM03813 type II SMA fibroblasts. AR42 (Fig. 2A) along with TSA (Fig. 2C) and 4PBA (Fig. 2D) markedly increased the number of SMN-positive gems in GM03813 fibroblasts in a dose-dependent manner relative to DMSO-treated GM03813 fibroblasts (Fig. 2E). AR19, on the other hand, had a marginal effect on subnuclear localization of SMN in these cells (Fig. 2B). Treatment of GM03813 cells with 1 μM TSA for 5 days resulted in marked toxicity (as assessed by SMN immunohistochemistry; data not shown); the other compounds tested did not show any toxicity at this dose. AR42, TSA and 4PBA increased the number of gems (Fig. 2G), the proportion of SMA cells containing gems (Fig. 2H) as well as the proportion of SMA cells containing multiple gems (Fig. 2I) in a dose-dependent manner. In fact, the number of gems approaches the number seen in carrier fibroblasts (GM03814; Fig. 2F) at the highest doses tested.

Chemical structures of the compounds tested.

Effect of AR42 on gem numbers in type II SMA fibroblasts. GM03813 type II SMA fibroblasts were treated with differing doses of AR42, AR19, 4PBA or TSA for 5 days (n = 3/dose/drug). The number of SMN-positive gems within 100 randomly selected nuclei was counted. Representative images are shown for GM03813 SMA fibroblasts treated with 1 μM AR42 (A), 1 μM AR19 (B), 1 μM 4PBA (C), 100 nM TSA (D) or DMSO (E) as well as for GM03814 carrier fibroblasts (F). The scale bars (yellow lines) represent 10 μm. The gem count analysis was expressed as (G) the number of gems per 100 nuclei, (H) the proportion of cells containing gems and (I) the proportion of cells containing multiple gems. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferroni post hoc test) difference between drug- and vehicle-treated GM03813 SMA cells.
To further investigate how SMN2 expression is regulated by AR42, we examined the effect of AR42 on SMN2 promoter activity using NSC34 motor neuron-like cells stably transfected with a β-lactamase reporter gene driven by a 3.4-kb fragment of the SMN2 promoter (clone 11)41. In response to exposure to 100 nM AR42, SMN2 promoter activity was increased by 54%; TSA also induced SMN2 promoter activity by 74% at this dose (Fig. 3A). Neither 4PBA nor AR19 affected SMN2 promoter activity at this dose. AR42 (Fig. 3B) increased SMN2 promoter activity in NSC34 cells in a dose-dependent manner with an estimated EC50 of 24 nM (95% confidence interval (CI) = 11.98–83.62 nM; R2 = 0.9372).

Effects of AR42 on SMN expression in cultured cells. (A) SMN2 promoter activity, expressed as the fluorescence ratio (F460:F530) relative to that for DMSO, in clone 11 NSC34 cells treated with 100 nM AR42, AR19, 4PBA or TSA for 19 h. (B) SMN2 promoter dose–response curve for AR42 treatment. (C) Changes in SMN-FL mRNA levels in GM03813 SMA fibroblasts treated with different doses of AR42, AR19, 4PBA and TSA for 5 days. The SMN-FL mRNA level in GM03814 healthy carrier fibroblasts was also measured. The SMN-FL mRNA levels were normalized to those of DMSO-treated GM03813 SMA fibroblasts (dashed line). (D) Changes in SMNΔ7 mRNA levels in GM03813 SMA fibroblasts treated with different doses of AR42, AR19, 4PBA and TSA for 5 days as well as in GM03814 healthy carrier fibroblasts. The SMNΔ7 mRNA levels were normalized to those of DMSO-treated GM03813 SMA fibroblasts (dashed line). (E) Changes in SMN protein levels in GM03813 SMA fibroblasts treated with different doses of AR42, AR19, 4PBA and TSA for 5 days as well as in GM03814 healthy carrier fibroblasts. The SMN protein levels are relative to those observed for β-actin. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferroni post hoc test) difference between drug- and vehicle-treated GM03813 SMA cells.
The effects of these compounds on the expression of SMN2 were examined in GM03813 type II SMA fibroblasts. Cells were treated with different doses of AR42, AR19, 4PBA and TSA for 5 days. Surprisingly, AR42 had no effect on either FL-SMN (Fig. 3C) or SMNΔ7 (Fig. 3D) mRNA levels even though these transcripts were induced in SMA fibroblasts treated with 4PBA or TSA. Treatment of GM03813 cells with AR19 did not increase expression of FL-SMN or SMNΔ7 mRNAs. Analysis of SMN protein levels in treated GM03813 type II SMA fibroblasts showed the same patterns as those for FL-SMN mRNA changes in response to drug treatment (Fig. 3E). Thus, AR42 does not upregulate SMN2 mRNA or protein expression but increases the subnuclear localization of SMN in SMA fibroblasts.
Administration of AR42 before onset of motor neuron loss improved the survival of and delayed disease end-stage in SMNΔ7 SMA mice
AR42 is orally bioavailable and can cross the blood–brain barrier42. To determine if AR42 treatment has a protective effect on the SMA phenotype, SMNΔ7 SMA mice were treated with AR42 (5 mg/kg/day; n = 15) or vehicle (n = 15) beginning at PND04. As a control, SMNΔ7 SMA mice were treated with the less potent analogue AR19 (5 mg/kg/day; n = 15). In a pilot dose determination trial, we found that neonatal mice treated with higher doses of AR42 (≥ 10 mg/kg/day) showed off-target toxicity like significant loss in body mass after dosing and reduced locomotion (data not shown). Bjornsson et al.43 also observed off-target effects in a mouse model for Kabuki syndrome that was treated with higher doses of AR42. As shown in Fig. 4A, oral administration of AR42 improved the survival of SMNΔ7 SMA mice by 27% (20.1 ± 1.6 days for AR42-treated mice as compared to 15.8 ± 0.4 days for vehicle-treated mice; p = 0.011, χ2 = 6.453). Treatment of SMNΔ7 SMA mice with AR19, however, did not significantly affect survival (14.5 ± 0.6 days for AR19-treated mice as compared to 15.8 ± 0.4 days for vehicle-treated mice; p = 0.132, χ2 = 2.264).

Effects of AR42 and AR19 on the lifespan and disease end-stage in SMNΔ7 SMA mice. (A, B) SMNΔ7 SMA mice were treated daily with AR42 (5 mg/kg/d; red dashed line) or AR19 (5 mg/kg/d; blue dotted line) beginning at PND04 and monitored for changes in lifespan (A) and disease end-stage, which is defined as the onset of body mass loss (B). AR42 increased survival by 27% (A; p = 0.011; Kaplan–Meier analysis with Mantel–Cox log rank post hoc test) and delayed the onset of body mass loss by 31% (B; p = 0.002; Kaplan–Meier analysis with Mantel–Cox log rank post hoc test). AR19, on the other hand did not affect lifespan or disease end-stage in SMNΔ7 SMA mice. (C, D) SMNΔ7 SMA mice were treated with AR42 (5 mg/kg/d) daily beginning at either PND04 (red dashed line) or PND09 (blue dotted line). Post-symptomatic—i.e. beginning at PND09, or after onset of motor neuron loss—treatment with AR42 did not positively affect survival (C) or onset of body mass loss (D) in these mice. In fact, post-symptomatic AR42 treatment resulted in earlier death of SMNΔ7 mice than vehicle (black line).
In the SMNΔ7 SMA mouse model, a phenotypic indicator of disease end-stage is the onset of loss of body mass13,44. The onset of body mass loss was delayed by 31% (Fig. 4B; 14.8 ± 1.0 days for AR42-treated mice as compared to 11.6 ± 0.3 days for vehicle-treated mice; p = 0.002, χ2 = 9.616) in SMNΔ7 SMA mice treated with AR42. Treatment of SMNΔ7 SMA mice with AR19, however, did not affect the onset of body mass loss in these mice (Fig. 4B; 11.3 ± 0.2 days for AR19-treated mice as compared to 11.6 ± 0.3 days for vehicle-treated mice; p = 0.239, χ2 = 1.389).
Therapeutic efficacy in SMA mouse models depends on the timing of treatment. For example, the DcpS inhibitor D15684445 and the butyrate prodrug VX56336 ameliorate the degenerative phenotype in SMNΔ7 SMA mice only if treatment begins before the onset of motor neuron loss. We, therefore, treated SMNΔ7 SMA mice with AR42 (n = 16) starting at postnatal day 9 (PND09) and did not observe an improvement in their survival (Fig. 4C); in fact, these mice died on average before vehicle-treated SMNΔ7 SMA mice (13.7 ± 0.7 days for AR42-treated mice at PND09 as compared to 15.8 ± 0.4 days for vehicle-treated mice; p = 0.043, χ2 = 4.103). Post-symptomatic treatment of SMNΔ7 SMA mice also negatively impacted disease end-stage (Fig. 4D; 10.5 ± 0.2 days for AR42-treated mice at PND09 as compared to 11.6 ± 0.3 days for vehicle-treated mice; p = 0.002, χ2 = 9.714). Taken together, these observations show therapeutic efficacy of AR42 in SMNΔ7 SMA mice when treatment with this drug begins prior to disease onset.
Effect of AR42 on the phenotype of SMNΔ7 SMA mice
As an indicator of growth rate in developing mice, we monitored the changes in body mass over time in SMNΔ7 SMA and non-SMA littermates treated with AR42 or vehicle. Consistent with previous findings using this colony of mice36,44,45,46,47,48, SMNΔ7 SMA mice had lower body masses than their age-matched non-SMA littermates regardless of treatment (Fig. 5A). However, the body masses of AR42-treated SMNΔ7 SMA were greater than age-matched vehicle SMNΔ7 SMA mice (Fig. 5A) especially at the late-stage of disease (i.e. PND15–17) but these differences were not statistically significant. When comparing the body masses between PND14 and PND04, AR42-treated SMNΔ7 SMA mice tended to show a small increase in the change in body mass when compared to vehicle-treated SMNΔ7 SMA mice but the difference was not statistically significant (Fig. 5B). We also observed a slight reduction in body mass of nonSMA mice treated with AR42 but the change was not statistically significant (p = 0.325).

Effect of AR42 on the body mass of SMNΔ7 SMA mice over time. (A) Body mass curves of SMNΔ7 SMA mice (solid shapes) or non-SMA (either carrier or normal; open shapes) littermates treated daily with either AR42 (5 mg/kg/d; circles) or vehicle (triangles). (B) Changes in body mass between PND14 and PND04 in SMNΔ7 SMA mice treated with AR42 or vehicle along with non-SMA littermates. The mean body masses at PND14 were expressed relative to those at PND04. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferroni post hoc test) difference when comparing SMNΔ7 SMA and non-SMA mice for each treatment group.
AR42 treatment before the onset of motor neuron loss improved the motor phenotype observed in SMNΔ7 SMA mice (Supplementary Movie). The effect of AR42 on motor impairment observed in the SMNΔ7 SMA mice was examined in further detail. SMNΔ7 SMA mice were treated with AR42 (5 mg/kg/d) or vehicle beginning at PND04. The controls for this experiment were unaffected (both carrier and normal pups) littermates; the motor phenotypes between carrier and normal mice were nearly identical and, hence, were combined in this experiment44. Since sex does not affect the motor phenotype in SMNΔ7 SMA mice44, both male and female pups were used. The success rate for the righting reflex was higher in SMNΔ7 SMA mice treated with AR42 than vehicle-treated SMNΔ7 SMA mice at PND07 and PND11 (Fig. 6A) but the righting reflex latencies were not altered by AR42 treatment (Fig. 6B). Vectorial movement is defined as locomotion along a single vector for a distance greater than the body length44. AR42 treatment markedly reduced vectorial movement latency in SMNΔ7 SMA mice relative to vehicle (Fig. 6C); in fact, the vectorial movement latency for AR42-treated SMNΔ7 SMA mice was similar to control littermates. The duration of vectorial movement tended to be higher in AR42-treated SMNΔ7 SMA mice than in vehicle-treated SMNΔ7 SMA mice at PND07 and PND11 but the difference was not statistically significant (Fig. 6D). AR42 treatment of SMNΔ7 SMA mice tended to increase spontaneous locomotor activity—the number of grids crossed in 60 s—relative to vehicle-treated SMNΔ7 SMA mice at PND11 and PND14 (Fig. 6E). AR42 treatment of SMNΔ7 SMA mice increased the pivoting response, i.e. the number of 90° pivots made in 60 s, relative to vehicle-treated SMNΔ7 SMA mice at all ages but the differences were not statistically significant (Fig. 6F). As we have observed in previous preclinical drug trials with the SMNΔ7 SMA mice36,48, there was a high degree of variability within the motor phenotype of each treatment group—especially at PND14—which could indicate strong and weak responders to AR42.

Effect of AR42 on the motor phenotype of SMNΔ7 SMA mice. SMNΔ7 SMA mice were treated either with AR42 (5 mg/kg/d) or vehicle beginning at PND04 and monitored at PND07, PND11 and PND14 for changes in motor behavior. Age-matched, non-SMA littermates were also assayed at each time point. (A) SMNΔ7 SMA mice showed impaired surface righting responses at PND07 and PND11 relative to age-matched control littermates. A greater proportion of AR42-treated SMNΔ7 SMA mice exhibited a successful surface righting response than vehicle-treated SMNΔ7 SMA mice. (B) SMNΔ7 SMA mice treated with either AR42 or vehicle had a higher righting reflex latency than control mice at both ages. The righting reflex latency time has an arbitrary cutoff of 61 s. (C) The latency of vectorial movement, or locomotion along a single vector for a distance greater than the body length, was higher in vehicle-treated SMNΔ7 SMA mice than control mice at PND07 and PND11. AR42 treatment markedly reduced vectorial movement latency in SMNΔ7 SMA mice when compared against vehicle-treated SMNΔ7 SMA mice. (D) Vectorial movement duration was reduced in vehicle-treated SMNΔ7 SMA mice when compared against age-matched control mice. AR42 tended to increase vectorial movement duration in SMNΔ7 SMA mice at PND07 and PND11. (E) SMNΔ7 SMA mice crossed fewer grids within 60 s, a measure of spontaneous locomotor activity, than control mice. AR42 treatment of SMNΔ7 SMA mice increased spontaneous locomotor activity relative to vehicle-treated SMNΔ7 SMA mice at PND11 and PND14 but the differences were not statistically significant. (F) The number of 90° pivots made in 60 s was reduced in vehicle-treated SMNΔ7 SMA mice when compared against age-matched control littermates. AR42 treatment of SMNΔ7 SMA mice increased the pivoting response relative to vehicle-treated SMNΔ7 SMA mice at all ages but the differences were not statistically significant. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to control mice and the number sign (#) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated SMNΔ7 SMA mice.
Effect of AR42 on motor neuron loss in the mouse lumbar spinal cord of SMNΔ7 SMA mice
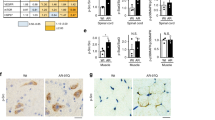
Significant motor neuron loss in the lumbar spinal cord was observed in the SMNΔ7 SMA mice at PND1113. Treatment of SMNΔ7 SMA mice with 4-PBA significantly rescued motor neuron loss at PND1136. Given the improvement of motor function in SMNΔ7 SMA mice treated with AR42, we examined the effect of AR42 on motor neuron loss in the spinal cords of SMNΔ7 SMA mice. We observed a 29.8% reduction in the number of motor neurons in the lumbar spinal cord of SMNΔ7 SMA mice at PND11 (Fig. 7), similar to the reduction in motor neuron counts we observed in previous studies36,45. AR42 treatment leads to an increase in motor neuron counts in SMNΔ7 SMA mouse lumbar spinal cords to counts similar to those observed in non-SMA mice at PND11.

Effect of AR42 on motor neuron loss in the lumbar spinal cords of SMNΔ7 SMA mice. SMNΔ7 SMA mice (n = 3/group) were treated with either AR42 or vehicle from PND04 until PND11. The number of Cresyl violet-positive, ventral motor neurons were counted in every 12th section of lumbar spinal cords of AR42-treated SMNΔ7 SMA mice (circles), vehicle-treated SMNΔ7 SMA mice (squares) or age-matched non-SMA littermates (triangles). AR42 increased lumbar spinal motor neuron counts in SMNΔ7 SMA mice to levels similar to those in nonSMA littermates. The data are presented as mean ± standard error with the average motor neuron counts/section shown for each individual mouse. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated SMNΔ7 SMA mice.
Effect of AR42 on SMN2 expression in the spinal cord of SMNΔ7 SMA mice
We wanted to determine the effect of AR42 on SMN expression in vivo in the spinal cords of SMNΔ7 SMA mice. These mice were treated with AR42 (5 mg/kg/d) or vehicle (n = 3/group) beginning at PND04, and treatment continued for 5 days (until PND08). Treatment of SMNΔ7 SMA mice with AR42 did not increase the levels of FL-SMN or SMNΔ7 mRNAs in the spinal cord (Fig. 8A). As shown in Fig. 8B, AR42 treatment did not significantly affect SMN protein levels in the spinal cord of treated SMNΔ7 SMA mice. SMN ELISA analysis also showed no change in SMN protein levels in response to AR42 treatment in SMNΔ7 SMA mice (Fig. 8C). Spliceosomal and U7 snRNP assembly are well established functions of SMN at the molecular level49 that are impaired in SMA mouse models50,51,52 and AR42 treatment did not affect snRNP assembly in spinal cord extracts from SMNΔ7 SMA mice (Fig. 8D).

Effect of AR42 on SMN protein levels and function in the spinal cords of SMNΔ7 SMA mice. SMNΔ7 SMA mice (n = 3/group) were treated with either AR42 or vehicle for 5 days beginning at PND04. (A) AR42 did not affect FL-SMN or SMNΔ7 mRNA levels in the spinal cords of SMNΔ7 SMA mice. (B) Total SMN protein levels in the spinal cord were not altered by AR42 treatment as shown by this representative immunoblot. (C) Quantification of human SMN protein using ELISA also showed no significant changes between AR42- and vehicle-treated SMNΔ7 SMA mice but SMN protein levels were lower in SMNΔ7 SMA mice when compared to age-matched non-SMA littermates. (D) In vitro assembly of U1 snRNPs was diminished in SMNΔ7 SMA spinal cord samples but AR42 treatment did not affect snRNP assembly. (E) The levels of the SMN-dependent transcripts Chodl, Cdkn1a and Tmem41b were not affected by AR42 treatment in SMNΔ7 SMA spinal cords. (F) AR42 significantly reduced the mRNA levels of the flip, but not the flop, isoform of Gria4 in SMNΔ7 SMA spinal cords. The levels of the synaptogenesis-associated transcripts Z+Agr and C1qb were not affected by AR42. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated carrier mice.
We examined the effect of AR42 on the levels of 3 different SMN-dependent transcripts: chondrolectin (Chodl), cyclin-dependent kinase inhibitor 1A (Cdkn1a) and transmembrane protein 41b (Tmem41b), the murine orthologue to the Drosophila gene stasimon53,54,55. Chodl levels were significantly lower in SMNΔ7 SMA spinal cords at PND08 and there were tendencies for increased Cdkn1a and reduced Tmem41b mRNA levels in these samples as well (Fig. 8E). AR42, however, did not affect the expression of these SMN-dependent transcripts in the SMNΔ7 SMA spinal cord.
In motor neurons, SMN deficiency leads to an early dysregulation of transcripts that are involved in synaptogenesis56. The effects of AR42 on the dysregulation of 4 of these synaptogenesis-associated transcripts—Z+ agrin (Z+Agr), complement component 1 B polypeptide (C1qb) as well as the AMPA-type ionotropic glutamate receptor 4 (Gria4) alternatively spliced flip and flop isoforms—in SMNΔ7 SMA spinal cords. We observed trends towards reduction of Z+Agr mRNA levels and increase of C1qb mRNA levels in SMNΔ7 SMA spinal cords at PND08; however, AR42 had no effect on their expression (Fig. 8F). At PND08, we observed a decrease in Gria4 flip mRNA levels but no change in the levels of the flop isoform. AR42 treatment further reduced Gria4 flip mRNA levels in the SMNΔ7 SMA spinal cord (Fig. 8F). Based on these observations, we conclude that AR42—like 4PBA and the butyrate-based prodrug VX56336—mediates its neuroprotective effects in the spinal cord independent of modifying SMN2 expression and SMN-dependent gene regulation.
Effects of AR42 on HDAC activity in SMNΔ7 SMA mice
As AR42 has very potent HDAC inhibitory activity37,38, spinal cord extracts from SMNΔ7 SMA mice treated with AR42 (5 mg/kg/d) or vehicle for 5 days were assayed for HDAC activity. Fluorimetric HDAC activity was lower in spinal cord samples from SMNΔ7 SMA mice treated with AR42 than those mice treated with vehicle (Fig. 9A). We also observed a marked increase in histone H3 acetylation at K9 in SMNΔ7 SMA spinal cords from AR42-treated mice (Fig. 9B). Interestingly, HDAC activity and histone acetylation were not different in spinal cords from vehicle-treated SMNΔ7 SMA mice and healthy controls.

Effect of AR42 on HDAC activity in the spinal cords of SMNΔ7 SMA mice. SMNΔ7 SMA mice (n = 3/group) were treated with either AR42 or vehicle for 5 days beginning at PND04. As a control, non-SMA mice (n = 3) were treated with vehicle for 5 days beginning at PND04. (A) Fluorimetric HDAC activity in treated spinal cord. Treatment of SMNΔ7 SMA mice with AR42 significantly reduced HDAC enzyme activity in the spinal cord. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated SMNΔ7 SMA mice. (B) Representative immunoblots of acetyl-histone H3 (K9) and total histone H3 levels in treated spinal cords. All band intensities were expressed relative to β-actin band intensities.
Effect of AR42 on Akt signaling in SMNΔ7 SMA mice
Previous work in cancer cells57 identified a link between AR42-mediated HDAC inhibition and Akt signaling. Based on these findings, we measured Akt and GSK3β—a downstream target of Akt—phosphorylation status in SMNΔ7 SMA mice treated with AR42 for 5 days. Phosphorylation of Akt at S473 in SMNΔ7 SMA mice was elevated relative to unaffected control mice, but the differences were not statistically significant (p = 0.064; Fig. 10A, B). Treatment with AR42 did not significantly increase Akt phosphorylation at serine 473 (S473) in SMA spinal cord relative to vehicle treated SMNΔ7 mice. Interestingly, the levels of total Akt protein were diminished in the spinal cords of SMNΔ7 SMA mice relative to control mice and AR42 treatment reduced total AKT protein levels to those observed in control mice although the changes were not statistically significant (Fig. 10C). The Akt (phospho-S473) versus total Akt ratio was significantly higher in the spinal cords of SMNΔ7 SMA mice treated with AR42 than in vehicle-treated SMNΔ7 SMA mice as well as compared against control mice (Fig. 10D).

Effect of AR42 on AKT and GSK3β phosphorylation in the spinal cords of SMNΔ7 SMA mice. SMNΔ7 SMA mice (n = 3/group) were treated with either AR42 or vehicle for 5 days beginning at PND04. As a control, non-SMA mice (n = 3) were treated with vehicle for 5 days beginning at PND04. (A) Representative immunoblots of phosphorylated Akt (S473), total Akt, phosphorylated GSK3β (S9) and total GSK3β levels in treated spinal cords. All band intensities were expressed relative to GAPDH band intensities. (B) There is a trend for increased phosphorylation of Akt at S473 in SMNΔ7 SMA mouse spinal cords that was not affected by AR42 treatment. (C) Total Akt protein levels were significantly increase in SMA spinal cords but AR42 treatment reduced total Akt protein levels to control. (D) The Akt (phospho-S473) versus total Akt ratio was significantly higher in the spinal cords of SMNΔ7 SMA mice treated with AR42. (E) Phosphorylation of GSK3β at S9 was significantly increased in SMNΔ7 SMA mouse spinal cords and treatment with AR42 further elevated GSK3β at S9. (F) The levels of total GSK3β protein were not altered by SMA genotype nor by AR42 treatment. (G) The GSK3β (phospho-S9) versus total GSK3β ratio was significantly higher in the spinal cords of SMNΔ7 SMA mice treated with AR42. The data are presented as mean ± standard error. The asterisk (*) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated carrier mice and the number symbol (#) denotes a statistically significant (p ≤ 0.05; one-way ANOVA with Bonferonni post hoc test) difference when compared to vehicle-treated SMA spinal cords.
SMNΔ7 SMA mouse spinal cords showed elevated phosphorylation of GSK3β at serine 9 (S9) when compared against control mice (Fig. 10A, E). Treatment of SMNΔ7 SMA mice with AR42 further increased GSK3β phosphorylation at S9 in the spinal cord (Fig. 10E). Total GSK3β levels were not affected by SMA status or by AR42 treatment (Fig. 10F). The GSK3β (phospho-S9) versus total GSK3β ratio was significantly higher in the spinal cords of SMNΔ7 SMA mice treated with AR42 than in control mice (Fig. 10G). Taken together, our data demonstrate that AR42 increases Akt and GSK3β phosphorylation in the spinal cords of SMNΔ7 SMA mice.
Discussion
In this study, we demonstrate in vivo efficacy of a novel, orally bioavailable HDAC inhibitor, AR42, in a mouse model for SMA37,38,42. AR42 is a 4PBA-tethered TSA derivative37,38. AR42 functions as pan-inhibitor of class I HDACs, i.e. HDAC1, HDAC2, HDAC3 and HDAC858. Similar to our findings that 4PBA and BA prodrugs ameliorate the survival and phenotype of SMNΔ7 SMA36,59, other HDAC inhibitors including VPA60, TSA26,34,35 and SAHA28 have been found to improve survival in mouse models for SMA. Treatment of SMA mice with JNJ-26481585 does not significantly improve their survival but there is a trend for amelioration of their motor phenotype31. While many of these studies showed increased SMN expression in vivo in response to HDAC inhibitor treatment26,28,35,60, we did not observe any detectable changes in SMN mRNA or protein levels in SMNΔ7 SMA mice treated with AR42. Consistent with our findings, Liu et al.34 also demonstrated that the beneficial effects of TSA treatment in SMA mice are independent of SMN upregulation. Moreover, in a clinical trial, SMA patients treated with VPA do not show increased SMN expression even though VPA treatment results in increased histone acetylation61. In addition to their neuroprotective roles, HDAC inhibitors like SAHA and TSA exert extra-neuronal protective actions in SMA mice like increasing muscle microvasculature62 and reducing levels of atrogene transcripts in affected muscle63.
As observed with AR42, some small molecules which increase SMN2 promoter activity do not necessarily increase FL-SMN transcript levels or SMN protein levels. This disconnect may be a consequence of the SMN2 promoter screening assay itself as the construct used may not fully represent the endogenous promoter activity. Alternatively, AR42 may be acting at the post-transcriptional/translational levels of SMN regulation. Compounds including ML372, LDN-75654 and cuspin-1 upregulate SMN expression post-transcriptionally, when validated in SMA patient-derived cell lines64,65,66. Flunarizine as well as the C5-substituted, 2,4-diaminoquinazolines D156884 and D157495 increase SMN localization to subnuclear gems without markedly increasing SMN expression67,68. D156844 and D157495 significantly improve the survival and motor phenotypes of SMA mice45,69,70. While SMN expression was not significantly altered in the spinal cord of SMA mice treated with these compounds, the subcellular localization of SMN in SMA motor neurons in vivo may have been affected. By extension, AR42 may increase the subcellular localization of SMN in neurons in vivo without increasing SMN expression. It is equally possible that the protective mode of action for AR42 in SMA mice may be independent of SMN2 gene regulation.
Since upregulation of SMN expression is not the primary mode of action for AR42 in SMA mice, we determined that the protective effects of AR42 may be related to differential phosphorylation of Akt and GSK3β. The Akt pathway is neuroprotective in neurons when its activity is increased71. Previous studies have shown that Akt phosphorylation is reduced in the spinal cords of symptomatic SMA mice72,73. In this study, we show that phosphorylation of Akt at S473 is increased in SMNΔ7 SMA mouse spinal cords and AR42 further potentiates that elevation in Akt phosphorylation at S473.
One of the primary targets of Akt activation is GSK3β. Inhibition of GSK3α/β activity with alsterpaullone increase SMN protein levels in vitro by reducing the rate of degradation in SMA fibroblasts and in Smn-deficient mESC-derived motor neurons74. BIP-135, a GSK3α/β inhibitor, increases survival of SMNΔ7 SMA mice by 15%75. In this study, we demonstrate that AR42 treatment increases GSK3β phosphorylation at S9 in the SMNΔ7 SMA mouse spinal cord. Phosphorylation of GSK3β at S9 by Akt inhibits the activity of GSK3β76. GSK3β inactivation by increasing its phosphorylation at S9 is essential for the development of and maintenance of neuronal polarity in neurons77,78,79. AR42 may protect the surviving SMA motor neurons from degeneration by enhancing inactivation of GSK3β through phosphorylation by Akt.
AR42 has shown strong potential as a therapeutic option in preclinical animal studies as well as in clinical trials for different forms of cancer. AR42 shows strong efficacy as a tumor growth inhibitor in multiple in vivo models for cancer progression42,80,81,82,83,84. Tumor-induced loss of muscle mass and function, i.e. cachexia, is diminished by AR4285. AR42 has recently demonstrated clinical efficacy in phase I trials for acute myeloid leukemia, multiple myeloma, T-cell lymphomas, NF2 schwannomas and meningiomas86,87,88,89. In addition to its role as a potential cancer therapeutic, AR42 also improves hippocampal memory deficits present in a transgenic mouse model for Kabuki syndrome43.
While AR42 treatment does increase survival and delay spinal motor neuron loss in SMNΔ7 SMA mice, these mice still display a motor phenotype. Previous studies have shown that SMA mice have additional deficits in the spinal sensory-motor circuit90,91,92,93. It is possible that AR42 improves motor neuron survival in SMA, which extends survival and improves motor behavior, but does not significantly ameliorate these sensory-motor circuit deficits. Previous work has shown that coadministration of small molecule circuit modifier—4-aminopyridine—and a neuroprotectant agent—pifithrin-α—provided significant therapeutic benefit to SMA mice94. Combination therapeutic regimens have previously shown additive therapeutic benefit in SMA animal models48,95,96,97. Future studies will explore the utility of AR42 as part of a multi-modal and multi-indication therapy for SMA as well as for other motor neuron diseases.
In conclusion, we have demonstrated that the class I HDAC inhibitor AR42 significantly improves the survival and motor phenotype in SMNΔ7 SMA mice in a SMN-independent manner when administered presymptomatically. AR42 increases AKT signaling in SMA spinal cords which may provide a neuroprotective mechanism of action for this compound. Future studies will characterize the regulation of AKT signaling in SMA motor neurons to determine a novel protective therapeutic strategy for SMA. While the currently approved therapies14,15,16,17 have significantly improved the prognosis for SMA patients, they do not fully ameliorate the disease phenotype in SMA patients. SMN2-independent therapies could further augment motor function in SMA patients. There are two biologics—apitegromab (SRK-015) and reldesemtiv (CK-2127107)—targeting the improvement of muscle function that are currently undergoing clinical trials in SMA patients98,99. Small molecule HDAC inhibitors like 4PBA36, VX-56336 and AR42 (this study) offer another SMN-independent protective strategy that could be used in concert with SMN2 inducers to maximize therapeutic outcomes in SMA patients. The additive effects of HDAC inhibition on the efficacy of SMN2 inducers has been recently demonstrated by the enhanced neuroprotective effects of valproic acid and splice-correcting antisense oligonucleotides96.
Methods
Drug compounds
AR42 (OSU-42; HDAC-42) and AR19 (OSU-19) were obtained from Arno Therapeutics, Inc. (Parsippany, NJ). 4-Phenylbutyrate (4PBA) was obtained from Lancaster Synthesis Inc. (Ward Hill, MA). Trichostatin A (TSA), dimethyl sulfoxide (DMSO), methyl cellulose (molecular mass = 41,000 g/mol) and Tween-80 were obtained from Sigma-Aldrich (St. Louis, MO).
Fibroblast cell culture
GM03813 fibroblasts100 (Coriell Cell Repositories; Camden, NJ) were derived from a type II SMA patient with deletion of both SMN1 alleles and 3 copies of SMN2101,102. GM03814 fibroblasts100 (Coriell Cell Repositories) were derived from the carrier mother of GM03813 with 1 copy of SMN1 and 5 copies of SMN2101,102. All fibroblast lines were maintained in Dulbecco’s modified essential medium (DMEM; Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (EqualFetal; Atlas Biologicals, Fort Collins, CO), 2 mM L-glutamine (Life Technologies) and 1% penicillin/streptomycin (Life Technologies).
Drug treatment of fibroblasts
For immunofluorescence, cells were seeded onto gelatinized glass coverslips at a density of 4000 cells/cm2. For mRNA expression analysis, fibroblasts were plated onto 6-well plates at a density of 3368 cells/cm2. Cells were plated onto 60-cm tissue culture-grade dishes at a density of 6667 cell/cm2 for protein analysis. Cells were treated with one of the following compounds (n = 3/dose): AR42 (10 nM–1 μM), AR19 (10 nM–1 μM), 4PBA (10 nM–1 μM), TSA (1–100 nM) or vehicle (DMSO). Test compounds were added to the medium at a 1:1000 dilution. Medium was changed daily for 5 days with fresh compound added daily.
Immunofluorescence and gem count analysis
Immunostaining of fibroblast cells was accomplished as described previously103,104 using the mouse anti-SMN monoclonal antibody (mAb) (MANSMA2 (8F7); Developmental Studies Hybridoma Bank, Iowa City, IA105). SMN immunostaining within the nuclei of treated fibroblasts was visualized using a DMRXA2 epifluorescence microscope (Leica Microsystems Inc., Buffalo Grove, IL) with an ORCA-ER cooled camera (Hamamatsu, Hamamatsu City, Japan) and Volocity 6.1.1 software (Perkin-Elmer, Waltham, MA). For gem counting, the following parameters were measured in 100 randomly selected nuclei: the number of gems, the number of cells with gems and the number of cells with more than 1 gem.
SMN2 promoter assay
The clone 11 cell line (Vertex Pharmaceuticals,41), which contains a β-lactamase reporter gene driven by the 3.4 kb SMN2 promoter, was used in this assay. Cells were maintained in a humidified chamber at 37 °C and 5% CO2 with DMEM containing 5% EquaFETAL, 2 mM L-glutamine and 1% penicillin/streptomycin. They were seeded onto a black-walled, clear bottom 96-well tissue culture plates (Santa Cruz Biotechnology) at a density of 5 × 104 cells/cm2. Drug compounds (n = 4/dose) were added to the medium using a 96-pin replicator (pin diameter = 1.19 mm; V&P Scientific, Inc., San Diego, CA) and plates were incubated for 19 h. 20 µL of 6X CCF2-AM dye (GeneBlazer In Vivo Detection Kit, Life Technologies) were added to each of the assay wells and plates were incubated at room temperature for 2 h before the plates are read on a fluorescence plate reader (λex = 405 nm, λem = 530 nm and λem = 460 nm; Victor X4, Perkin Elmer). The 460 nm:530 nm fluorescence ratios were plotted against compound concentrations and used to generate a dose response curve for the SMN2 promoter assay.
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cell pellets or tissues treated with drug using RNeasy Mini columns (QIAGEN, Germantown, MD) according to manufacturer’s directions. cDNA was prepared from 0.5 to 1 μg total RNA using the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) as per manufacturer’s instructions. Target transcripts were amplified via PCR from cDNA diluted 200–400-fold using the QuantiTect SYBR Green PCR kit (QIAGEN). The following primer sets (Integrated DNA Technologies; Coralville, IA) for the target transcripts were used: full-length SMN (FL-SMN) 67 (SMNex6F) 5′-ccatatgtccagattctcttgatga-3′, (SMNex78R) 5′-atgccagcatttctccttaattta-3′; SMNΔ767 (SMNex6F), (SMNex68R) 5′-atgccagcatttccatataatagc-3′; chondrolectin (Chodl) (F) 5′-ttcatgggtctcttcaggttg-3′, (R) 5′-acctttaccagtggaatgacg-3′; cyclin-dependent kinase inhibitor 1A (Cdkn1a;55 (F) 5′-gacattcagagccacaggcacc-3′, (R) 5′-gagcgcatcgcaatcacggcgc-3′; transmembrane protein 41b (Tmem41b;54 (F) 5′-gaacgaaaagccttgtgcagaagc-3′, (R) 5′-ttcaccctctcttcctcactaagctg-3′; Z+ agrin (Z+Agr;55) (F) 5′-tgtcctgggggcttctctgg-3′, (R) 5′-gctgggatctcattggtcagctc-3′; complement component 1 B polypeptide (C1qb) (F) 5′-aggttttctccatgtgtcctg-3′, (R) 5′-ctctccaaactcaccaaggtc-3′; AMPA-type ionotropic glutamate receptor 4 (Gria4) flip isoform56 (F) 5’-ggtgaatgtggacccaagga-3’, (R) 5′-gctacgttgctcaggctcaag-3′ and Gria4 flop isoform56 (F) 5′-ggaggtgactccaaggacaaga-3′, (R) 5′-ccgccaaccagaatgtagaag-3′. Primers for the human reference transcripts β-actin (ACTB), large ribosomal protein P0 (RPLP0) and glyceraldehyde 3-phosphate dehydrogenase (GAPD) as well as for the mouse reference transcripts phosphoglycerate kinase (Pgk1), ribosomal protein L13a (Rpl13a) and β-glucuronidase (Gusb) were purchased from Real Time Primers LLC (Elkins Park, PA). Quantitative PCR was performed in a 384 well plate on a 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA). Each sample was assayed in triplicate.
The relative transcript levels were calculated using the efficiency-adjusted 2−ΔΔCt method106,107. The PCR efficiency (E) for each primer set was calculated from the slope of a Ct versus log10(cDNA serial dilution) curve (E = 10[−1/slope])108. ΔCt,adjusted is the difference between the adjusted Ct (Ct,measured × E) for the target transcript and the geometric mean of the adjusted Ct values for the three reference genes109 and ΔΔCt is defined as the difference between the ΔCt for the SMA sample and the ΔCt for the control sample. The reference transcripts were ACTB, RPLP0 and GAPD for human cells and were Pgk1, Rpl13a and Gusb for mouse samples.
Immunoblot
Fibroblasts treated with drug for 5 days or dissected spinal cords from animals treated with drugs for 5 days were homogenized in lysis buffer (0.1% Triton X-100 and Complete Protease Inhibitor cocktail (Roche Life Sciences, Indianapolis, IN) dissolved in PBS, pH 7.4). These samples were resolved through polyacrylamide gels containing 0.1% SDS via electrophoresis and transferred onto PVDF membranes via electroblotting as described previously45. For the detection of SMN protein, 100 μg of tissue protein extract was added to each lane of a midi-gel; 10 μg of protein extract were added to each lane of a mini-gel for all other proteins to be detected. Immunoblotting was completed as described in45. The following primary antibodies were used in this study: mouse anti-SMN mAb (1:500; MANSMA2,105 or 1:2000, clone 8, BD Biosciences), mouse anti-β-actin mAb (1:20,000; clone AC-15, Sigma-Aldrich), rabbit anti-histone H3 polyclonal antibody (pAb) (1:1000; Cell Signaling Technology, Beverly, MA), rabbit anti-acetyl-histone H3 (K9) mAb (1:1000; clone C5B11, Cell Signaling Technology), rabbit anti-Akt pAb (1:1000; Cell Signaling Technology), rabbit anti-phospho-Akt (S473) mAb (1:1000; clone 193H12, Cell Signaling Technology), rabbit anti-GSK3β mAb (1:1000; clone 27C10, Cell Signaling Technology), rabbit anti-phospho-GSK3β (S9) mAb (1:1000; clone 5B3, Cell Signaling Technology) and rabbit anti-GAPDH pAb (1:10,000; Sigma-Aldrich). The horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (1:5000) were obtained from Rockland Immunochemicals, Inc. (Gilbertsville, PA). Band intensities were measured using ImageJ 1.45 s (National Institutes of Health, Bethesda, MD) or C-DiGit (Licor, Lincoln, NE). The full-length images for the immunoblots were provided in the Supplementary Information.
Animals and ethical statement
SMNΔ7 SMA mice (SMN2+/+; SMNΔ7+/+;mSmn−/−)13 were generated from male and female carrier mice of the genotype SMN2+/+; SMNΔ7+/+;mSmn+/− (line 4299; FVB.Cg-Tg(SMN2*delta7)4299Ahmb Tg(SMN2)89Ahmb Smn1tm1Msd; Jackson Laboratories, Bar Harbor, ME, #005025). As diet can affect the survival and phenotype of these mice46 as well as responsiveness to certain drugs35,47, the mice were fed the Harlan-Teklad 22/5 Rodent Diet for the experiments in this study. Neonatal offspring were genotyped using a PCR-based assay on genomic DNA from tail biopsies as described previously13,44. In these experiments, SMA mice are defined by the genotype SMN2+/+;SMNΔ7+/+;mSmn−/− while nonSMA mice are either carrier (SMN2+/+;SMNΔ7+/+;mSmn+/−) or normal (SMN2+/+;SMNΔ7+/+;mSmn+/+) mice. Survival was defined by the following early removal criteria: greater than 20% loss of body mass within a period of 24 h, prolonged periods (greater than 10 min) of lethargy, aberrant observed respiration, non-responsiveness to tactile stimulation and loss of surface righting reflex after PND14. Those mice reaching these early removal criteria were ethically euthanized.
All animal experiments were conducted at the Ohio State University. They were conducted in accordance with the protocols described in the National Institutes of Health Guide for the Care and Use of Animals and were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee. These studies were completed in accordance with ARRIVE guidelines.
Drug formulations and administration
AR42 and AR19 were dissolved in an aqueous solution containing 0.5% methyl cellulose and 0.1% Tween-80. All aqueous solutions were filter sterilized prior to injections. Beginning at either 4 or 9 days after birth (PND04 or PND09), SMNΔ7 SMA mice and their non-SMA littermates were treated with either AR42 (5 mg/kg/d), AR19 (5 mg/kg/d) or vehicle (0.5% methyl cellulose and 0.1% Tween-80 in ddH2O) by oral administration using a curved 18-gauge feeding needle (Harvard Apparatus, Holliston, MA) as described previously110. Drugs were administered to mice until the last SMA pup in the litter perished.
Behavioral analysis
AR42-treated SMNΔ7 SMA mice were assessed for changes in vectorial movement duration, spontaneous locomotor activity and pivoting activity as described in44. All the measures were collected from treated mice at PND07, PND11 and PND14 using Stopwatch+ (Center for Behavioral Neuroscience, Atlanta, GA). Vectorial movement duration was measured as the amount of time each mouse was crawling (PND07) or walking (PND11 and PND14) within the viewing timeframe was collected. For spontaneous locomotor activity, each pup was placed in the center of a gridded (with 28 2.5-cm2 grids) arena and the number of grids crossed in 60 s was counted. For pivoting, each pup was placed in the center of a gridded arena and the number of times the pup turned 90 °C (pivots) during a 60-s time frame was counted. To minimize the stress on the pup, all motor phenotype assays were conducted in a single session.
SMN enzyme-linked immunosorbent assay (ELISA)
Quantification of SMN protein levels in spinal cord extracts was measured using the SMN (human) Enzyme Immunometric Assay from Enzo Life Sciences (Farmingdale, NY) as described previously111 except that 40 μg of spinal cord extract were used for each sample. SMN concentrations were expressed as pg SMN per mL extract.
snRNP assembly
In vitro snRNP assembly assays were performed with extracts from SMNΔ7 SMA mice treated with AR42 or vehicle for 5 days. Preparation of mouse tissue extracts and snRNP assembly experiments were carried out essentially as previously described45,50. The amount of immunoprecipitated U1 snRNAs was quantified using a STORM 860 Phosphorimager (GE Healthcare, Piscataway, NJ) and the ImageQuant version 4.2 software.
HDAC fluorimetric assay
HDAC activity was measured using the Fluor de Lys fluorimetric assay (Enzo Life Sciences, Inc., Farmingdale, NY) according to manufacturer’s directions. SMNΔ7 SMA mice treated with AR42 or vehicle for 5 days beginning at PND04 and 25 μg of spinal cord protein extract were used for this assay. Fluorescence was measured using a SpectraFluor Plus fluorescence reader (TECAN, Morrisville, NC) using a λex = 360 nm and a λem = 465 nm.
Spinal cord histology
SMNΔ7 SMA mice and carrier littermates were treated with AR42 or vehicle beginning at PND04 until PND11. Treated mice were anesthetized with 2.5% Avertin and then transcardially perfused with ice-cold PBS followed by 4% paraformaldehyde in Sørensen’s phosphate buffer (100 mM Na2HPO4 and 100 mM NaH2PO4, pH 7.4). The lumbar spinal cords were postfixed with 4% paraformaldehyde in Sørensen’s phosphate buffer overnight at 4 °C followed by cryoprotection with 30% sucrose in ddH2O overnight at 4 °C. The lumbar spinal cords were sectioned transversely at a thickness of 25 μm using the MultiBrain® Technology by NeuroScience Associates (Knoxville, TN). Every 12th section block was mounted onto a glass slide and stained with 1% Cresyl violet as described previously36. Motor neurons of the lumbar spinal cord—identified using the Allen Spinal Cord Atlas112 as a reference—in each 12th section were counted automatically using the Cell Counter (K De Vos, University of Sheffield) plugin of ImageJ113.
Statistical analysis
Data were expressed as means ± standard error and were analyzed using one-way ANOVA with a Bonferonni post hoc test or unpaired t-tests. Kaplan–Meier analysis was performed on lifespan and onset of body mass loss data using the Mantel–Cox log rank post hoc test. Dose–response curves were generated using the four-parameter logistic formula. Goodness of fit was assessed with R2 and normality testing (Shapiro–Wilk). All statistical analyses were performed with SPSS v22.0 or SigmaPlot v12.
Data availability
All data pertaining to this study are presented within the manuscript. Further inquiries related to data availability should be directed to M.E.R.B.
References
Crawford, T. O. & Pardo, C. A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110 (1996).
Tisdale, S. & Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 35, 8691–8700 (2015).
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 96, 6307–6311 (1999).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Butchbach, M. E. R. Genomic variability in the survival motor neuron genes (SMN1 and SMN2): Implications for spinal muscular atrophy phenotype and therapeutics development. Int. J. Mol. Sci. 22, 7896 (2021).
Didonato, C. J. et al. Cloning, characterization and copy number of the murine survival motor neuron gene: Homolog of the spinal muscular atrophy-determining gene. Genome Res. 7, 339–352 (1997).
Viollet, L. et al. cDNA isolation, expression and chromosomal localization of the mouse survival motor neuron gene (SMN). Genomics 40, 185–188 (1997).
Schrank, B. et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U. S. A. 94, 9920–9925 (1997).
Hsieh-Li, H. M. et al. A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66–70 (2000).
Monani, U. R. et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in SMN−/− mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339 (2000).
Michaud, M. et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 38, 125–135 (2010).
Le, T. T. et al. SMNΔ7, the major product of the centromeric survival motor neuron gene (SMN2), extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857 (2005).
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017).
Mercuri, E. et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018).
Baranello, G. et al. Risdiplam in type 1 spinal muscular atrophy. N. Engl. J. Med. 384, 915–923 (2021).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Lunke, S. & El-Osta, A. The emerging role of epigenetic modifications and chromatin remodeling in spinal muscular atrophy. J. Neurochem. 109, 1557–1569 (2009).
Chang, J. G. et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. U. S. A. 98, 9808–9813 (2001).
Andreassi, C. et al. Phenylbutyrate increases SMN expression in vitro: Relevance for treatment of spinal muscular atrophy. Eur. J. Hum. Genet. 12, 59–65 (2004).
Brahe, C. et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet. 13, 256–259 (2005).
Brichta, L. et al. Valproic acid increases the SMN2 protein level: A well-known drug as potential therapy for spinal muscular atrophy. Hum. Mol. Genet. 12, 2481–2489 (2003).
Brichta, L., Holker, I., Haug, K., Klockgether, T. & Wirth, B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann. Neurol. 59, 970–975 (2006).
Harahap, I. S. K. et al. Valproic acid increases SMN2 Expression and modulates SF2/ASF and hnRNPA1 expression in SMA fibroblast cell lines. Brain Dev. 34, 213–222 (2012).
Sumner, C. J. et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 54, 647–654 (2003).
Avila, A. M. et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117, 659–671 (2007).
Hahnen, E. et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem. 98, 193–202 (2006).
Riessland, M. et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet. 19, 1492–1506 (2010).
Riessland, M., Brichta, L., Hahnen, E. & Wirth, B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum. Genet. 120, 101–110 (2006).
Garbes, L. et al. Lbh589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 18, 3645–3658 (2009).
Schreml, J. et al. Severe SMA mice show organ impairment that cannot be rescued by therapry with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 21, 652 (2013).
Kernochan, L. E. et al. The role of histone acetylation in SMN gene expression. Hum. Mol. Genet. 14, 1171–1182 (2005).
Evans, M. C., Cherry, J. J. & Androphy, E. J. Differential regulation of the SMN2 gene by individual HDAC Proteins. Biochem. Biophys. Res. Commun. 414, 25–30 (2011).
Liu, H., Yazdani, A., Murray, L. M., Beauvais, A. & Kothary, R. The SMN-independent beneficial effects of trichostatin A on an intermediate mouse model of spinal muscular atrophy. PLoS ONE 9, E101225 (2014).
Narver, H. I. et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 64, 465–470 (2008).
Butchbach, M. E. R. et al. Protective effects of butyrate-based compounds on a mouse model for spinal muscular atrophy. Exp. Neurol. 279, 13–26 (2016).
Lu, Q., Wang, D. S., Chen, C. S., Hu, Y. D. & Chen, C. S. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J. Med. Chem. 48, 5530–5535 (2005).
Lu, Q. et al. Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J. Med. Chem. 47, 467–474 (2004).
Liu, Q. & Dreyfuss, G. A novel nuclear structure containing the survival of motor neurons protein. Embo J. 15, 3555–3565 (1996).
Coovert, D. D. et al. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214 (1997).
Jarecki, J. et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 14, 2003–2018 (2005).
Jacob, A. et al. Preclinical validation of AR42, a novel histone deacetylase inhibitor as treatment for vestibular schwannomas. Laryngoscope 122, 174–189 (2012).
Bjornsson, H. T. et al. Histone deactylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci. Transl. Med. 6, 256–135 (2014).
Butchbach, M. E. R., Edwards, J. D. & Burghes, A. H. M. Abnormal motor phenotype in the SMNΔ7 mouse model of spinal muscular atrophy. Neurobiol. Dis. 27, 207–219 (2007).
Butchbach, M. E. R. et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 19, 454–467 (2010).
Butchbach, M. E. R. et al. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 391, 835–840 (2010).
Butchbach, M. E. R., Singh, J., Gurney, M. E. & Burghes, A. H. M. The effect of diet on the protective action of D156844 observed in spinal muscular atrophy mice. Exp. Neurol. 256, 1–6 (2014).
Harris, A. W. & Butchbach, M. E. R. The effect of the DcpS inhibitor D156844 on the protective action of follistatin in mice with spinal muscular atrophy. Neuromuscul. Disord. 25, 699–705 (2015).
Li, D. K., Tisdale, S., Lotti, F. & Pellizzoni, L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 32, 22–29 (2014).
Gabanella, F. et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 9, E921 (2007).
Zhang, Z. et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600 (2008).
Tisdale, S. et al. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3’-End formation of histone mRNAs. Cell Rep. 5, 1187–1195 (2013).
Bäumer, D. et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. Plos Genet. 5, E1000773 (2009).
Lotti, F. et al. An SMN-dependent U12 splicing event essential for motor circuit formation. Cell 151, 440–454 (2012).
Ruggiu, M. et al. A role for SMN Exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol. Cell. Biol. 32, 126–138 (2012).
Zhang, Z. et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 110, 19348–19353 (2013).
Chen, C. S., Weng, S. C., Tseng, P. H., Pin, H. L. & Chen, C. S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280, 38879–38887 (2005).
Tng, J. et al. Achiral derivatives of hydroxamate AR-42 potently inhibit class I HDAC enzymes and cancer cell proliferation. J. Med. Chem. 63, 5956–5971 (2020).
Edwards, J. D. & Butchbach, M. E. R. Effect of the butyrate prodrug pivaloyloxymethyl butyrate (An9) on a mouse model for spinal muscular atrophy. J. Neuromuscul. Dis. 3, 511–516 (2016).
Tsai, L. K., Tsai, M. S., Ting, C. H. & Li, H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J. Mol. Med. 86, 1243–1254 (2008).
Renusch, S. R. et al. Spinal muscular atrophy biomarker measurements from blood samples in a clinical trial of valproic acid in ambulatory adults. J. Neuromuscul. Dis. 2, 119–130 (2015).
Somers, E. et al. Increasing SMN levels using the histone deacetylase inhibitor SAHA ameliorates defects in skeletal muscular microvasculature in a mouse model of severe spinal muscular atrophy. Neurosci. Lett. 544, 100–104 (2013).
Bricceno, K. V. et al. Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy Mice. Hum. Mol. Genet. 21, 4448–4459 (2012).
Abera, M. B. et al. Ml372 blocks SMN ubiquitination and improves spinal muscular atrophy pathology in mice. JCI Insight 1, e88427 (2016).
Rietz, A. et al. Discovery of a small molecule probe that post-translationally stabilizes the survival motor neuron protein for the treatment of spinal muscular atrophy. J. Med. Chem. 60, 4594–4610 (2017).
Letso, R. R., Bauer, A. J., Lunn, M. R., Yang, W. S. & Stockwell, B. R. Small molecule screen reveals regulation of survival motor neuron protein abundance by Ras proteins. ACS Chem. Biol. 8, 914–922 (2013).
Gentillon, C., Connell, A. J., Kirk, R. W. & Butchbach, M. E. R. The effects of C5-substituted 2,4-diaminoquinazolines on selected transcript expression in spinal muscular atrophy cells. PLoS ONE 12, E0180657 (2017).
Sapaly, D. et al. Small-molecule flunarizine increases SMN protein in nuclear Cajal bodies and motor function in a mouse model of spinal muscular atrophy. Sci. Rep. 8, 2075 (2018).
Van Meerbeke, J. P. et al. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum. Mol. Genet. 22, 4074–4083 (2013).
Gogliotti, R. G. et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 22, 4084–4101 (2013).
Yu, F., Sugawara, T., Maier, C. M., Hseih, L. B. & Chan, P. H. AKT/BAD signaling and motor neuron survival after spinal cord injury. Neurobiol. Dis. 20, 491–499 (2005).
Biondi, O. et al. IGF-1R reduction triggers neuroprotective signaling pathways in spinal muscular atrophy mice. J. Neurosci. 35, 12063–12079 (2015).
Branchu, J. et al. Shift from extracellular signal-related kinase to AKT/cAMP response element-binding protein pathway increases survival-motor-neuron expression in spinal-muscular-atrophy-like mice and patient cells. J. Neurosci. 33, 4280–4294 (2013).
Makhortova, N. R. et al. A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 7, 544–552 (2011).
Chen, P. C. et al. Identification of a maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitor, BIP-135, that prolongs the median survival time of Δ7 SMA KO mouse model of spinal muscular atrophy. ACS Chem. Neurosci. 3, 5–11 (2012).
Cross, D. A. E., Alessi, D. R., Cohen, P., Andjelkovich, M. & Hemmings, B. A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 (1995).
Hur, E. M. & Zhou, F. Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551 (2010).
Jiang, H., Guo, W., Liang, X. & Rao, Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: Critical roles of GSK-3β and its upstream regulators. Cell 120, 123–135 (2005).
Yoshimura, T. et al. GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149 (2005).
Lu, Y. S. et al. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology 46, 1119–1130 (2007).
Lucas, D. M. et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS ONE 5, E10941 (2010).
Sargeant, A. M. et al. OSU-HDAC42, a histone deacetylase inhibitor, blocks prostate tumor progression in a transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 68, 3999–4009 (2008).
Yang, Y. T. et al. A rationally designed histone deacetylase inhibitor with distinct antitumor activity against ovarian cancer. Neoplasia 11, 552–563 (2009).
Bush, M. L. et al. AR42, a novel histone deaceetylase inihibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro Oncol. 13, 983–999 (2011).
Tseng, Y. C. et al. Preclinical investigation of the novel histone deacetylase inhibitor AR-42 in the treatment of cancer-induced cachexia. J. Natl. Cancer Inst. 107, Djv274 (2015).
Liva, S. G. et al. Phase I study of AR-42 and decitabine in acute myeloid leukemia. Leuk. Lymphoma 61, 1484–1492 (2020).
Sborov, D. W. et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 58, 2310–2318 (2017).
Collier, K. A. et al. A phase 1 trial of the histone deacetylase inhibitor AR-42 in patients with neurofibromatosis type 2-associated tumors and advanced solid malignancies. Cancer Chemother. Pharmacol. 87, 599–611 (2021).
Welling, D. B. et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investig. Otolaryngol. 6, 1008–1019 (2021).
Ling, K. K. Y., Lin, M. Y., Zingg, B., Feng, Z. & Ko, C. P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE 5, E15457 (2010).
Mentis, G. Z. et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69, 453–467 (2011).
Ling, K. K. Y., Gibbs, R. M., Feng, Z. & Ko, C. P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 21, 185–195 (2012).
Fletcher, E. V. et al. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci. 20, 905–916 (2017).
Simon, C. M. et al. Chronic pharmacological increase of neuronal activity improves sensory-motor dysfunction in spinal muscular atrophy mice. J. Neurosci. 41, 376–389 (2021).
Kray, K. M., Mcgovern, V. L., Chugh, D., Arnold, W. D. & Burghes, A. H. M. Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic Δ7 SMA mice. Neurobiol. Dis. 159, 105488 (2021).
Marasco, L. E. et al. Counteracting chromatin effects of a splicing-correcting antisense oligonucleotide improves its therapeutic efficacy in spinal muscular atrophy. Cell 185, 2057–2070 (2022).
Dumas, S. A. et al. A combinatorial approach increases SMN level in SMA model mice. Hum. Mol. Genet. 31, 2984–3000 (2022).
Barrett, D. et al. A randomized phase 1 study, pharmacokinetic and pharmacodynamic study of the novel myostatin inhibitor apitegromab (SRK-015): A potential treatment for spinal muscular atrophy. Adv. Ther. 38, 3203–3222 (2021).
Rudnicki, S. A. et al. Reldesemtiv in patients with spinal muscular atrophy: A phase 2 hypothesis-generating study. Neurotherapeutics 18, 2130 (2021).
Scudiero, D. A. et al. Alzheimer disease fibroblasts are hypersensitive to the lethal effects of a DNA-damaging chemical. Mutat. Res. 159, 125–131 (1986).
Stabley, D. L. et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genomic Med. 3, 248–257 (2015).
Stabley, D. L. et al. Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromuscul. Disord. 27, 439–446 (2017).
Mack, S. G., Cook, D. J., Dhurjati, P. & Butchbach, M. E. R. Systems biology investigation of camp modulation to increase SMN levels for treatment of spinal muscular atrophy. PLoS ONE 9, E115473 (2014).
Thurmond, J. et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J. Med. Chem. 51, 449–469 (2008).
Young, P. J., Le, T. T., Man, N. T., Burghes, A. H. M. B. & Morris, G. E. The relationship between SMN, the spinal muscular atrophy protein, and nuclear coiled bodies in differentiated tissues and cultured cells. Exp. Cell Res. 256, 365–374 (2000).
Schmittgen, T. D. & Livak, K. J. Analyzing real-Time PCR Data by the comparative Ct method. Nat. Protoc. 3, 1101–1108 (2008).
Yuan, J. S., Wang, D. & Stewart, C. N. Jr. statistical methods for efficiency adjusted real-time PCR quantification. Biotechnol. J. 3, 112–123 (2008).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, E45 (2001).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 1–12 (2002).
Butchbach, M. E. R., Edwards, J. D., Schussler, K. R. & Burghes, A. H. M. A novel method for oral delivery of compounds to the neonatal SMNΔ7 model of spinal muscular atrophy. J. Neurosci. Methods 161, 285–290 (2007).
Man, N. T. et al. A two-site ELISA can quantify upregulation of SMN protein by drugs for spinal muscular atrophy. Neurology 71, 1757–1763 (2008).
Henry, A. M. & Hohmann, J. G. High-Resolution gene expression atlases for adult and developing mouse brain and spinal cord. Mamm. Genome 23, 539–549 (2012).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We would like to thank Arno Therapeutics, Inc. for generously providing AR42 and Vertex Pharmaceuticals, Inc. for generously providing the clone 11 reporter cell line. The SMN EIA kits were generously provided by Assay Designs, now Enzo Life Sciences, through their Kits for Charity program. Additionally, we would like to thank Dr. Glenn Morris for kindly providing the SMN mAb and the Nemours Biomolecular Core Laboratory for access to the 7900HT Fast Real-Time PCR system. The study was supported by grants from Cure SMA (M.E.R.B. and A.H.M.B.), Miracles for Madison Fund (A.H.M.B.), National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS38650 to A.H.M.B. and R01NS102451 to L.P.), an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103463 and P30GM114736 to M.E.R.B.) and the Nemours Foundation (M.E.R.B.).
Author information
Authors and Affiliations
Contributions
M.E.R.B., L.P. and A.H.M.B. conceived and designed the experiments; M.E.R.B., A.W.H., C.J.L., A.J.C., R.W.K. and L.S. performed the experiments; M.E.R.B., C.J.L., L.P. and A.H.M.B. analyzed the data; all authors wrote the paper. All authors have read and approved submission of this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Video 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lumpkin, C.J., Harris, A.W., Connell, A.J. et al. Evaluation of the orally bioavailable 4-phenylbutyrate-tethered trichostatin A analogue AR42 in models of spinal muscular atrophy. Sci Rep 13, 10374 (2023). https://doi.org/10.1038/s41598-023-37496-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37496-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
