
Abstract
Defective gastrointestinal barrier function and, in turn, microbial translocation have been identified as significant contributors to persistent inflammation in antiretroviral (ARV)-treated people living with HIV. Metabolic supplementation of short-chain fatty acids (SCFAs), generally produced by the commensal microbiome, may improve these outcomes. Butyrate is a SCFA that is essential for the development and maintenance of intestinal immunity and has a known role in supporting epithelial integrity. Herein we assessed whether supplementation with the dietary supplement sodium butyrate would improve immune reconstitution and reduce inflammation in ARV-treated, simian immunodeficiency virus (SIV)-infected rhesus macaques. We demonstrate that butyrate supplementation does not significantly improve immune reconstitution, with no differences observed in systemic CD4+ T-cell frequencies, T-cell functionality or immune activation, microbial translocation, or transcriptional regulation. Our findings demonstrate that oral administration of sodium butyrate is insufficient to reduce persistent inflammation and microbial translocation in ARV-treated, SIV-infected macaques, suggesting that this therapeutic may not reduce co-morbidities and co-mortalities in treated people living with HIV.
Similar content being viewed by others


Introduction
Fully suppressive antiretroviral (ARV) therapy does not completely ameliorate inflammation in people living with HIV, with chronic inflammation a leading contributor to reduced life expectancy among these individuals1. Although time to treatment initiation, lifestyle, and environmental factors contribute to magnitude, increased inflammation is persistent and is associated with defined co-morbidities including hypertension, osteoporosis, hyperlipidemia, cancer, hepatitis, and renal disease1,2. Indicators of inflammation are varied, including circulating acute phase reactants and markers of coagulation (high-sensitivity C-reactive protein (CRP) and D-dimer), antimicrobial immunity (soluble CD14 (sCD14), soluble CD163, endogenous endotoxin-core antibody), and non-specific cytokine release (IL-6 and IL-18)2. Through persistent stimulation, chronic inflammation further contributes to a slow exhaustion of the adaptive lymphocyte population, resulting in poor vaccine responsiveness and susceptibility to opportunistic infections2,3. The design and implementation of adjunct therapies to address this inflammation will further improve the life expectancy and quality of life for people living with HIV.
Microbial translocation is a significant contributor to persistent inflammation and incomplete immune reconstitution in ARV-treated HIV infections4. In addition to the presence of circulating markers of antimicrobial immunity, translocation is characterized by the presence of circulating microbial products such as lipopolysaccharide (LPS), peptidoglycan, flagellin, and 16S or bacterial CpG DNAs5. Translocation can be visualized across the gastrointestinal epithelium, where breaches in epithelial integrity allow access to the underlying mucosa and into systemic circulation6,7. In the progressive SIV non-human primate model of HIV infection, administration of the LPS-binding compound sevelamer prevents plasma LPS and sCD14 accumulation and limits CRP and D-dimer elevation in acute, ARV-untreated infection8. Furthermore, the use of antibiotics cotrimoxazole and vancomycin have been shown to reduce, though not eliminate, adverse health effects in ARV-untreated HIV and SIV infection, respectively9,10. Though useful in treating the effects of translocation, the prolonged co-administration of antibiotics, sevelamer, and other pharmaceuticals is impractical in the setting of prolonged, non-curative, ARV therapy11.
The commensal microbiome is essential for the development and maintenance of the gastrointestinal epithelium and intestinal immunity in part through the production of diffusible metabolites, including SCFAs such as butyrate12,13. Within colonocytes, butyrate is an energy source and signals through peroxisome proliferator activated receptor gamma (PPARγ) to drive cellular energy metabolism towards b-oxidation, thereby limiting oxygen availability and restricting the growth of pathogenic facultative anaerobes14. Additionally, butyrate enhances histone H3 acetylation by inhibiting histone deacetylases (HDACs)15,16,17,18. The associated epigenetic modifications support the development of regulatory T-cells (Tregs)19,20,21 and the production of IL-22 from T-helper 22 (TH22) cells and innate lymphoid cells (ILCs)22. In part through the activity of IL-22, butyrate contributes to tight junction formation, intestinal-stem-cell epithelial regeneration, mucin production, the physiologic production of reactive oxygen species, and antimicrobial peptide production23,24,25. A low-abundance of butyrate-producing taxa has been described in some HIV cohorts26,27,28,29,30,31,32,33,34 and supplementation of treated HIV and SIV infections with prebiotics or probiotics31,35,36 has been shown to reduce microbial translocation and immune activation, improve CD4+ T-cell frequencies, and improve thymic output or reduce lymphoid fibrosis. As such, butyrate supplementation alone has been proposed as a natural and inexpensive therapeutic to reduce inflammation and improve immune reconstitution in treated infections30,37.
Herein, we demonstrate that oral butyrate supplementation in ARV-treated, SIV-infected macaques does not significantly improve immune reconstitution. Supplemented animals do not show improved systemic CD4+ T-cell recovery, do not exhibit alterations in T-cell functionality, nor do they exhibit evidence of improved intestinal repair or microbial translocation. Our findings demonstrate that sodium butyrate is not sufficient to improve immunodeficiency over and above the effects of ARVs themselves.
Results
Butyrate supplementation does not improve viral suppression or CD4+ T-cell recovery in ARV-treated, SIV-infected macaques
To investigate whether butyrate might improve immune reconstitution in treated SIV-infected Asian macaques, we initiated combination ARV treatment (emtricitabine, tenofovir disoproxil fumarate, and dolutegravir sodium) in chronically SIV-infected rhesus macaques 14 days prior to the administration of daily sodium butyrate (Table 1). ARV treatment was initiated prior to butyrate supplementation to ensure that suppression dynamics were comparable across the control and experimental groups. Following administration of ARVs, control (n = 6) and butyrate-supplemented (n = 7) animals were followed for 180 days, during which time blood and tissue lymphocytes were assessed for canonical markers of disease progression and immunological functionality. ARV initiation was associated with a rapid decline in plasma viral load in all animals (Fig. 1a). Although rates of viral decline and suppression varied across animals, viremia did not significantly differ between groups (two-way ANOVA). The exclusion of animals that did not effectively suppress viremia by study-end did not change the study outcome (data not shown). We next assessed the impact of butyrate administration on peripheral blood CD4+ T-cell counts and CD4+ T cell frequencies in tissues (Fig. 1b, c). As compared to baseline CD4+ T-cell values, ARV treatment with and without butyrate supplementation was associated with a significant recovery of CD4+ T-cells in the periphery, mesenteric lymph nodes (MLN), and bronchoalveolar lavage (BAL) by day 180 post-treatment and a trend towards recovery in rectal (RB) and jejunal (Jej) biopsies at day 90. However, no significant differences were noted by treatment status for any of the anatomic sites studied.

Butyrate supplementation of ARV treatment does not alter SIV suppression or target cell frequencies. (a) Longitudinal plasma SIV viral load in ARV-only (black) and ARV + butyrate (blue) animals, with individual animals denoted by individual symbols as identified in Table 1. (b) Circulating CD4+ T-cell count. (c–e) PBMC, RB, MLN, Jej, and BAL % CD4+ T-cells (c) and %CCR5 + CD4+ (d) and CD8 + (e) TM. (f, g) PBMC, RB, and MLN %Ki-67 + CD4+ (f) and CD8 + (g) TM. All timepoints are relative to the day of ARV initiation. Lines among datapoints represent means. Lines among p-values span timepoints considered significantly different, color-coded by treatment status. Significance methods as follows: Two-way ANOVA (a), unpaired and paired two-way t-tests as appropriate (b–g).
We next considered whether butyrate supplementation might influence the recovery of CD4+ memory T-cells (TM) expressing the SIV co-receptor CCR5. CCR5 frequencies increased from baseline to day 180 post-treatment in peripheral blood mononuclear cells (PBMCs) and the MLN for both groups. A significant increase was observed in RB for the butyrate-supplemented group only (Fig. 1d). For tissues sampled at day 90 post-treatment, CCR5 expression remained unchanged or decreased. To assess whether differences in CD4+ TM CCR5-expression might be mediated by residual viral replication, we also examined CCR5 expression on non-target, CD8 + TM. Here, a significant difference from baseline was noted only in PBMCs at day 180 post-treatment, with no significant differences observed between treatment groups (Fig. 1e).
Changes in CCR5 expression among tissue-based CD4+ T-cells may be the direct result of virus replication (or absence thereof) among CD4+ T-cells or, may be attributed to proliferation which might differ in the presence of butyrate. To assess these possibilities, we next measured Ki-67 expression among PBMC, RB, and MLN CD4+ and CD8 + TM on day 180 post-treatment (Fig. 1f, g). No differences were seen by treatment group, suggesting that the loss of direct infection was the main mechanism for CD4 recovery in both butyrate-untreated and -treated animals.
Butyrate supplementation does not alter markers of mucosal homing or residency in ARV-treated, SIV-infected macaques
We assessed whether butyrate-supplementation in ARV-treated, SIV-infected macaques might improve mucosal homing or retention by measuring expression of CD103, a highly expressed integrin in mucosal tissues38, and CD69, an often used marker of T-cell tissue residency39, in CD4+ and CD8 + TM across multiple tissue sites. Following ARV administration, T-cell CD103 expression significantly increased in multiple tissue sites as compared to baseline. Among CD4+ TM, CD103 expression increased only in PBMCs at day 90, irrespective of treatment group (Fig. 2a). A significant difference in CD4+ TM CD103 expression was noted between groups in the BAL at both baseline (two-way Student's t test, p = 0.032) and day 90 (p = 0.020); however, no longitudinal differences were noted for either group suggesting that these differences were independent of butyrate-supplementation. Among CD8 + TM, ARV-only animals exhibited an increase in CD103 expression from baseline to day 90 in PBMCs and to day 180 in the MLN and a decrease from day 0 to 90 in the BAL. CD8 + TM in butyrate-supplemented animals exhibited an increase from baseline to day 90 and 180 in PBMCs and to day 180 in RB and MLN (Fig. 2b). The significant increase in peripheral CD103 + memory T-cells in both groups at day 90 post-infection may reflect a redistribution of intestinal memory T-cells, contributing to the enhanced loss of CCR5-expressing cells (Fig. 1d) prior to complete viral suppression.

T-cell markers of intestinal homing and tissue residency remain unchanged with butyrate supplementation of ARV treatment. (a–f) PBMC, RB, MLN, Jej, and BAL %CD103 + (a, b) and CD69 + (c, d), CD69 + CD103 + (e, f) CD4+ (a, c, e) CD8 + (b, d, f) TM in ARV-only (black) and ARV + butyrate (blue) animals, with individual animals denoted by individual symbols as identified in Table 1. All timepoints are relative to the day of ARV initiation. Lines among datapoints represent means. Lines among p-values span timepoints considered significantly different, color-coded by treatment status. Asterisks denote timepoints where groups significantly differed. Significance assessed by unpaired and paired two-way t tests as appropriate.
When considering CD69, we observed significant declines from baseline to day 90 in PBMC CD4+ and CD8 + TM from ARV-only treated animals and in RB CD4+ and CD8 + TM from butyrate-supplemented animals—this decline extended to day 180 among PBMC CD8 + TM only (Fig. 2c, d). In contrast, we observed increased CD69 expression from baseline to day 180 for both groups in MLN CD4+ TM and for ARV-only animals in RB CD8 + TM. Among Jej CD4+ TM, we observed a significant difference between groups at day 90 (p = 0.042), with significantly lower CD69 expression in butyrate-supplemented animals (Fig. 2c). As both groups exhibited a trend in declining CD69 expression among Jej CD4+ TM from day 0 to 90, this significance may indicate that butyrate-supplementation is accelerating this decline. We additionally considered that double-positive CD69 + CD103 + T-cells might differ in responsiveness to ARV therapy. Here differences were observed only among BAL CD4+ TM and RB CD8 + TM and reflected the changes observed in CD103 alone (Fig. 2e, f). Between group differences were noted in BAL CD4+ TM at both baseline (p = 0.0310) and day 90 post-treatment (p = 0.0129), suggesting that these differences were, in part, independent of butyrate supplementation. From day 0 to 180 post-treatment, a significant increase in CD69 + CD103 + was observed for ARV + butyrate RB CD8 + TM alone; however, no differences were noted between groups at day 180 post-treatment.
Butyrate supplementation does not differentially affect T-cell functionality in ARV-treated, SIV-infected macaques
To investigate whether butyrate supplementation improved T-cell function, we assessed at day 180 post-treatment whether homeostatic proliferation may be altered by measuring IL-2, whether proinflammatory cytokine capacity was altered by measuring IFNg and TNFa, and whether antimicrobial function was altered by measuring IL-17. No significant differences were observed between treatment groups for either CD4+ or CD8 + TM isolated from PBMCs, RB, or MLN (Fig. 3a–h).

T-cell function does not improve with butyrate supplementation of ARV treatment. (a–h) PBMC, RB, and MLN %IL-2+ (a, b), IFNγ+ (c, d), TNFα+ (e, f), and IL-17 + (g, h) CD4+ (a, c, e, g) and CD8 + (b, d, f, h) TM in ARV-only (black) and ARV + butyrate (blue) animals, with individual animals denoted by individual symbols as identified in Table 1. (i) %FOXP3 + RB and MLN CD4+ TM. All timepoints are relative to the day of ARV initiation. Lines among datapoints represent means. Significance assessed by unpaired two-way t tests.
An imbalance of Treg frequencies impedes immune reconstitution in treated HIV and SIV infections40,41. As butyrate signaling promotes the development of regulatory lymphocytes19,20,21, we considered that butyrate supplementation might specifically influence frequencies of intestinal Tregs. When we investigated frequencies of RB and MLN CD4+ Tregs (FOXP3 + CD4+ TM) however, we saw no significant differences between groups (Fig. 3i). These results indicate that butyrate supplementation did not improve post-therapeutic T-cell function in SIV-infected macaques.
Butyrate supplementation does not improve intestinal integrity nor microbial translocation in ARV-treated, SIV-infected macaques
In HIV and SIV infection, epithelial damage and microbial translocation are evident and only partially mitigated by ARV therapy3,4,6. As SCFAs promote colonic epithelial integrity12 we next determined whether butyrate supplementation altered biomarkers of epithelial integrity in the context of ARV treatment. Measured by enzyme-linked immunosorbent assay (ELISA), no significant differences in circulating intestinal fatty-acid binding protein 2 (iFABP2) nor zonulin were observed for either group from baseline to day 180 post-treatment; we did not observe a difference between groups at either timepoint (Fig. 4a). To determine whether butyrate supplementation ameliorated microbial translocation, we next assessed the presence of Escherichia coli in colonic tissue by immunohistochemistry (IHC) at day 180 post-treatment. E. coli was evident in colonic tissues at levels above that observed for a healthy, uninfected batch control; however, no significant differences in translocation were evident between ARV-only and ARV + butyrate animals (Fig. 4b, c). As microbial translocation is not limited to E. coli7,42,43, we further assessed circulating levels of sCD14 as a surrogate biomarker of microbial translocation44. We saw a surprising increase in sCD14 for both groups from baseline to day 180; however, sCD14 levels were comparable between groups at both assessed timepoints (Fig. 4d).

Butyrate supplementation of ARV treatment neither promotes intestinal repair nor reduces microbial translocation. (a) Plasma iFABP2 (left) and zonulin (right) concentrations in ARV-only (black) and ARV + butyrate (blue) animals, with individual animals denoted by individual symbols as identified in Table 1. (b) Representative IHC images of day 180 colons stained for E. coli from an ARV-only (left) or ARV + butyrate (right) animal. (c) Translocation index of Colon sections stained for E. coli as in (b). Dashed line represents the colonic translocation index of a single, healthy macaque stained as a batch control. (d) Plasma sCD14 concentrations. (e) Heatmap depicting relative colonic transcript abundance (columns) in macaques (rows) at day 180 as assessed by NanoString. Animal treatment status (left, as in a) and relative expression values (right), as indicated. Data clustered by Euclidian distance with Ward's minimum distance linkage. (f) PCA considering treatment-group and within-animal relative frequency of Colonic transcripts at day 180 as assessed in (e). (g) Relative expression of PPARG and NOS2 in the Colon and Jej, at day 180 as assessed in (e). (h) Normalized histone H3 acetylation expression as measured by ELISA in the Colon and Jej at day 180. (i) Fold-change frequencies of ASVs identified as differentially abundant in response to butyrate supplementation. Data show ARV + butyrate:ARV-only frequency fold-change, color-grouped by phylum and individually shaded by family as indicated. (j, k) Alpha (j) and Beta (k) diversity estimates at days 0 and 180. Alpha diversity estimates include observed (left) and Shannon (right) richness. Beta diversity assessed by unweighted UniFrac. All timepoints are relative to the day of ARV initiation. Lines among p values span timepoints considered significantly different, color-coded by treatment status. Solid lines among datapoints represent means. Dashed lines among datapoints represent a SIV-batch control. Significance methods as follows: unpaired and paired two-way t-tests as appropriate (a, c–d, g–h, and j), MetaLonDa (i), and adonis (k).
In part, butyrate supports epithelial integrity and intestinal immunity by transcriptional control, both directly through its interaction with PPARg45 and indirectly by global inhibition of histone H3 deacetylase15,16,17,18. To assess whether butyrate supplementation altered the colonic transcriptional profile, we quantified 770 immunological transcripts by NanoString46. No differences in expression profile were evident between groups at day 180 post-treatment (Fig. 4e) and transcriptional profiles did not cluster together by group by Ward's linkage nor by principal component analysis (PCA; Fig. 4f). Transcripts mapped to immunological pathways by ingenuity pathway analysis (IPA) as expected, but there were no differences in activation status (z-score) between the two groups (Supplementary Table S1). We similarly did not see any differences among jejunal transcripts (Supplementary Fig. S1 and Supplementary Table S1).
Despite the absence of effect on immunological transcript expression, we sought to determine whether butyrate supplementation was able to effectively modulate either specific transcriptional regulation or global epigenetic reprogramming in the context of treated, chronic SIV infection. In its role as a regulator of b-oxidation and intestinal hypoxia, butyrate is known to signal through PPARg, leading to the altered expression of nitric oxide synthase 2 (NOS2)14. No differences in either PPARG or NOS2 were observed between ARV-only and ARV + butyrate animals in either colonic or jejunal homogenates at day 180 post-treatment (Fig. 4g). As expected, given the absence of other effects in our animals, no differences in histone H3 acetylation were observed in either colonic or jejunal homogenates at day 180 post-treatment (Fig. 4h).
In therapeutic models of murine colitis, butyrate supplementation enhances inflammation resistance and is accompanied by modest shifts in the intestinal microbiome47,48. To investigate whether butyrate supplementation might influence the microbiome in the context of ARV-treated chronic SIV infection, we characterized the fecal microbiome by 16S Illumina sequencing and identified longitudinal amplicon sequence variant (ASV) perturbations utilizing MetaLonDA. Several ASVs declined over the treatment period, concentrated particularly among Bacteroidetes Prevotellaceae and Firmicutes Ruminococcaceae (Fig. 4i). These isolated differences did not translate into a difference in either alpha or beta diversity at day 180 post-treatment (Fig. 4j, k). Collectively, our findings indicate that oral butyrate supplementation does not improve upon ARV-mediated immune reconstitution or inflammation reduction.
Discussion
ARV therapy has significantly improved the lifespan and quality of life for people living with HIV and yet, prolonged treatment is not without complication. Non-AIDS related co-morbidities are now a leading cause of mortality for people living with HIV and are attributed to inflammation1 stemming, in part, from deficiencies in gastrointestinal immunity and epithelial integrity49. Microbiome-derived SCFAs such as butyrate are essential for the development and maintenance of gastrointestinal immunity and epithelial integrity12,13. Butyrate-producing bacteria are reported at lower frequencies in people living with HIV regardless of treatment status26,27,28,29,30,31,32,33,34 and the loss of butyrate production and butyrate-producing genes correlates with residual inflammation31,37. Although the use of prebiotic and probiotic therapies improves immune reconstitution and reduces inflammation in treated people living with HIV31,50 and infected macaques35,36, it is unclear whether butyrate directly contributes to these outcomes. Herein, we demonstrate that oral butyrate supplementation in ARV-treated macaques is insufficient to improve immune reconstitution and epithelial integrity or to reduce microbial translocation.
Butyrate supplementation has been shown to prevent or reverse inflammation in murine models of intestinal bacterial infection and colitis47,48,51. Our data suggest that the benefits of prebiotic and probiotic taxa in treated lentiviral infections31,35,36 may be multi-factorial and/or independent of butyrate activity. In addition to SCFAs, probiotic taxa synthesize many metabolites useful for host immunity and epithelial integrity and additionally compete with pathobionts for intestinal niches12,13. These functions may be needed to supplement the activity of butyrate. Conversely, the short-term use of antibiotics known to deplete butyrate-producing taxa—clinically indicated for the emergence of secondary and opportunistic infections—does not significantly worsen clinical indices of inflammation in treated people living with HIV52 nor in untreated macaques10 which may suggest that butyrate production is saturated throughout progressive lentiviral infections. Importantly, frequencies of butyrate-producing bacteria alone do not dictate net butyrate production, with the presence/absence of non-butyrate producing taxa directly contributing to variability in butyrate concentrations and saturation thresholds in vitro53. These complex interactions may limit the efficacy of monotypic therapeutics. Our work did not address the necessity of butyrate signaling in contributing to immune reconstitution in either ARV treatment alone or in the context of probiotic therapeutics. Another possibility is that absorbance of butyrate administered orally limited its potential effectiveness within the large intestine.
Butyrate influences intestinal immunity by two predominant mechanisms—signaling through PPARγ to drive β-oxidation14 and the inhibition of HDACs to epigenetically regulate transcriptional programming15,16,17,18. We did not observe a butyrate-effect in either the intestinal transcriptome, PPARG or NOS2, or in H3 acetylation (Fig. 4). Although lentiviral infections have been shown to specifically alter these parameters54,55,56,57, disruptions to butyrate-responsive pathways may not be the primary roadblock to complete immune restoration. Lentiviral infections induce widespread epigenetic changes58,59, many of which persist or rise anew after the initiation of ARV therapy60,61,62,63. Although HDAC inhibitors have been successfully used to reactivate viral reservoirs in latency reversal studies64,65,66, sodium butyrate treatment was not associated with increased viremia in our study (Fig. 1). The timing of ARV administration may also contribute to unique reconstitution hurdles. Early ARV initiation can limit many complications, including immune activation, inflammation, CD4+ T-cell loss, and gastrointestinal damage3,67; however, perturbations in the intestinal microbiome and microbial translocation remain evident37,68,69.
The administration of sodium butyrate in healthy mice is associated with a significant shift in beta diversity, with some evidence for a loss of butyrate-producing species47,48. Although the predominant loss of Ruminococacceae ASVs in our butyrate-supplemented animals (Fig. 4i) complements these previous observations, we did not observe significant shifts in beta diversity (Fig. 4j). In addition to an absence of immunological perturbations (Figs. 1, 2, 3, 4), these findings suggest that there remains an impediment to butyrate signaling in ARV-treated, SIV-infected non-human primates which cannot be overcome by supplementation alone. This impediment may originate from within the microbiome itself, with ratios of butyrate producers to non-producers contributing to butyrate saturation thresholds53. However, as butyrate-enhanced reductions in intestinal permeability and microbial translocation are at least partially independent of the microbiome48—directed largely through HIF1a (hypoxia-inducible factor 1-alpha) and PPARg signaling14,48—non-efficacy in our supplemented animals may reflect an uncharacterized disruption in these host-intrinsic pathways. Lastly, there remains the possibility that sodium butyrate was unable to reach sufficient concentrations within the intestinal lumen of our ARV-treated macaques, which could be remedied in future studies with the use of the butyrate prodrug tributyrin70.
In summary, we demonstrate that sodium butyrate supplementation of ARV therapy in chronically SIV-infected macaques is insufficient to improve immune reconstitution. Butyrate treatment was well-tolerated but unable to alter cellular and transcriptional measures of immunity and did not suppress microbial translocation. Further work is needed to determine the utility of supplemental butyrate to improved outcomes in the context of ARV therapy, with and without adjunct probiotics.
Methods
Animals and vancomycin treatment
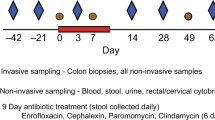
Study design and data reporting in this manuscript follows the recommendations in the (ARRIVE) guidelines. Thirteen SIV-infected male rhesus macaques (Macaca mulatta), aged 6–15, were assigned to an ARV-only (n = 6) or ARV + Butyrate treatment (n = 7) group as in Table 1, with sample size based on previous studies of experimental manipulations of disease progression in the macaque model. Groups were stratified by weight and genotype (Mamu-A*001, -A*002, -B*008, and -B*017) and animals sampled as mixed populations. All animals were infected with SIVmac239X for greater than 136 days. Animals were treated with 1 mL/kg/day s.c. of emtricitabine (40 mg/kg; Hangzhou API Chem), tenofovir disoproxil fumarate (5.1 mg/kg; Hangzhou API Chem), and dolutegravir sodium (2.5 mg/kg; Hangzhou API Chem) solubilized in 15% (w/w) Kleptose (Roquette) in water71. Two weeks after ARV treatment initiation, ARV + Butyrate animals received sodium butyrate (Bodybio) as a single oral tablet (600 mg) mixed with food treats. Dose was chosen based on the commercial availability of over-the-counter tablets within the desired range of 500–1000 mg/day—a range which corresponds to 50–100% of the lowest daily demand reported in the adult intestinal lumen72. Respective treatments were continued throughout the duration of the study. The NIAID Division of Intramural Research Animal Care and Use Program, as part of the NIH Intramural Research Program, approved all experimental procedures (animal study protocol LVD 26). The Program complies with all applicable provisions of the Animal Welfare Act and other federal statutes and regulations relating to animals.
Animals were housed and cared for at the NIH Animal Center, under the supervision of the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited Division of Veterinary Resources and as recommended by the Office of Animal Care and Use Nonhuman Primate Management Plan. Husbandry and care met the standards set forth by the Animal Welfare Act, Animal Welfare Regulations, as well as The Guide for the Care and Use of Laboratory Animals (8th Edition). The physical conditions of the animals were monitored daily. Animals in this study were exempt from contact social housing due to scientific justification, per institutional animal care and use committee (IACUC) protocols, and were housed in non-contact, social housing where primary enclosures consisted of stainless-steel primate caging. Animals were provided continuous access to water and offered commercial monkey biscuits twice daily as well as fresh produce, eggs and bread products twice weekly and a foraging mix consisting of raisins, nuts and rice thrice weekly. Enrichment to stimulate foraging and play activity was provided in the form of food puzzles, toys, cage furniture, and mirrors or television.
Plasma viral RNA
Viral load assessed as previously described10.
Sample collection
Samples were collected as previously published10,36,55,73. Blood, BAL, stool, and biopsies from colon, mesenteric lymph nodes, jejunum, and rectum were collected longitudinally or at necropsy (day 180 post-treatment). Sampling occurred in random order. Neither the investigators nor the animal handlers were blinded to group allocation to ensure multi-lateral supervision of design and palliative treatment. Animals were sedated with Ketamine HCl at 10 mg/kg intramuscular (i.m.) for longitudinal blood sampling or with Telazol at 3–4 mg/kg i.m. for tissue timepoints. For jejunal biopsies, animals were further anesthetized with isoflurane gas by intubation, to effect. Successful anesthetization was monitored by response to stimuli.
Euthanasia was initiated at experimental endpoint (day 180 post-treatment) using protocols consistent with the American Veterinary Medical Association (AVMA) guidelines. Animals were first sedated with Telazol at 4 mg/kg i.m., followed by Pentobarbital at 80 mg/kg to achieve euthanasia. No animals met clinical endpoint criteria as defined by: (a) loss of 25% body weight from baseline weight when assigned to the protocol, (b) major organ failure or medical conditions unresponsive to treatment, (c) complete anorexia for 4 days or an inability to feed or drink sufficient nutrients to maintain body weight without assistance for 7 days, (d) distress vocalization unresponsive to treatment or intervention for 7 days, or (e) tumors arising from other than experimental means that grew in excess of 10% of body weight, impaired movement, or ulcerated.
For BAL collection, silicone tubing was directed into the trachea with the assistance of a laryngoscope, whereupon warmed normal saline was instilled and subsequently aspirated for collection. For longitudinal RBs, fecal material was removed from the rectum and biopsies obtained with biopsy forceps. Longitudinal MLN biopsies were obtained by laparoscopy and longitudinal jejunal biopsies by video-guided endoscopy, with samples collected by biopsy forceps. 10 intestinal pinch biopsies were obtained per animal for longitudinal assessments. Biopsies collected at necropsy were directly accessed immediately post-mortem. Biopsies were maintained in RPMI prior to processing.
Approximately 1 mL of macaque stool was collected fresh from each animal by inserting a sterile swab 2 cm into the rectum and spinning to collect available sample. Collected feces were snap-frozen and stored at − 80 °C until accession.
Biopsies were maintained in RPMI-1640 medium for transport and rinsed twice with PBS prior to processing. Plasma was isolated from blood by centrifugation. Mononuclear cells were isolated from blood by Ficoll gradient centrifugation and from BAL and tissue biopsies by straining/grinding samples through a 0.22 μm cell strainer. Three of 10 intestinal pinch biopsies were transferred to soil-grinding Precellys tubes (Bertin Technologies, France) and homogenized in 1 mL TRIzol (ThermoFisher Scientific, USA) at room temperature on a Precellys 24 homogenizer at 5000 revolutions per minute (RPM) in 4 successive 20 s intervals. TRIzol-preserved homogenates were immediately transferred to − 80 °C for storage.
Immune phenotyping and functional assessment
Polychromatic flow cytometry and cell sorting were performed on stained mononuclear cells as previously described36, using a BD LSRII (FACSDiva v9.0). Cell were stimulated for functional assessment by overnight culture with phorbol myristate acetate (2.5 ng/mL) and ionomycin (1 μg/mL), in the presence of brefeldin A (1 μg/mL). Antibodies against the following antigens were used for staining at predetermined concentrations: CCR5 (clone 3A9), CD20 (2H7), CD28 (CD28.2), CD3 (SP34-2), CD45 (D058-1283), CD69 (FN50), Ki-67 (B56), and TNFa (MAb11) from BD; CD103 (BLy7), CD4 (OKT4), CD8 (SK1), IFNg (e450), and IL-17 (eBio64DEC17) from Thermo Fisher-Scientific; CD95 (DX2) and IL-2 (MQ1-17H12) from Biolegend; and FOXP3 (3G3) from Miltenyi.
Cell viability was assessed using the Live/Dead Aqua Fixable Dead Cell Stain (Thermo Fisher-Scientific). CD4+ and CD8 + TM were defined as CD95 + singlet, clean, live, CD3 + lymphocytes with positive/negative gating based on clearly grouped populations, historical-determined expression, and the use of internal controls (Supplementary Fig. S2). A threshold of 100 collected events in the parent population was utilized for all subset expression analyses (FlowJo 9.9.6).
ELISAs
Concentrations of iFABP2, zonulin, and sCD14 were quantified from plasma and acetylated histone H3 was quantified from colon intestinal homogenate histone extracts using commercially available ELISA kits (My Biosource #MBS740424, Alpco #30-ZONSHU-E1, R&D #DC140, and abcam #ab115102, respectively), according to the manufacturers’ protocols. Histone extracts were normalized to 200–400 ng/uL by BCA protein assay (Millipore). All samples were assessed as technical duplicates. iFABP2 and sCD14 were independently confirmed (data not shown).
RNA extraction, quantification, and assessment
For intestinal transcript quantification TRIzol-preserved samples were thawed and treated with 200 μL chloroform to separate nucleic acid into an aqueous phase. Following separation, Total RNA was isolated from the aqueous phase using the MagMAX-96 total RNA isolation kit (ThermoFisher Scientific) per the manufacturer's protocol. RNA concentration and purity (A260/280 ≥ 1.8) were assessed by spectrophotometer prior to cDNA generation and normalized to 50–100 ng/μL in PCR-grade H2O. For transcript quantification by NanoString, preparation, hybridization, and detection of RNA samples were carried out by following the NanoString manufacturer’s instructions (NanoString Technologies) using the nCounter NHP Immunology Panel. Subsequent analyses were performed using the nSolver (v4.0.70) analysis system (NanoString Technologies). Reads from NanoString were normalized to internal positive and negative controls and housekeeping genes. For visualization by heatmap, transcript data were clustered by Euclidian distance with Ward's minimum distance linkage. Transcriptome variability was visualized by PCA using the ggbiplot package (v0.55) in RStudio (v1.1.463) using R (v3.6.2). NanoString quantified transcripts were further ascribed to canonical gene expression pathways by IPA (Qiagen v01-19-02). The relative expression of individual transcripts PPARG and NOS2 was assessed from the NanoString generated counts, normalized to endogenous glucuronidase beta (GUSB).
16S isolation and analysis
16S rDNA was isolated and sequenced as previously described73. Raw Illumina FASTQ files were first demultiplexed using a custom Python script. Returned paired-end FASTQ reads were filtered and processed using the DADA2 package (v1.14.1) in RStudio using R to infer ASVs at a 99% identity threshold using the Silva database (v132). Before quality trimming 3,050,651 reads were included in 26 samples with an average of 117,333 reads per sample. Reads were trimmed to 225 bp (forward) or 200 bp (reverse) and filtered to exclude sequences with degenerate bases (N), more than 2 expected errors (maxEE), or chimerism. DADA2 quality trimming resulted in 2,056,542 reads for all the samples with an average of 79,098 reads per sample. 2 samples with less than 1000 reads were omitted from further analysis. ASVs identified as non-Bacteria, mitochondria (Rickettsiales Mitochondria), and Cyanobacteria were removed from further consideration as were resultant genera at less than 3% prevalence or phyla with no genera diversity. Longitudinally, differentially abundant taxa were identified by MetaLonDa (v1.1.8) in RStudio using R. Fully analyzed 16S miSeq data (n = 24) are deposited in the NCBI Sequence Read Archive (SRA) under project number PRJNA779879.
Immunohistochemistry
Five µm paraffin sections were incubated at 60 °C for 60 min, dewaxed in xylene and rehydrated in ethanol. Ag retrieval was performed in pH 6.0 0.00356 M citric acid solution in a steamer for 45 min. Slides were blocked in Intercept® (PBS) Blocking Buffer (LI-COR). Slides were stained with polyclonal Rabbit Anti-E. coli (Dako #B0357) and Spectral DAPI (AKOYA Biosciences). Images were acquired on a Leica SP8 inverted confocal microscope equipped with HyD hybrid detectors. Between 1 and 2 stained tissue slides of each sample were scored blindly by 4 individuals on a scale of 0 (no E. coli present) to 3 (present throughout the sample). Reported scores reflect the mean scores of both slides and individual scorers.
Statistical analyses
Longitudinal viral load was assessed by two-way ANOVA with Bonferroni’s multiple-comparison test (Prism v9.0, GraphPad Software Inc.). T-cell phenotype and function, ELISA analyte concentration, translocation index, individual transcript expression, histone H3 acetylation levels, and alpha diversity indices were assessed by paired and unpaired, two-way t-tests (Prism), as appropriate. Bulk transcript expression acquired by Nanostring was assessed within nSolver by t-test and included the Benjamini–Yekutieli false discovery rate. IPA canonical pathway enrichment and z-score determined by IPA using default parameters. Longitudinally, differentially abundant bacterial ASVs were identified by MetaLonDA. Differences in unweighted Unifrac beta-diversity were assessed by the adonis function in RStudio using R. No data that met minimum threshold requirements as outlined in the Methods were excluded.
Data availability
16S MiSeq data are deposited in the NCBI SRA under Project No. PRJNA779879. Other data available from this study are available from the corresponding author upon reasonable request.
References
Hunt, P. W., Lee, S. A. & Siedner, M. J. Immunologic biomarkers, morbidity, and mortality in treated HIV infection. J. Infect. Dis. 214(Suppl 2), S44-50. https://doi.org/10.1093/infdis/jiw275 (2016).
Cai, C. W. & Sereti, I. Residual immune dysfunction under antiretroviral therapy. Semin. Immunol. https://doi.org/10.1016/j.smim.2021.101471 (2021).
Deleage, C. et al. Impact of early cART in the gut during acute HIV infection. JCI Insight. https://doi.org/10.1172/jci.insight.87065 (2016).
Brenchley, J. M. et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371. https://doi.org/10.1038/nm1511 (2006).
Sandler, N. G. & Douek, D. C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 10, 655–666. https://doi.org/10.1038/nrmicro2848 (2012).
Estes, J. D. et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6, e1001052. https://doi.org/10.1371/journal.ppat.1001052 (2010).
Klase, Z. et al. Dysbiotic bacteria translocate in progressive SIV infection. Mucosal Immunol. 8, 1009–1020. https://doi.org/10.1038/mi.2014.128 (2015).
Kristoff, J. et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J. Clin. Invest. 124, 2802–2806. https://doi.org/10.1172/JCI75090 (2014).
Bourke, C. D. et al. Cotrimoxazole reduces systemic inflammation in HIV infection by altering the gut microbiome and immune activation. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aav0537 (2019).
Ortiz, A. M. et al. Experimental microbial dysbiosis does not promote disease progression in SIV-infected macaques. Nat. Med. 24, 1313–1316. https://doi.org/10.1038/s41591-018-0132-5 (2018).
Sandler, N. G. et al. Sevelamer does not decrease lipopolysaccharide or soluble CD14 levels but decreases soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol levels in individuals with untreated HIV infection. J. Infect. Dis. 210, 1549–1554. https://doi.org/10.1093/infdis/jiu305 (2014).
Allaire, J. M. et al. The intestinal epithelium: Central coordinator of mucosal immunity. Trends Immunol. 39, 677–696. https://doi.org/10.1016/j.it.2018.04.002 (2018).
Lopez, C. A., Kingsbury, D. D., Velazquez, E. M. & Baumler, A. J. Collateral damage: Microbiota-derived metabolites and immune function in the antibiotic era. Cell Host Microbe 16, 156–163. https://doi.org/10.1016/j.chom.2014.07.009 (2014).
Byndloss, M. X. et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. https://doi.org/10.1126/science.aam9949 (2017).
Vidali, G., Boffa, L. C., Bradbury, E. M. & Allfrey, V. G. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc. Natl. Acad. Sci. USA 75, 2239–2243. https://doi.org/10.1073/pnas.75.5.2239 (1978).
Candido, E. P., Reeves, R. & Davie, J. R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14, 105–113. https://doi.org/10.1016/0092-8674(78)90305-7 (1978).
Sealy, L. & Chalkley, R. The effect of sodium butyrate on histone modification. Cell 14, 115–121. https://doi.org/10.1016/0092-8674(78)90306-9 (1978).
Boffa, L. C., Vidali, G., Mann, R. S. & Allfrey, V. G. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J. Biol. Chem. 253, 3364–3366 (1978).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. https://doi.org/10.1038/nature12721 (2013).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. https://doi.org/10.1038/nature12726 (2013).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. https://doi.org/10.1126/science.1241165 (2013).
Yang, W. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11, 4457. https://doi.org/10.1038/s41467-020-18262-6 (2020).
Keir, M., Yi, Y., Lu, T. & Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 217, e20192195. https://doi.org/10.1084/jem.20192195 (2020).
Schulthess, J. et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 50, 432–445. https://doi.org/10.1016/j.immuni.2018.12.018 (2019).
Kumar, A. et al. The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J. Immunol. 182, 538–546. https://doi.org/10.4049/jimmunol.182.1.538 (2009).
Dillon, S. M. et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 7, 983–994. https://doi.org/10.1038/mi.2013.116 (2014).
Vujkovic-Cvijin, I. et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 5, 193ra191. https://doi.org/10.1126/scitranslmed.3006438 (2013).
Vujkovic-Cvijin, I. et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat. Commun. 11, 2448. https://doi.org/10.1038/s41467-020-16222-8 (2020).
Lozupone, C. A. et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 14, 329–339. https://doi.org/10.1016/j.chom.2013.08.006 (2013).
Dillon, S. M. et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS 31, 511–521. https://doi.org/10.1097/QAD.0000000000001366 (2017).
Serrano-Villar, S. et al. The effects of prebiotics on microbial dysbiosis, butyrate production and immunity in HIV-infected subjects. Mucosal Immunol. 10, 1279–1293. https://doi.org/10.1038/mi.2016.122 (2017).
McHardy, I. H. et al. HIV Infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome 1, 26. https://doi.org/10.1186/2049-2618-1-26 (2013).
Mutlu, E. A. et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 10, e1003829. https://doi.org/10.1371/journal.ppat.1003829 (2014).
Vazquez-Castellanos, J. F. et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 8, 760–772. https://doi.org/10.1038/mi.2014.107 (2015).
Klatt, N. R. et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J. Clin. Invest. 123, 903–907. https://doi.org/10.1172/JCI66227 (2013).
Ortiz, A. M. et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated, SIV-infected macaques. Mucosal Immunol. 9, 458–467. https://doi.org/10.1038/mi.2015.75 (2016).
Tanes, C. et al. Gut microbiome changes associated with epithelial barrier damage and systemic inflammation during antiretroviral therapy of chronic SIV infection. Viruses. https://doi.org/10.3390/v13081567 (2021).
Strauch, U. G. et al. Integrin alpha E(CD103)beta 7 mediates adhesion to intestinal microvascular endothelial cell lines via an E-cadherin-independent interaction. J. Immunol. 166, 3506–3514. https://doi.org/10.4049/jimmunol.166.5.3506 (2001).
Sathaliyawala, T. et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197. https://doi.org/10.1016/j.immuni.2012.09.020 (2013).
Favre, D. et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 5, e1000295. https://doi.org/10.1371/journal.ppat.1000295 (2009).
Favre, D. et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2, 32ra36. https://doi.org/10.1126/scitranslmed.3000632 (2010).
Ericsen, A. J. et al. Microbial translocation and inflammation occur in hyperacute immunodeficiency virus infection and compromise host control of virus replication. PLoS Pathog. 12, e1006048. https://doi.org/10.1371/journal.ppat.1006048 (2016).
Nganou-Makamdop, K. et al. Translocated microbiome composition determines immunological outcome in treated HIV infection. Cell 184, 3899–3914. https://doi.org/10.1016/j.cell.2021.05.023 (2021).
Vinton, C. L. et al. Biomarkers of cellular stress do not associate with sCD14 in progressive HIV and SIV infections in vivo. Pathog. Immunol. 5, 68–88. https://doi.org/10.20411/pai.v5i1.363 (2020).
Alex, S. et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell Biol. 33, 1303–1316. https://doi.org/10.1128/MCB.00858-12 (2013).
Geiss, G. K. et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 26, 317–325. https://doi.org/10.1038/nbt1385 (2008).
Jiminez, J. A., Uwiera, T. C., Abbott, D. W., Uwiera, R. R. E. & Inglis, G. D. Butyrate supplementation at high concentrations alters enteric bacterial communities and reduces intestinal inflammation in mice infected with Citrobacter rodentium. mSphere. https://doi.org/10.1128/mSphere.00243-17 (2017).
Fachi, J. L. et al. Butyrate protects mice from Clostridium difficile-induced colitis through an HIF-1-dependent mechanism. Cell Rep 27, 750–761. https://doi.org/10.1016/j.celrep.2019.03.054 (2019).
Sim, J. H., Mukerji, S. S., Russo, S. C. & Lo, J. Gastrointestinal dysfunction and HIV comorbidities. Curr. HIV/AIDS Rep. 18, 57–62. https://doi.org/10.1007/s11904-020-00537-8 (2021).
d’Ettorre, G. et al. Probiotic supplementation promotes a reduction in T-cell activation, an increase in Th17 frequencies, and a recovery of intestinal epithelium integrity and mitochondrial morphology in ART-treated HIV-1-positive patients. Immunol. Inflamm. Dis. 5, 244–260. https://doi.org/10.1002/iid3.160 (2017).
Kelly, C. J. et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671. https://doi.org/10.1016/j.chom.2015.03.005 (2015).
Department of Health and Human Services.
Clark, R. L. et al. Design of synthetic human gut microbiome assembly and butyrate production. Nat. Commun. 12, 3254. https://doi.org/10.1038/s41467-021-22938-y (2021).
George, M. D., Sankaran, S., Reay, E., Gelli, A. C. & Dandekar, S. High-throughput gene expression profiling indicates dysregulation of intestinal cell cycle mediators and growth factors during primary simian immunodeficiency virus infection. Virology 312, 84–94. https://doi.org/10.1016/s0042-6822(03)00207-1 (2003).
Flynn, J. K. et al. Luminal microvesicles uniquely influence translocating bacteria after SIV infection. Mucosal Immunol. 14, 937–948. https://doi.org/10.1038/s41385-021-00393-8 (2021).
Lusic, M., Marcello, A., Cereseto, A. & Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 22, 6550–6561. https://doi.org/10.1093/emboj/cdg631 (2003).
Benkirane, M. et al. Activation of integrated provirus requires histone acetyltransferase: p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 273, 24898–24905. https://doi.org/10.1074/jbc.273.38.24898 (1998).
Jochems, S. P. et al. DNA methylation changes in metabolic and immune-regulatory pathways in blood and lymph node CD4+ T cells in response to SIV infections. Clin. Epigenet. 12, 188. https://doi.org/10.1186/s13148-020-00971-w (2020).
Turner, A. W. & Margolis, D. M. Chromatin regulation and the histone code in HIV latency. Yale J. Biol. Med. 90, 229–243 (2017).
Corley, M. J. et al. Abrupt and altered cell-type specific DNA methylation profiles in blood during acute HIV infection persists despite prompt initiation of ART. PLoS Pathog. 17, e1009785. https://doi.org/10.1371/journal.ppat.1009785 (2021).
Jefferys, S. R. et al. Epigenomic characterization of latent HIV infection identifies latency regulating transcription factors. PLoS Pathog. 17, e1009346. https://doi.org/10.1371/journal.ppat.1009346 (2021).
Moron-Lopez, S. et al. The genome-wide methylation profile of CD4+ T cells from individuals with human immunodeficiency virus (HIV) identifies distinct patterns associated with disease progression. Clin. Infect. Dis. 72, e256–e264. https://doi.org/10.1093/cid/ciaa1047 (2021).
Yates, K. B. et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 22, 1020–1029. https://doi.org/10.1038/s41590-021-00979-1 (2021).
Archin, N. M. et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485. https://doi.org/10.1038/nature11286 (2012).
Rasmussen, T. A. et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 1, e13-21. https://doi.org/10.1016/S2352-3018(14)70014-1 (2014).
Sogaard, O. S. et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 11, e1005142. https://doi.org/10.1371/journal.ppat.1005142 (2015).
Sereti, I. et al. Persistent, albeit reduced, chronic inflammation in persons starting antiretroviral therapy in acute HIV infection. Clin. Infect. Dis. 64, 124–131. https://doi.org/10.1093/cid/ciw683 (2017).
Sortino, O. et al. Impact of acute HIV infection and early antiretroviral therapy on the human gut microbiome. Open Forum Infect. Dis. 7, ofz367. https://doi.org/10.1093/ofid/ofz367 (2020).
Rocafort, M. et al. Evolution of the gut microbiome following acute HIV-1 infection. Microbiome 7, 73. https://doi.org/10.1186/s40168-019-0687-5 (2019).
Heidor, R., Ortega, J. F., de Conti, A., Ong, T. P. & Moreno, F. S. Anticarcinogenic actions of tributyrin, a butyric acid prodrug. Curr. Drug Targets 13, 1720–1729. https://doi.org/10.2174/138945012804545443 (2012).
Del Prete, G. Q. et al. Short communication: Comparative evaluation of coformulated injectable combination antiretroviral therapy regimens in simian immunodeficiency virus-infected rhesus macaques. AIDS Res. Hum. Retroviruses 32, 163–168. https://doi.org/10.1089/AID.2015.0130 (2016).
Banasiewicz, T., Domagalska, D., Borycka-Kiciak, K. & Rydzewska, G. Determination of butyric acid dosage based on clinical and experimental studies: A literature review. Prz. Gastroenterol. 15, 119–125. https://doi.org/10.5114/pg.2020.95556 (2020).
Ortiz, A. M. et al. Antiretroviral therapy administration in healthy rhesus macaques is associated with transient shifts in intestinal bacterial diversity and modest immunological perturbations. J. Virol. https://doi.org/10.1128/JVI.00472-19 (2019).
Acknowledgements
We would like to acknowledge Heather Kendall, JoAnne Swerczek, Richard Herbert, and all the veterinary staff at the NIH animal center for their excellent veterinary care. We thank the NIAID Microbiome Program, NIAID Bioinformatics and Computational Biosciences Branch, and NCI Quantitative Molecular Diagnostic Core for technical and analytical assistance.
Funding
Open Access funding provided by the National Institutes of Health (NIH). Funding for this study was provided in part by the Division of Intramural Research/NIAID/NIH. The content of this publication does not necessarily reflect the views or policies of DHHS, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
A.M.O., J.S., C.A.L., P.J.B., C.A., K.B., J.K.F., C.L.V., A.R.R., and J.M.B. performed experiments. J.M.B. and A.M.O. conceived and designed the study. A.M.O., K.B., C.A., H.D.H., and J.M.B. analyzed data. H.D.H. provided valued technical assistance and discussions. All authors contributed to writing the paper and approved the final version of this manuscript to be published and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ortiz, A.M., Simpson, J., Langner, C.A. et al. Butyrate administration is not sufficient to improve immune reconstitution in antiretroviral-treated SIV-infected macaques. Sci Rep 12, 7491 (2022). https://doi.org/10.1038/s41598-022-11122-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11122-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
